

Abstract
Aims
Although endothelial cell senescence is known to play an important role in the development of cardiovascular pathologies, mechanisms that attenuate this process have not been extensively investigated. The aim of this study was to investigate whether SIRT6, a member of the sirtuin family of NAD+-dependent protein deacetylases/ADP-ribosyltransferases, protects endothelial cells from premature senescence and dysfunction, and if so which is its mode of action.
Methods and results
mRNA expression analysis demonstrated comparable levels of SIRT1 and SIRT6 transcripts in endothelial cells derived from different vascular beds and significantly higher levels of SIRT6 in these cells relative to those in haematopoietic progenitor cells. SIRT6 depletion by RNA interference in human umbilical vein endothelial cells (HUVEC) and aortic endothelial cells reduced cell proliferation, increased the fraction of senescence-associated-β-galactosidase-positive cells, and diminished the ability of the cells to form tubule networks on Matrigel. Further examination of SIRT6-depleted HUVEC demonstrated higher intercellular-adhesion molecule-1 (ICAM-1) and plasminogen-activator inhibitor-1 mRNA, lower levels of endothelial nitric oxide synthase mRNA and protein, higher ICAM-1 surface expression, and up-regulation of p21. Fluorescence microscopy of SIRT6-depleted HUVEC stained with anti-phospho-histone H2A.X and anti-telomere-repeat-binding-factor-1 antibodies showed evidence of increased nuclear DNA damage and the formation of telomere dysfunction-induced foci.
Conclusion
This work demonstrates that the presence of SIRT6 in endothelial cells confers protection from telomere and genomic DNA damage, thus preventing a decrease in replicative capacity and the onset of premature senescence. These findings suggest that SIRT6 may be important to maintain endothelial homeostatic functions and delay vascular ageing.
Keywords: Sirtuin, Telomere, Ageing, Senescence, Endothelial cells
1. Introduction
Cellular senescence is a damage and stress response which locks up mitotically competent cells into a permanent form of growth arrest. Senescent cells undergo distinct changes in gene expression that impair cellular functions.1 In recent years, this phenomenon has been increasingly linked to the process of ageing and the development of cardiovascular pathologies.2 In endothelial cells, the loss of replicative capacity resulting from the senescent state militates against the integrity of the endothelium and impairs successful angiogenesis. Furthermore, the endothelial senescent phenotype is pro-inflammatory, pro-atherosclerotic, and pro-thrombotic.3
Sirtuins are a group of evolutionarily-conserved NAD+-dependent protein deacetylases and ADP-ribosyltransferases, implicated in the regulation of energy metabolism, stress responses, cell survival, and lifespan.4 In mammals, there are seven sirtuins which localize to different cellular compartments and regulate the function of a large variety of protein substrates. SIRT1, the most studied mammalian homologue, has been shown to protect endothelial cells from premature senescence5 and to regulate angiogenesis6 and vascular tone.7
More recently, a role for SIRT6 as a cytoprotective protein has begun to emerge. SIRT6 has been implicated in DNA repair,8,9 telomere maintenance,10 attenuation of inflammation,11 glucose homeostasis,12 and inhibition of obesity-induced metabolic dysfunction.13 However, its role in vascular cell biology has so far not been explored. Here, we demonstrate that SIRT6 is highly expressed by endothelial cells derived from different vascular beds and that its depletion by RNA interference (RNAi) induces nuclear DNA damage, telomere dysfunction, inhibition of cell replication, and a senescent phenotype.
2. Methods
2.1. Cells and culture conditions
Cryopreserved human umbilical vein endothelial cells (HUVEC), human aortic endothelial cells (HAEC), or human dermal microvascular endothelial cells) were supplied by Lonza (Slough, UK) and grown in their recommended media (EGM-2 or EGM-2MV) as previously described.14 Cord blood CD34+ cells were supplied by Stem Cell Technologies (London, UK) and cultured for 4 days in Stemspan medium (Stem Cell Technologies) supplemented with 2% human umbilical cord blood plasma, 40 ng/mL thrombopoietin (Insight Biotechnology, Wembley, UK), 50 ng/mL stem cell factor, 100 ng/mL Flt3 ligand, and 10 ng/mL interleukin-3 (all from R&D Systems, Abingdon, UK), to expand the haematopoietic progenitor cells (HPC) as previously described.15 Outgrowth endothelial cells (OEC) were derived from mononuclear cells isolated from umbilical cord blood. To this end, low-density mononuclear cells were purified by Ficoll-Paque (Pharmacia Biotec) discontinuous gradient centrifugation and cultured on fibronectin-coated 24-well plates (4 × 106 cells per well) in EGM-2 fortified with 10% foetal calf serum and 20 ng/mL VEGF-A. After four days in culture, non-adherent cells were removed and the remaining cells were fed with fresh medium every 3 days until a confluent monolayer was obtained (normally after 3 weeks in culture). OEC tested positive for Ulex europaeus lectin 1, VE-cadherin, and CD146, and negative for CD14, thus confirming their endothelial phenotype. All cultures were maintained at 37°C in a humidified incubator under 5% CO2/95% air. Unless otherwise indicated, experiments with endothelial cells were performed on early passage cultures.
2.2. Transfections
First- to third-passage HUVEC or HAEC were transfected at 70% confluence for 24 h with 20 nM siRNAs targeting human SIRT6 (On-TARGETplus SMART pool, Dharmacon) (S6-siRNA) or with a non-targeting control pool (NT-siRNA), using Hyperfect reagent (Qiagen). In short-term silencing experiments, once the transfection complexes were removed, the cultures were maintained in normal fresh medium for a further 24 h before analysis. In long-term silencing experiments 24 h after the first round of transfection, the cells were harvested by trypsinization and then subjected to two additional 24 h rounds of transfection, initiated at the time of seeding and separated by a 24 h intervening period of recovery in fresh medium. Following the last transfection, the cultures were maintained in fresh medium for a further 72 h period prior to analysis.
2.3. RNA expression analysis
Cellular RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA expression was measured by quantitative polymerase chain reaction (Q-PCR) as previously described,15 using the following gene-specific TaqMan probes from the Assay on Demand gene expression collection (Applied Biosystems, Warrington, UK): SIRT1 (Hs01009006_m1), SIRT6 (Hs00213036_m1), ICAM-1 (intercellular-adhesion molecule-1; Hs99999152_m1), eNOS (endothelial nitric oxide synthase; Hs00167166_m1), PAI-1 (plasminogen activator inhibitor-1; Hs01126606_m1), and TATA Box binding protein (TBP, Hs99999910_m1). Relative mRNA expression levels were calculated by the comparative cycle threshold (CT) method, using the CT values obtained for TBP as internal references.
2.4. Western blotting
Endothelial cells (∼7.5 × 105 cells) were washed with ice-cold phosphate buffered saline and scrapped of the dish into 100 μL lysis buffer containing 1% sodium dodecyl sulphate, 1 mM phenylmethylsulphonyl fluoride, Protease Inhibitor Cocktail (Sigma), and 25 mM Tris–HCl, pH 6.8. The resultant lysates were further disrupted by sonication for 10 s at an amplitude of 35%, using a VCX500 Ultrasonic Processor (Sonics & Materials, Inc.) and then centrifuged at 14 000g for 5 min to remove particulate material. Proteins (∼20 μg) in samples of the supernatant fraction were separated by LDS-10% polyacrylamide gel electrophoresis under reducing conditions and transferred to PVDF or nitrocellulose membranes (Invitrogen, Paisley, UK). SIRT6 was identified by immunoblotting using a rabbit polyclonal antibody raised against the C-terminus of human SIRT6 (Cell Signaling Technology, Danvers, MA, USA) at a 1:1000 dilution. p21 was detected with the rabbit monoclonal antibody 12D1 (Cell Signaling) at a 1:1000 dilution. Mouse monoclonal antibodies obtained from BD Transduction Laboratories (Oxford, UK) were used at 1:500 dilution to detect eNOS (clone 3/eNOS) and p16 (clone G175–405). To control for variations in protein loading, membranes were stripped and re-probed with rabbit IgGs against actin (Sigma-Aldrich) or tubulin (Cell Signaling Technology) as indicated, at a 1:3000 dilution. Immunoreactive bands were detected using horseradish peroxidase-labelled anti-rabbit or anti-mouse IgGs (Cell Signaling) and enhanced chemoluminescence. The relative intensity of the bands was quantified by scanning densitometry, using the Image J software (available at http://rsb.info.nih.gov/).
2.5. Bromodeoxyuridine incorporation
HUVEC were transfected in Lab-Tek II chamber slides (Nunc) as described above and then incubated for 24 h with 10 μM bromodeoxyuridine (BrdU) in normal medium. Cells were fixed with 4% paraformaldehyde for 10 min at 4°C, washed with PBS, and then treated with 2 N HCl for 10 min followed by 10 min neutralization with 0.1 M sodium tetraborate buffer, pH 8.5. After washing with PBS, slides were incubated for 30 min with 5% BSA in PBS containing 0.5% Tween 20 to block non-specific binding sites and then stained with an FITC-conjugated anti-BrdU mouse monoclonal antibody (clone PRB1, eBioscience Ltd, Hatfield, UK) at a 1:100 dilution. After antibody staining slides were washed extensively in PBS, they were counterstained with 150 nM 4′,6′-diamino-2-phenylindole (DAPI) in PBS and mounted with ProLong Gold antifade reagent (Invitrogen). Cells were viewed under a Nikon Eclipse 80i fluorescence microscope, using a ×20 objective and fluorescent filters appropriate for fluorescein and DAPI. The percentage of BrdU-labelled cells was determined by counting at least 400 nuclei in three randomly selected fields per slide.
2.6. Determination of SA-β-gal activity
Cytochemical staining for SA-β-gal was performed at pH 6, using the chromogenic substrate 5-bromo-4-chloro-3-indoly-β-d-galactopyranoside as previously described.16 Quantitative determination of SA-β-gal activity was carried out by flow cytometry, using the fluorigenic substrate 5-dodecanoylaminofluorescein di-β-d-galactopiranoside (C12FDG). Briefly, cell cultures were pretreated with 100 nM bafilomcyin A1 for 1 h followed by incubation with 33 μM C12FDG for a further hour. Cells were then harvested by trypsinization and analysed in an FC500 MPL flow cytometer, using the MXP/CXP software (Beckman Coulter) as previously described.16
2.7. Capillary tube network formation
Transfected cells were seeded on Matrigel-coated 96-well plates (3 × 104 cells per well) in 0.1 mL of EGM-2. After 24 h of incubation, tubule formation was viewed under an Axiovert 25CFL inverted microscope (Karl Zeiss, Germany) at ×40 magnification. Images were captured under phase contrast, using a Nikon DS-2Mv digital camera, and analysed using the Image J programme as previously described.17 Tubule length was determined by drawing a line along each tubule and measuring the line in pixels. Branch points were manually counted.
2.8. Detection of γH2AX foci and telomere dysfunction-induced foci
DNA damage foci and telomere dysfunction-induced foci (TIFs) were examined by immunofluorescence microscopy essentially as previously described.18 All incubation steps were carried out at room temperature. In brief, HUVEC grown on cover slips were fixed for 10 min with 4% paraformaldehyde in PBS, followed by permeabilization with 0.25% Triton X-100 in PBS for 10 min. Cells were subsequently washed with PBS, blocked for 1 h with 1% BSA (Sigma-Aldrich) in PBS containing 0.1% Tween-20, and then incubated for 1 h with an anti-telomere repeat binding factor-1 (TRF-1) mouse monoclonal antibody (clone TRF-78, Abcam) at a 1:1000 dilution, followed by a goat anti-mouse IgG-Alexa Fluor 488 conjugate (Invitrogen) at a 1:1000 dilution. Prior to incubation with the second primary antibody, cells were washed extensively in PBS/0.1% Tween-20, post-fixed with 4% paraformaldehyde for 15 min, washed again with PBS, and then incubated for a further 15 min with 25 mM glycine in PBS to inactivate the paraformaldehyde. Detection of γH2AX was carried out by incubation with a rabbit polyclonal antibody raised against a synthetic phosphopeptide mapping to residues surrounding Ser139 of human histone H2A.X (Cell Signaling) at a 1:100 dilution, followed by Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen) at a 1:5000 dilution. After antibody staining, cover slips were washed extensively in PBS/01% Tween-20 and nuclei were counterstained with DAPI, followed by mounting as described above. Samples were viewed with a Nikon Eclipse 80i microscope equipped with a Nikon S Fluor ×40/1.3NA oil objective and fluorescence filter sets appropriate for DAPI, fluorescein, and Texas Red (UV-2E/C, B-2E/C, and G-2E/C, respectively). Forty to 50 Z-stack fluorescence images were captured at 0.2 μm intervals with a Hamamatsu Orca 285 digital camera, using the Volocity 3D image analysis software (Perkin Elmers, Inc., version 5.5). High-resolution images were deconvolved using the Volocity Restoration module. To determine co-localization in three dimensions, Z-stacks were converted to voxels (volume pixels) and further analysed with the Volocity Co-localization module after image thresholding. The average green, red, and co-localized fluorescence (expressed as voxels per cell) and the percentage of TIF-positive cells (cells with five or more sites of co-localization) were determined by analysing at least 250 nuclei in 10 randomly selected fields per treatment.
2.9. Statistical analysis
Data are reported as means ± SD. Statistical analysis was performed using Graph Pad Prism (release 5). Results were evaluated by t-test or one- or two-way analysis of variance followed by Bonferroni's post hoc tests as appropriate. A value of P < 0.05 was considered to denote statistical significance.
3. Results
3.1. SIRT6 is expressed in endothelial cells derived from diverse vascular beds
To assess whether SIRT6 might play a relevant role in endothelial cells, we first compared the mRNA expression profile of this gene in different endothelial cell types, using the expression of SIRT1 in HPC as a reference. Figure 1 shows that HPC, OEC, and endothelial cells derived from different vascular beds express comparable levels of SIRT1. In contrast, SIRT6 levels were significantly higher in OEC than in HPC (Figure 1A) and in mature endothelial cells (Figure 1B). Furthermore, in the latter levels of SIRT1 and SIRT6 transcripts were comparable. These results suggested that in endothelial cells, SIRT6 serves an important function. Western blot analysis of HUVEC and HAEC cultures revealed that SIRT6 was also readily detectable at the protein level and that there was a moderate decrease in expression in late-passage cultures (Figure 1C). These results raised the possibility that SIRT6 may play a role in endothelial cell senescence.
Figure 1.

Expression of SIRT6 in different endothelial cell types. (A) and (B) SIRT1 and SIRT6 mRNA expression profiles. Results are expressed relative to SIRT1 levels in HPC. *P < 0.05, **P < 0.01 vs. SIRT6 in HPC; n = 3–6. (C) SIRT6 protein levels in early- and late-passage cells. Representative immunoblots are shown on the left; quantification is shown on the right. Relative SIRT6 levels were calculated as the ratio of the intensity of the SIRT6 bands to the intensity of the corresponding actin or tubulin bands and results are expressed as a percentage of the level of SIRT6 measured in early-passage cells. *P < 0.05; n = 3.
3.2. Silencing of SIRT6 reduces endothelial cell proliferation and induces premature senescence
To investigate the role of SIRT6 in endothelial cell senescence, we first silenced this gene in HUVEC, using RNAi. Reduction of SIRT6 protein levels in silenced cells was confirmed by western blotting (Figure 2A). Q-PCR analysis confirmed that this treatment did not affect other members of the sirtuin family (data not shown). SIRT6-silenced cultures showed decreased rates of proliferation and a reduction in the cell fraction passing through the S-phase (Figure 2B and C) but no increased caspase 3/7 activity, a measure of apoptotic cell death (Supplementary material online, Figure S1). Importantly, SIRT6-silenced cultures also displayed an increase in the proportion of SA-β-gal+ cells (Figure 3A). Comparison between untreated cells and cells transfected with the NT control siRNA pool demonstrated that this effect could not be attributed to stresses to which the cells could have been subjected during the transfection procedure (Figure 3A).
Figure 2.

Depletion of SIRT6 inhibits HUVEC replication. (A) Western blot analysis of SIRT6 protein expression in cells left untreated or harvested 24 h after transfection with either NT or S6 siRNAs. A representative immunoblot is shown on the left; quantification is shown on the right; n = 3. Relative SIRT6 levels were calculated as the ratio of the intensity of the SIRT6 bands to the intensity of the corresponding actin bands and results are expressed as a percentage of the level of SIRT6 measured in the untreated samples. (B) Growth curves and (C) BrdU labelling of transfected cultures; n = 3–4. **P < 0.01 and ***P < 0.001 vs. NT siRNA; n.s., non-significant.
Figure 3.
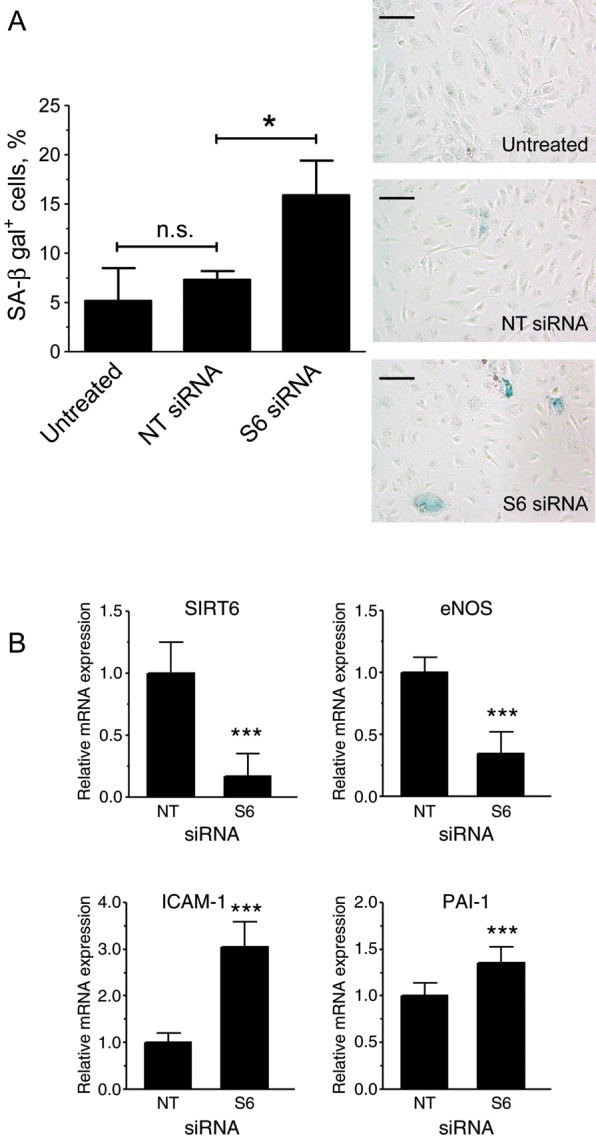
Depletion of SIRT6 in HUVEC induces premature senescence. (A) SA-β-gal staining after three serial transfections; quantification by flow cytometry is shown on the left (n = 4). Representative bright-field images of stained cultures are shown on the right. Bars = 100 μm. (B) mRNA expression analysis (n = 9). *P < 0.05 and ***P < 0.001; n.s., non significant.
To further substantiate these findings, we repeated the siRNA transfections in HAEC. In agreement with the results in HUVEC, also in these cells silencing of SIRT6 inhibited cell proliferation and increased the proportion of SA-β-gal+ cells (Supplementary material online, Figure S2). Taken together, these results demonstrate that, in endothelial cells, silencing of SIRT6 inhibits cell proliferation and induces premature senescence.
3.3. Silencing of SIRT6 induces a pathological phenotype
Endothelial cell senescence leads to changes in gene expression reminiscent of a pathological phenotype. Consistent with this notion, Q-PCR analysis showed that SIRT6-silenced HUVEC displayed higher levels of PAI-1 and ICAM-1 mRNA and lower levels of eNOS mRNA (Figure 3B). Similarly, testing for ICAM-I and eNOS by flow cytometry and immunobloting, respectively, showed corresponding changes at the protein level (Supplementary material online, Figure S3 and Figure 6).
Figure 6.

Depletion of SIRT6 affects eNOS and p21 protein levels. Western blot analysis of SIRT6, eNOS, and p21 protein expression in cells transfected with NT or S6 siRNAs. A representative immunoblot is shown on the left, and quantification is shown on the right. Relative protein levels were calculated as the ratio of the intensity of the indicated bands to the intensity of the corresponding tubulin bands and are expressed as a percentage of the values measured in the NT controls. *P < 0.05, **P < 0.01, and ***P < 0.001; n = 4.
3.4. Silencing of SIRT6 impairs capillary tube network formation
Since senescence is associated with impaired angiogenic functions,3 we examined whether SIRT6 depletion impinged on the ability of HUVEC or HAEC to form capillary-like networks in vitro. As shown in Figure 4, SIRT6-silenced cells formed less developed tubule networks, with both mean tubule length and the number of branch points significantly diminished.
Figure 4.

Depletion of SIRT6 inhibits in vitro angiogenesis. (A) Representative phase-contrast photomicrographs of tubules formed on Matrigel by NT and S6 siRNA-transfected HUVEC and HAEC. Bars = 200 μm. (B) Comparison of tubule parameters (length and branch points) by computer-assisted image analysis. Results are expressed as a percentage of the values measured in the NT controls. *P < 0.05; n = 5–6.
3.5. Silencing of SIRT6 in HUVEC induces DNA damage and telomere dysfunction without causing intracellular oxidative stress
To elucidate the mechanism by which depletion of SIRT6 reduced proliferation and induced premature senescence, we examined whether this treatment resulted in the accumulation of γH2AX foci, which are formed at sites of DNA damage and dysfunctional telomeres.19 Figure 5 shows that in SIRT6-depleted HUVEC, there was a significant increase in γH2AX foci. In addition, we found that as the frequency of γH2AX foci increased, the appearance of TIFs, i.e. sites where γH2AX foci co-localized with TRF-1 signals,19 also increased. These findings demonstrate that, in endothelial cells, SIRT6 attenuates DNA damage and telomere dysfunction.
Figure 5.

Depletion of SIRT6 promotes DNA damage and telomere dysfunction. (A) Representative deconvolved images of control (NTsiRNA)- and SIRT6 (S6 siRNA)-depleted HUVEC immunostained with antibodies against γH2AX (red) and TRF-1 (green) and counterstained with DAPI (blue). Arrows in the merged images point to sites of co-localization. The right-most small panel shows an enlarged view of the boxed area in the merged image. Bars = 5 μm. (B and C) Quantification of γH2AX foci and TIFs expressed as mean fluorescence voxels/cell. (D) Quantification of TIFs expressed as the percentage of cells with five or more TIFs/cell. Data represent the average of three experiments with >250 cells scored in each case. *P < 0.05, **P < 0.01.
Since oxidative stress is known to be a major cause of DNA and telomere damage in endothelial cells,20 we evaluated whether depletion of SIRT6 led to an increase in reactive oxygen species (ROS). We used flow cytometry with two different redox-sensitive probes: H2DCF-DA, which is oxidized predominantly by intracellular peroxides, peroxynitrite, and the hydroxyl radical, and DHE, which reacts with superoxide. As shown in Supplementary material online, Figure S4, ROS levels did not increase as a consequence of SIRT6 depletion.
3.6. Silencing of SIRT6 in HUVEC activates the p21 pathway
To further elucidate the downstream signalling mechanism by which SIRT6 depletion-induced senescence in endothelial cells, protein levels of the cyclin-dependent kinase inhibitors p21 and p16 were examined by western blotting. This analysis showed higher levels of p21 expression in SIRT6-depleted cells compared with controls (Figure 6). In contrast, p16 levels were undetectable under all the conditions tested (data not shown).
4. Discussion
The present study demonstrates that SIRT6 is highly expressed in endothelial cells where it confers protection from both genomic DNA damage and telomere dysfunction. These findings are consistent with studies in fibroblasts, embryonic stem cells, and tumour cell lines which have implicated SIRT6 in DNA repair mechanisms8,9,21 and in the maintenance of telomere stability.10
A major finding of this study is the demonstration that siRNA-mediated depletion of SIRT6 inhibits endothelial cell replication and leads to the onset of senescence. Induction of senescence by SIRT6 silencing was not only observed in HUVEC, a model system that is commonly used to study this process, but also in HAEC, an endothelial cell type that ages in vivo. In human endothelial cells, the senescent phenotype is pro-atherosclerotic, pro-thrombotic, and anti-angiogenic.3 Consistent with this notion, depletion of SIRT6 led to an increase in the mRNA levels of the pro-thrombotic factor PAI-1 and also in the mRNA and protein levels of the pro-inflammatory adhesion molecule ICAM-1. Furthermore, depletion of SIRT6 resulted in a reduction in eNOS expression and diminished the ability of the cells to form capillary-like tube networks in vitro. Taken together, these findings indicate that SIRT6 plays a prominent role in the maintenance of endothelial cell homeostatic functions. Our results also showed that the degree of SIRT6 depletion induced by RNAi (>75% on average) was generally larger than the magnitude of its functional effects. This raises the possibility that endothelial cells may have additional compensatory mechanisms, including redundancy in the function of other sirtuins, which could have attenuated the effects of the loss of SIRT6. In addition, the possibility that residual SIRT6 enzymatic activity might account for this discrepancy, or that the consequences of depletion take longer to become fully manifested, cannot not be ruled out at present.
Work on fibroblasts and keratinocytes suggests that the mechanism by which SIRT6 modulates senescence is cell-type-specific.10,11 In fibroblasts, depletion of this protein induces senescence by de-stabilizing telomeres through effects on telomeric chromatin,10 whereas in keratinocytes the effect involves a reduction in the attenuation of NFκB-mediated signalling.11 In endothelial cells, senescence can occur through several mechanisms, with genomic DNA damage and telomere dysfunction being pathophysiologically relevant (reviewed in Erusalimsky and Kurz20). Our work shows that both these processes are affected by SIRT6 in these cells. Whether interplay between NFκB and SIRT6 also plays a role in this case, or whether there might be differences in the regulation of senescence by SIRT6 in different endothelial cell types, remains to be established.
A major cause of DNA and telomere damage-induced senescence in endothelial cells is oxidative stress (reviewed in Erusalimsky and Kurz20). This raised the possibility that SIRT6 depletion affected the oxidative balance of the cell. However, experiments using two different redox-sensitive probes showed that ROS levels did not increase when SIRT6 was depleted, indicating that in this case, that was not the cause of senescence. An alternative possibility that remains to be explored is that SIRT6-induced histone deacetylation controls the access of DNA repair proteins to both the telomere and genomic DNA or that it regulates the DNA repair machinery in some other way; this would explain why its depletion leads to an increase in DNA and telomere damage.
It is generally accepted that the senescence response triggered by genomic DNA damage and telomere dysfunction is mediated by the same signalling events known to be involved in the classical DNA damage response. This response involves the activation of the ATM/ATR and Chk1/Chk2 protein kinase family members, the phosphorylation of the histone variant H2AX to form DNA damage foci and TIFs, the stabilization of p53 through phosphorylation, the increase in the expression of its downstream transcriptional target p21, and finally inhibition of the cyclin E-CDK2 complex by the latter.3 Thus, in our experiments, the detection of γH2AX foci and the up-regulation of the CDK inhibitor p21 indicate that in endothelial cells, SIRT6 depletion induces senescence via activation of the classical DNA damage response and the p21 pathway. The tumour-suppressor p16, which inhibits the cyclin D-CDK4/6 complexes, is another important mediator of senescence. Although the role of the p16 pathway in endothelial cells has remained controversial, some studies suggest that it is also involved in the response to telomere damage.20 In this context, it is noteworthy that, in the present study, we could not detect an up-regulation of p16 as a result of SIRT6 depletion.
Previous studies have shown that SIRT1 regulates endothelial homeostasis by acting at multiple levels, including inhibition of senescence by deacetylation of p535 and LKB1,22 and regulation of angiogenic functions via deacetylation of FOXO16 and NOTCH1.23 In contrast, SIRT1 has so far not been implicated in the regulation of genomic integrity in endothelial cells. Thus, the present study raises the possibility that, in these cells, SIRT1 and SIRT6 collaborate at different levels to maintain endothelial homeostasis, with SIRT6 playing primarily a cytoprotective role, regulating chromatin functions and DNA repair, and SIRT1, fine-tuning intracellular signalling networks. Further work on the nature of the signalling events regulated by SIRT6 will be required to verify this matter.
A salient observation of the present study is that although SIRT1 expression levels were found to be comparable in HPC, OEC, and mature endothelial cells, SIRT6 expression was higher in the latter. This raises the possibility that SIRT6 fulfils also a role in the establishment and/or maintenance of the endothelial phenotype.
The present study may have important implications in the search of strategies to counteract the development of age-associated vascular pathologies. Studies in mice have shown that SIRT6 deficiency produces premature ageing phenotypes,8 whereas SIRT6 overexpression causes a moderate increase in the lifespan of male mice.24 In this context, our findings that depletion of SIRT6 in endothelial cells induces a senescent phenotype suggest that increasing the levels or activity of this protein may be a relevant approach to delay vascular ageing.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by a Cardiff Metropolitan University Vice-Chancellor's Doctoral Scholarship (A.U.).
Supplementary Material
References
- 1.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 2.Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40:634–642. doi: 10.1016/j.exger.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009;106:326–332. doi: 10.1152/japplphysiol.91353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakagawa T, Guarente L. Sirtuins at a glance. J Cell Sci. 2011;124:833–838. doi: 10.1242/jcs.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 9.McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009;1:109–121. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, et al. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. 2010;9:162–173. doi: 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- 14.Kurz DJ, Hong Y, Trivier E, Huang HL, Decary S, Zang GH, et al. Fibroblast growth factor-2, but not vascular endothelial growth factor, upregulates telomerase activity in human endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:748–754. doi: 10.1161/01.ATV.0000069624.55424.61. [DOI] [PubMed] [Google Scholar]
- 15.Ahluwalia M, Donovan H, Singh N, Butcher L, Erusalimsky JD. Anagrelide represses GATA-1 and FOG-1 expression without interfering with thrombopoietin receptor signal transduction. J Thromb Haemost. 2010;8:2252–2261. doi: 10.1111/j.1538-7836.2010.03970.x. [DOI] [PubMed] [Google Scholar]
- 16.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 17.Donovan D, Brown NJ, Bishop ET, Lewis CE. Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis. 2001;4:113–121. doi: 10.1023/a:1012218401036. [DOI] [PubMed] [Google Scholar]
- 18.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 19.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 20.Erusalimsky JD, Kurz DJ. Endothelial cell senescence. Handb Exp Pharmacol. 2006;176(Pt 2):213–248. doi: 10.1007/3-540-36028-x_7. [DOI] [PubMed] [Google Scholar]
- 21.Mao Z, Hine C, Tian X, Van MM, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY, et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106:1384–1393. doi: 10.1161/CIRCRESAHA.109.215483. [DOI] [PubMed] [Google Scholar]
- 23.Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–238. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.