

Abstract
Several lines of evidence indicate that the etiology of late-onset Alzheimer’s disease (LOAD) is complex, with significant contributions from both genes and environmental factors. Recent research suggests the importance of epigenetic mechanisms in defining the relationship between environmental exposures and LOAD. In epidemiologic studies of adults, cumulative lifetime lead (Pb) exposure has been associated with accelerated declines in cognition. In addition, research in animal models suggests a causal association between Pb exposure during early life, epigenetics, and LOAD. There are multiple challenges to human epidemiologic research evaluating the relationship between epigenetics, LOAD, and Pb exposure. Epidemiologic studies are not well-suited to accommodate the long latency period between exposures during early life and onset of Alzheimer’s disease. There is also a lack of validated circulating epigenetics biomarkers and retrospective biomarkers of Pb exposure. Members of our research group have shown bone Pb is an accurate measurement of historical Pb exposure in adults, offering an avenue for future epidemiologic studies. However, this would not address the risk of LOAD attributable to early-life Pb exposures. Future studies that use a cohort design to measure both Pb exposure and validated epigenetic biomarkers of LOAD will be useful to clarify this important relationship.
Keywords: DNA methylation, epigenetics, epidemiology, Late-onset Alzheimer’s disease, lead exposure, Pb
ALZHEIMER’S DISEASE
General Alzheimer’s Disease Epidemiology
Alzheimer’s disease (AD) is a highly prevalent, progressive, and fatal neurodegenerative disease associated with aging. Clinical manifestation of AD includes progressive memory impairment and a gradual difficulty performing normal activities. A small percentage of cases, termed early-onset AD (EOAD), experience disease onset prior to age 60. EOAD cases are attributed to highly penetrant genetic mutations in amyloid pathway genes including amyloid precursor protein (APP) on chromosome 21, presenilin 1 (PSEN1) on chromosome 14, and presenilin 2 (PSEN2) on chromosome 1 [1, 2]. These mutations lead to the accumulation of β-amyloid plaques, a pathological hallmark of AD.
Termed late-onset AD (LOAD), the majority of AD cases are sporadic and symptoms manifest after age 60. Numerous low-penetrant genetic risk factors conferring a modest increase in risk of disease have been identified for LOAD, the most studied of which is the apolipoprotein ε4 allele (APOE-ε4). The global population prevalence of APOE-ε4 is 22%, while approximately 60% of LOAD cases carry at least one allele [3, 4]. Large, multi-center genome-wide association studies (GWAS) estimate the population attributable risk for APOE variants is 19-35% [5]. GWAS have identified additional polymorphisms associated with LOAD risk including genes for ABCA7, BIN1, CD2AP, CD33, CLU, CR1, EPHA1, MS4A, and PICALM [6-9], each associated with small increases in population attributable risk (PAR) ranging from 2-9.3% with a combined non-APOE PAR of 31-35%. Additional APOE ε4 dose adjustment reveals 50% of the PAR for LOAD is accounted for by known single nucleotide polymorphisms (SNPs) [8]. While these variants are important both for risk assessment and identification of novel mechanisms of pathogenesis, they are neither necessary nor sufficient for the development of LOAD.
Twin studies are an important epidemiologic tool for estimating the relative contribution of genetics and the environment in disease development. Incomplete twin concordance and variable age of onset supports a significant role for non-genetic factors in LOAD etiology. Among monozygotic twin (MZ) pairs, approximately 45-67% of twin pairs are concordant for LOAD [10-12]. Heritability of liability based on twin studies is estimated to be 58-79% [10, 12]. Linkage analysis reveals age at LOAD onset is partially genetically linked to regions on chromosomes 4 (208 cM) and 10 (139 cM) [13]. However, among a group of MZ pairs in which both twins develop the disease, differences in age of onset range from 4 to 16 years [10]. Both genetic and environmental factors likely contribute to LOAD development and time course.
Association studies have identified several non-genetic risk factors for LOAD, including depression [14], hypertension [15, 16], stroke [17], diabetes [15], hypercholesterolemia [15], obesity [18], head trauma [19], smoking [15, 20, 21], and having greater than 6 siblings [22]. Protective factors, those that reduce the risk of developing LOAD or delay the onset of LOAD, include physical activity [23, 24], social engagement [25], mental activity [25, 26], education (via the cognitive reserve hypothesis) [23, 25], statin use [27], non-steroidal anti-inflammatory drug (NSAID) use [23], moderate alcohol consumption [23, 28], coffee consumption [23], past vaccinations [29], and childhood residence in the suburbs relative to the city [22]. In particular, nutrition may play a protective role in LOAD onset. Consumption of one meal/week of fish rich in omega-3 fatty acids reduced the risk of developing AD by 60% in the Chicago Health and Aging Project [30]. Individuals with plasma vitamin E less than or equal to 21.0 μmol/L had a higher risk of incident dementia than individuals with plasma levels greater than or equal to 25.5 μmol/L [31]. The natural plant polyphenols curcumin and green tea epigallocatechin gallate (EGCG) have anti-oxidant and neuroprotective properties that may be protective against LOAD [32]. EGCG reduces APP translation through modulation of the intracellular iron pool in vitro neuroblastoma cell culture [33] and AD transgenic mice exposed in vivo to EGCG show reduced Aβ plaque density [34]. In an additional AD transgenic mouse model study, curcumin suppressed inflammation and oxidative damage in the brain and lowered levels of soluble Aβ and plaques [35]. Anthropometric measures of shorter adult knee height and arm span may reflect nutritional deficits in childhood [36] and women in the lowest quartile of arm span in the Cardiovascular Health Cognition cohort study had 1.5 times elevated risk of dementia [37].
Proposed environmental exposures associated with LOAD include aluminum [38, 39], copper [38, 40], zinc [38], mercury [41], lead (reviewed below), iron [32], pesticides [42, 43], solvents [44], electromagnetic field [45], and particulate matter in air pollution [46]. Environmental exposure studies have been underrepresented in the AD literature, likely due to the challenges of retrospective exposure assessment in older adults.
LEAD (PB) EXPOSURE
Overview of Pb Exposure as a Risk Factor
Zawia and colleagues have published a series of experimental studies on rodents and primates demonstrating that Pb exposure in early life results in late-life neuropathological changes similar to those of AD (reviewed elsewhere in this issue of Current Alzheimer’s Research). This work, coupled with the recognition that exposure to Pb in the general population until recently has been high, has heightened interest in the epidemiology of Pb exposure and neurodegenerative disease. We discuss trends in Pb exposure and epidemiologic studies that provide evidence for the role of Pb as a risk factor for AD.
Pb Exposure Epidemiology
One of the greatest environmental health successes of our society was the regulatory action to reduce what had been decades of high Pb exposure in the US [47]. Between 1976 and 1991, the mean blood Pb levels for people in the US dropped 78% from 12.8 μg/dL to 2.8 [48]. Since 1991 the standard elevated blood Pb level defining the need for action from Pb poisoning in children has been set to 10 μg/dL. The previously elevated mean blood Pb level of 12.8 μg/dL is a sobering testament to the high levels of Pb exposure endured by the general US population and other countries in the recent past.
Hazardous public health impacts remain despite low mean population blood Pb levels. One issue is that the general reduction in Pb exposure is not universal. Pockets of high Pb exposure remain in certain sectors of the US population where housing was constructed prior to 1950 and at which time leaded paint was used [49, 50], or where plumbing pipes and solder containing Pb have not yet been replaced [51]. Fly ash from municipal waste incineration contains high levels of heavy metals including Pb [52]. In the high temperatures of the trash incineration process, Pb is converted to the volatile PbCl2 compound, which can contaminate surrounding areas [52]. It is estimated that the US has hundreds of defunct Pb battery recycling sites [53] and Pb/zinc mines and smelters. The surrounding soil and water of these industrial sites are often contaminated with high levels of Pb, leading to human exposure [54], and many of these sites are designated Superfund sites on the National Priorities List by the US Environmental Protection Agency (EPA) [55, 56]. In many developing countries the combustion of leaded gasoline continues and industrial emissions of Pb have been increasing [57, 58]. Groups of people continue to experience high blood Pb levels based on their occupation or residential proximity to these hazards.
The pharmacodynamics of Pb in the human body makes past exposure to this heavy metal relevant to current and future health outcomes. Pb dust is inhaled or ingested and absorbed through the lung epithelia or gastrointestinal tract respectively. Pb is taken up by divalent metal transporters in the gut, binds tightly to heme molecules, and circulates throughout the body via blood. A small percentage of circulating Pb is highly toxic because it is free and bioavailable in the plasma [59]. Plasma Pb contributes to both soft tissue Pb as well as bone Pb deposition [60]. Pb can occupy both Ca2+ sites in the hydroxyapatite structure of bones [61] and greater than 95% of the adult body burden of Pb is stored in bones [61]. Given that cortical bone turns over at a slow rate of approximately 2% per year in healthy adults, Pb can be stored for decades in bone [62-64]. Storage in bone is not a permanent Pb detoxification mechanism as Pb can have direct effects on the cellular components of bone [65], and bone Pb can be mobilized in times of higher bone turnover such as during pregnancy, lactation, and osteoporosis [66]. Individuals born in the US prior to the Pb phase out in the 1970s may have accumulated elevated bone Pb stores that become mobilized in later life.
Biomarkers of Pb Exposure
Whole blood Pb is the most common biomarker of Pb exposure. The half-life of Pb in blood is relatively short, approximately 35 days [60]. This biomarker is best used for quantifying recent environmental exposures and mobilization of endogenous Pb [67]. Similarly, soft tissues also have relatively high turnover of Pb with a mean half-life of approximately 40 days, but soft tissue Pb quantification is invasive and not typically used for epidemiologic studies [60].
An expert panel on adult Pb toxicity convened by the US Centers for Disease Control concluded that bone Pb levels were the best biomarker of cumulative Pb exposure [68]. Spongy trabecular bone, such as that found in the patella, has an intermediate half-life of 5-15 years in adults [69]. More dense cortical bone, as in the tibia, has a much longer half-life of 10-30 years [70]. Thus, epidemiologic studies measuring Pb in bone can quantify a subject’s life history of cumulative Pb exposure.
Bone Pb levels can be measured either in vivo using Cd109 K-shell X-ray Fluorescence (KXRF) [68] or by direct, chemical measurement of Pb in excised total joint replacement or post-mortem bone samples [71]. Measurements with KXRF technology are painless and non-invasive, with minimal radiation exposure [69]. The KXRF instrument uses low-level gamma radiation to provoke emission of fluorescent photons from a subject’s tibia and patella [72]. The photons are detected and quantified over a spectrum of wavelengths from which the characteristic emission profile of Pb can be extracted [69]. Post-mortem bone samples can be acid digested and quantified for Pb levels using inductively coupled plasma mass spectrometry (ICP-MS) [73].
There are several approaches to predicting cumulative Pb exposure in the absence of direct bone Pb measures. The Park model incorporates blood Pb and information on subject demographics, medical history, and metabolic parameters to predict cumulative Pb exposure [74]. The Gorell system predicts cumulative Pb exposure based on blood Pb levels and physiologically based pharmacokinetic (PBPK) models incorporating industrial hygienist rated occupational Pb exposure for each job over the duration worked [75]. Both strategies have been validated with bone Pb measures.
Pb as a Neurotoxicant and Risk Factor for LOAD
Pb is a well-known neurotoxicant in children. Even at relatively low (subclinical) levels, epidemiologic studies demonstrate that childhood Pb exposure affects IQ and behavior with major impacts on IQ and functioning [47, 76].
A growing body of toxicological and population-based research indicates that cumulative environmental Pb exposure is neurotoxic in adults as well [77]. Pb exposure is a significant risk factor for accelerated declines in cognition [78, 79], an effect that a recent CDC panel concluded was likely causal [80]. The Veteran’s Affairs Normative Aging Study (NAS) is a longitudinal cohort of men free of disease when recruited in 1963. Based on data from repeated measures of bone Pb, blood Pb, and cognitive tests in the NAS, there are significant associations between high Pb exposure and decreased cognition. The cognitive domains associated with increased Pb exposure differ depending on the time of exposure. In a cross-sectional analysis, higher blood Pb was associated with reduced ability to recall and define words, identify line-drawn objects, and difficulty with a perceptual comparison test [81]. Both higher blood and bone Pb were associated with decreased spatial copying skill [81]. Higher bone Pb was associated with reduced pattern memory [81]. Longitudinal analyses confirm that the Pb associated declines in cognitive function are greater than changes observed with normal aging alone [82]. Also in the NAS, functional genetic polymorphisms in the δ-aminolevulinic acid dehydratase (ALAD) and hemochromatosis (HFE) genes modify the association between Pb and cognition measured by the Mini-Mental State Exam (MMSE), where variant carriers have more pronounced cognitive deficits associated with Pb exposure [83, 84]. Future research may study the ecological association between geographic regions with elevated Pb exposure and prevalence of LOAD.
Pb is also a risk factor for increased hippocampal gliosis measured by magnetic resonance spectroscopy in the NAS [85], an abnormality associated with LOAD development. Molecular epidemiology studies show cumulative Pb exposure is associated with an increased risk of amyotrophic lateral sclerosis [86-88] and Parkinson’s Disease [89], suggesting that Pb exerts a significant neurodegenerative effect. This effect may have specificity through epigenetic change as a pathogenic mechanism.
Toxicological studies are consistent with the epidemiologic research. Early life Pb exposure in animal models is associated with latent APP pathway dysregulation. Rats exposed to Pb in early life showed increased expression of APP mRNA and elevated Aβ aggregation without changes in α-, β-, or γ-secretases at 20 months of age [90, 91]. Similarly, primates exposed to Pb during the first 2 months of life only had significant adverse brain changes at 23 years of age when compared to their unexposed counterparts [92]. Pb exposed primates had increased amyloidogenesis, senile plaque deposition, and up-regulation of key proteins in the amyloid processing pathway, such as APP and beta-site APP-cleaving enzyme 1 (BACE1) [92].
OVERVIEW OF EPIGENETICS
Literally meaning “above the genome,” the epigenome comprises the heritable changes in gene expression that occur in the absence of changes to the DNA sequence itself. Epigenetic mechanisms include chromatin folding and attachment to the nuclear matrix, packaging of DNA around nucleosomes, covalent modifications of histone tails, and DNA methylation. The influence of regulatory small RNAs and micro RNAs on gene transcription is also increasingly recognized as a key mechanism of epigenetic gene regulation [93]. Epigenetic mechanisms are important in growth and cellular differentiation [94]. Epigenetic change can be stochastic [95] or internally orchestrated as part of aging [96]. Longitudinal change in global and gene-specific DNA methylation clusters within families, suggesting that there is genetic control of methylation status [97]. Inappropriate epigenetic changes are associated with many diseases including cancers [98], Rett syndrome [99], Beckwith-Wiedemann syndrome [100] and other imprinting disorders. Environmental signals can trigger epigenetic responses and may be an important mechanism by which environmental exposures are associated with disease [101]. Furthermore, epigenetic mechanisms may play an important role in the developmental origins of adult health and disease (DOHaD) by providing a mechanism underlying the latent effects of adverse fetal, infant, and childhood environments on late-life chronic disease [102-104].
Epigenetic Epidemiology and Alzheimer’s Disease
Epigenetic epidemiology is the study of the effects of heritable epigenetic changes on the occurrence and distribution of diseases in populations [105]. This research includes both trans-generational and intra-individual cellular epigenetic inheritance systems. Epigenetic changes are associated with epidemiologic risk factors such as aging [106, 107] and environmental exposures [101], as well as psychiatric outcomes [108] and neurodegeneration [109].
Evidence for the role of epigenetics in AD pathogenesis is found in human studies of various tissues, animal models, and cell culture [110-112]. Global changes associated with AD have been observed in DNA methylation, miRNAs, and histone modifications. A human post-mortem case-control study identified global DNA hypomethylation in the entorhinal cortex of AD subjects by quantifying the percentage of positive 5-methylcytosine neuronal nuclear immunoreactivity [113]. Within a single MZ twin pair discordant for AD, DNA from the temporal neocortex neuronal nuclei was hypomethylated in the AD twin compared to their cognitively normal twin using similar methods to the previous study [114]. An AD case-control study in the post-mortem human parietal lobe cortex revealed differential regulation of miR-NAs including miR-204, miR-211, and miR-44691 using a custom μParaflo array [115]. Age-matched AD cases have increased neuronal global phosphorylation of histone 3 relative to controls determined by immunolabeling in the hippocampus, a histone modification that suggests mitotic activation [116].
Given that epigenetics play an important regulatory role in gene expression, epigenetic dysregulation of important AD tau and amyloid processing pathway genes may point to a potential mechanism for AD disease progression. In experiments where neuroblastoma cells were cultured under low folate and vitamin B12 conditions, PSEN1 and BACE1 were hypomethylated, mRNA expression of BACE1 and PSEN1 was significantly induced, and Aβ production was increased [117]. Addition of S-adenosyl methionine (SAM) was able to restore BACE1 and PSEN1 expression to baseline levels, though DNA methylation reversal was incomplete [117]. An additional study using human neuroblastoma cells and male rat brain tissue shows APP mRNA expression is repressed by thyroid hormone (T3) sensitive histone modifications [118]. Treatment with T3 decreases H3K4 methylation and H3 acetylation at the APP promoter, leading to APP silencing that was reversed with histone deacetylase (HDAC) and histone lysine demethylase inhibitors [118].
There have been several candidate-gene methylation studies in LOAD cases and controls. In a post-mortem brain study of 26 controls and 44 LOAD cases with varying degrees of disease severity, no differences were seen in DNA methylation in regions associated with Microtubule Associated Protein Tau (MAPT), PSEN1, and APP, nor were differences detected between frontal cortex and hippocampal DNA [119]. Investigation of 6 familial AD frontal cortex and cerebellum brain samples revealed no methylation at the APP promoter in any case in either brain region [120]. These studies were limited by a candidate-gene approach and highlight the need for genome-wide assessment of DNA methylation.
It is critical that epigenetic epidemiology studies of AD epigenetics consider age as an independent predictor of epigenetic change as age-specific epigenetic drift has been observed at AD related loci among healthy normal controls. In a set of control parietal cortex samples, the promoter of APP was hypomethylated in individuals greater than 70 years of age relative to younger subjects [121]. DNA methylation upstream of the MAPT gene also varied with age in the control parietal cortex and was associated with an age-related decline in MAPT gene expression [122]. Specifically, MAPT promoter CpG dinucleotides located in the Sp1 transcriptional activator binding site were hypermethylated with age, while CpG dinucleotides located within the GCF transcriptional repressor binding region were hypomethylated with age [122]. Another study of postmortem cerebral cortex in 125 subjects ranging from 17 weeks of gestation to 104 years of age measured methylation by MethyLight PCR at candidate tag loci for 50 genes selected for their relevance to LOAD, CNS differentiation, and cancer. CpG sites in the promoters of eight genes showed robust linear increases in DNA methylation across the lifespan [123]. An additional study examined prefrontal cortex samples across a 30 year age range and noted that the average DNA methylation in promoters of MTHFR and APOE increased by 6.8% across the age range, while control samples decreased by 10.6% with age [124]. Given the likely epigenetic drift, clinical samples should be carefully matched on age.
Epigenetics and Heavy Metals, with a Focus on Pb
Epigenetic alterations have been observed following exposure to environmental metals [125], including arsenic, nickel, chromium, cadmium, and Pb. Perhaps the heavy metal most studied in the field of cancer epigenetic epidemiology is arsenic. In a population-based study of 351 individuals with bladder cancer, elevated toenail arsenic measurements are associated with increased tumor sample promoter methylation of RASSF1A and PRSS3 tumor suppressor genes [126]. Nickel, chromium, and cadmium epigenetics research has largely been in toxicologically based in vitro experiments. A cell line of human lung bronchoepithelial cells treated with nickel chloride show global histone modification changes including decreased H2A, H2B, H3, and H4 acetylation and increased H3K9 dimethylation [127]. When the same cell line is treated with chromium, the cells exhibit increased H3K9 dimethylation at the MLH1 gene promoter region, which correlates with decreased MLH1 mRNA expression [128]. Cadmium exposure in a rat liver cell line initially reduces DNA methyltransferase activity and global DNA methylation, but after 10 weeks of prolonged exposure, the cells show significant increases in DNA methyltransferase and global DNA methylation above the baseline [129].
Evidence suggests that Pb, in particular, may play a role in epigenetics throughout the life course. In a study of 103 mother-infant pairs, maternal cumulative Pb exposure was inversely associated with offspring umbilical cord genomic DNA methylation of Alu retrotransposable elements [130]. Similarly, bone Pb levels were inversely associated with peripheral blood genomic DNA methylation of LINE-1 retrotransposons in 517 elderly men from the NAS [131]. Individuals exposed to extremely high levels of Pb (51-100 μg/dL blood Pb) had higher methylation in the promoter of the p16 tumor suppressor gene [132]. Research is needed to expand this early epidemiologic work on global and candidate gene DNA methylation to more comprehensively understand specific pathways influenced by Pb exposure in humans.
Animal studies have investigated the relationship between Pb exposure and epigenetics. Early life exposure to Pb in primates causes dysregulation of biological pathways important to LOAD pathogenesis in late life and is associated with reduced DNA methyltransferase 1 (DNMT1) activity [92]. Rat pheochromocytoma cells exposed to Pb show dose dependent decreases in global methylation and decreases in APP promoter methylation at 4 CpG sites [133]. These changes were associated with increases in APP mRNA and Aβ protein levels [133]. Toxicological and epidemiological studies suggest that Pb exposure may be associated with epigenetic change, but further research is needed.
DATA INTEGRATING ALZHEIMER’S DISEASE, EPIGENETICS, AND PB EXPOSURE
Alzheimer’s and Pb Exposure are Associated with Changes in One-Carbon Metabolism, the Substrate for DNA Methylation
De novo and maintenance DNA methylation is dependent on available methyl (-CH3) groups. One-carbon metabolism reactions are reversible and deficiencies in methyl donors can cause DNA hypomethylation. For example, mice given diets deficient in the methyl donor choline show lower global brain methylation [134] and elevated expression of APP, consistent with promoter hypomethylation [135]. Epidemiologic studies indicate that AD patients have altered circulating levels of one-carbon metabolism members including homocysteine (HCY), SAM, folate, and vitamin B12. Elevated HCY is associated with increased risk of developing AD and increased rate of disease progression among individuals with the disease. Prospective data from the Framingham Heart Study show that each standard deviation increase in log transformed plasma total HCY levels was associated with an adjusted relative risk of dementia of 1.8 (95% CI: 1.3-2.5) eight years after the HCY measurement [136]. AD patients in the Oxford Project to Investigate Memory and Ageing have increased serum HCY relative to cognitively normal control subjects (n=164) and the individuals with the greatest disease progression over the subsequent three years had the highest original HCY levels [137]. SAM is a methyl-donor molecule that is hydrolyzed to form HCY, the substrate for DNA methylation. AD patients also have decreased cerebrospinal fluid SAM relative to cognitively normal controls [138].
Several proteins in the one-carbon metabolism cycle may be disturbed by Pb exposure because elemental Pb reacts with free sulfhydryl groups on proteins. HCY metabolism may be directly inhibited by Pb binding to the sulfhydryl group in HCY. Furthermore, HCY is transsulfurated into cysteine by cystathionine β-synthase (CBS) and CBS has two sulfhydryl groups with which Pb can react. There is also evidence for Pb’s involvement in methionine processing. Rats developmentally treated with Pb have impaired long-term potentiation (LTP), memory, and synaptic plasticity. Co-treatment with SAM and Pb increases LTP relative to Pb treatment alone and reduces circulating blood Pb levels [139]. Similarly, neuroblastoma cells exposed to Pb experience viability loss, glutathione antioxidant depletion, membrane lipid peroxidation, DNA damage, and apoptosis; pre-treatment with a methionine derivative reduces these harmful effects [140].
Pb exposure and HCY levels are linked in cross-sectional epidemiologic studies. In the Baltimore Memory and Aging Project involving greater than 1,000 adults, higher blood Pb was associated with higher HCY [141]. Analyses from the 1999-2002 National Health and Nutrition Examination Survey (NHANES) showed HCY was strongly associated (OR=1.92) with peripheral arterial disease (PAD) [142]. Subsequent analysis showed the original association was actually due to confounding from smoking, blood Pb and cadmium levels, and impaired renal function [142]. This suggests that the association between HCY and chronic disease may be driven by environmental exposures.
Animal Model Studies Linking Pb Exposure, Epigenetics, and Amyloidogenesis
A series of rat and primate model studies conducted by the Zawia research group collectively demonstrate that early life Pb exposure reduces DNA methyltransferase activity and specifically alters the regulation of many AD pathway related genes including APP and BACE1 that are known to be CpG rich. Rats exposed to Pb from post natal day (PND) 1 through PND 20 experienced a transient increase in APP mRNA expression in cortical brain tissue, which returned to basal levels at 1 year, and later resurged at 20 months of age in the absence of continued exposure [91]. The observed late-life rise in APP mRNA was accompanied by elevated Aβ, suggesting that early life Pb exposure may have long-term effects on amyloidogenesis in late life [91]. In a follow-up study on the same tissues, investigators noted the effects on Aβ formation and aggregations were not due to changes in protein levels of APP processing secretases [90]. In a third study using the early-life exposed rat brain tissues, elevated oxidative DNA damage measured by cerebral 8-hydroxy-2’-deoxyguanosine (8-oxo-dG) was observed in the exposed animals [143]. Local 8-oxo-dG is associated with hypomethylation at adjacent CpG sites [144]. Direct oxidation of 5-methylcytosine to 5-hydroxymethylcytosine may be part of active DNA demethylation [145]. Analogous primate experiments by Zawia et al., are consistent with these rodent findings. Primates exposed in early life to Pb had elevated levels of the Aβ peptide, 8-oxo-dG DNA, and mRNA from APP and BACE1 on autopsy 23 years later relative to controls, suggesting Pb is involved in LOAD-like pathology [92]. Brain tissue from these exposed primates also had 20% reduced DNA methyltransferase 1 activity [92] and lower methylation at the promoter of APP [146]. In vivo animal model studies spanning multiple organisms support an integrated role of Pb exposure and epigenetics in amyloidogenesis.
Challenges to LOAD Epidemiologic Research Integrating Epigenetics and Pb Exposure
Human epidemiologic research integrating LOAD, environmental exposure to Pb, and epigenetics faces many challenges. Clinical criteria for AD include progressive impairment in memory in the absence of motor, sensory, or coordination deficits [147]. However, the standard of diagnosis for AD requires the pathologic post-mortem identification of Aβ plaques and tau neurofibrillary tangles. Epidemiologic studies can take advantage of predictive and diagnostic biomarkers, including a panel of plasma signaling proteins [148], cerebrospinal fluid protein analyses [149], magnetic resonance imaging (MRI) volumetric and structural measures [150], and positron emission tomography (PET) neuroimaging of metabolic rate and Aβ pathology [151, 152]. However, these research methods require additional validation to become routine early detection methods [153].
Another concern in environmental epidemiology is that the length of time between exposure and disease onset. Barker first introduced the hypothesis that early-life conditions could be linked to late life chronic disease, otherwise known as the developmental origins of health and disease (DOHaD) hypothesis [154]. Fetal or childhood exposures have been associated with adverse health outcomes including impaired glucose tolerance [155] and hypertension [156, 157]. Indeed, several early life events related to growth, metabolism, and cognitive reserve have been associated with LOAD [158]. AD risk is increased with limited education and income, and both factors are associated with poor early life environment and growth [159]. Middle life risk factors including obesity [160], limited physical activity [161], and diabetes [162] are shared between AD and cardiovascular disease. Low birth weight and intrauterine growth restriction are related to metabolism, fat distribution, and insulin resistance at mid-life and it has been suggested that these early-life events may be associated with AD as well [163-165]. However, LOAD is a chronic disease of old age and a prospective developmental exposure study could not feasibly follow an early life cohort for 75 years with our current late stage diagnostic measures. Additionally, retrospective exposure assessment is difficult. The human body has efficient detoxification and clearance mechanisms for many toxicants and many chemicals do not bioaccumulate in the human body. There is an acute need to develop biomarkers that correspond to prior toxicologic exposures.
Finally, an additional roadblock is that brain specific epigenetic measurements are only possible post-mortem. Molecular epidemiology research of toxicant induced disease is strengthened when performed with relevant tissue samples. Brain tissue collection is invasive and not possible longitudinally on live subjects. Model animal research and epidemiology studies of human pre-mortem available tissues such as skin, blood, colon, etc. are necessary to fill in stages of disease tissue not available through end of life epidemiologic brain banks.
Potential Approaches to Study Pb Exposure, Epigenomics, and Alzheimer’s Disease Epidemiology
To best understand the relationship between Pb exposure (both early-life and later life) and LOAD, studies should take advantage of available biomarkers of Pb and technologic advances in epigenetic measurements. Bone Pb levels are a strong predictor of negative health outcomes including elevated risks for hypertension [166-168], ischemic heart disease [169], and mortality [170], but the relationship between cumulative Pb exposure and LOAD has not been assessed. Cumulative Pb exposure of LOAD subjects can be measured either non-invasively in vivo using K-x-ray fluorescence [68] or by direct measurement of Pb in bone samples [71]. At Alzheimer’s Disease Research Centers (ADRCs) where LOAD subjects consent to brain tissue donation on autopsy, it would be most ideal to directly measure Pb in samples of cranial bone obtained at the time of brain harvesting. Measurement of Pb in the cranium is highly correlated with a weighted average of skeletal Pb levels, as well as the level of Pb in tibia bone [171], the latter being the bone most commonly measured in epidemiologic studies of chronic Pb toxicity [68]. Sampling cranial bone Pb would make it possible to concurrently study LOAD epidemiology, brain tissue epigenetics and cumulative Pb exposure in post-mortem case-control studies.
Circulating epigenetic biomarkers would be useful to conduct case-control studies of Pb exposure (by in vivo KXRF) with live subjects. Post-mortem Alzheimer’s disease brain tissue epigenetic studies are expanding, but use of this tissue collected at end of life is not feasible to track within individual changes over time as in longitudinal epidemiological aging cohort studies. Biologically-available biomarkers would allow for repeated epigenetic measures throughout the disease course. Epigenetic markers in white blood cells (WBC) have been used as biomarkers in other diseases. Global DNA methylation has been associated with several cancers, myelodysplastic syndrome, and schizophrenia and thus does not appear to be a disease specific biomarker. Gene-specific methylation data and risk factor methylation data are more limited and results are inconsistent [172]. Larger, prospective cohort studies are needed to determine whether WBC gene-specific epigenetics will be informative with AD and with Pb exposure Fig. (1). Upon epigenetic biomarker development, cohort studies could integrate and target distinct age groups. Birth cohorts could investigate the role of in utero and postnatal Pb exposure on AD biomarkers to test the hypothesis suggested by animal research [146] that early life is a critical window for Pb’s influence on developmental reprogramming. Mid-life cohorts could focus on later exposure periods and could incorporate traditional AD risk factors such as hypertension status and education achieved. Late-life cohorts would involve the best AD and mild cognitive impairment (MCI) diagnostic tools and study the role of cumulative lifetime Pb exposure.
Fig. (1).
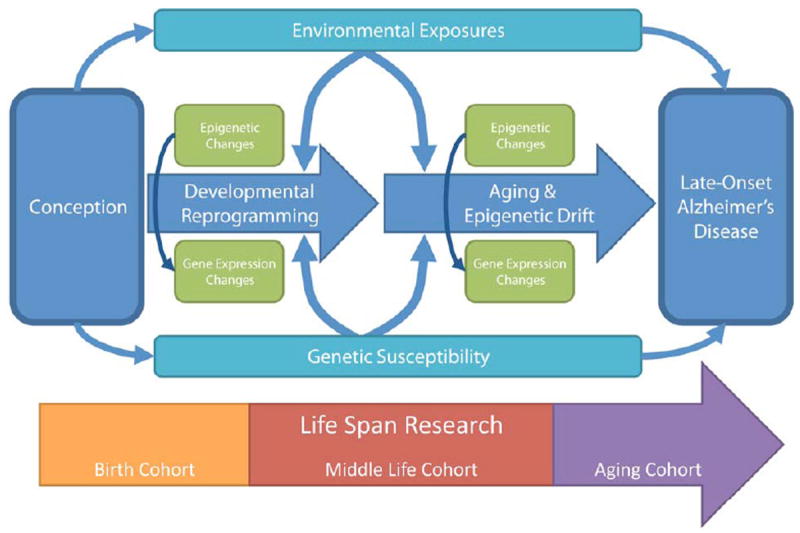
Conceptual diagram describing the relationship between environmental exposures, including to the heavy metal lead, with the development of late-onset Alzheimer’s disease. There is a complex interplay of genetics and epigenetic programming. Epidemiologic cohort studies can be designed to study different stages in the life course leading to disease development.
Finally, epidemiologic data needs to be incorporated with epigenetic studies on ADRC brain bank tissues. Epigenetic changes are associated with age [124], sex [173], exposures [174], and diseases [175]. Alzheimer’s disease specific epigenetic change may need to be extracted from a noisy background of age-specific epigenetic drift, sex-specific epigenetic marks, co-morbidity disease changes, and a lifetime of environmental exposures. The majority of existing studies of brain epigenetics focus on CpG islands and the application of array-based approaches that only cover a portion of the genome, largely in genic regions. Rapid advances in technology and reduction in costs have made new approaches using next-generation sequencing (NGS) feasible for larger sample sizes. These new approaches have been lauded as unbiased but criticized as relative (rather than quantitative) measures of DNA methylation. The depth of genome coverage will be able to provide the large amount of information needed to detect subtle changes from multiple sources. Integration of these data with ongoing studies of biomarkers in other neurodegenerative diseases and in non-diseased aging populations will help elucidate the specific epigenetic changes associated with LOAD, providing a foundation for prevention and treatment of this disease.
Acknowledgments
The authors would like to thank Zishaan Farooqui and Muna S. Nahar for manuscript editing assistance. This study was supported by the MADRC’s pilot fund, the University of Michigan NIEHS Center (P30ES017885), and the Michigan CTSA (UL1RR024986). Ms. Bakulski was supported by the Interdisciplinary Studies in Public Health and Aging NIA T32 (PI: Hu; T32AG027708). Funding for Dr. Dana C. Dolinoy was supported by ES017524.
ABBREVIATIONS
- AD
Alzheimer’s Disease
- LOAD
Late-Onset Alzheimer’s Disease
- EOAD
Early-Onset Alzheimer’s Disease
- GWAS
Genome Wide Association Study
- ADRC
Alzheimer’s disease research center
- NGS
Next generation sequencing
- NAS
Normative Aging Study
- HCY
Homocysteine
- 8-oxo-dG
8-Hydroxy-2’-Deoxyguanosine
- PND
Post Natal Day
- KXRF
K-shell X-ray Fluorescence
- DOHaD
Developmental Origins of adult Health and Disease
- WBC
White Blood Cell
- MCI
Mild Cognitive Impairment
- SNP
Single nucleotide polymorphism
- EGCG
Epigallocatechin gallate
- SAM
S-adenosyl methionine
- Pb
Lead
Footnotes
CONFLICT OF INTEREST
The authors have no conflict of interest associated with this manuscript.
Publisher's Disclaimer: DISCLAIMER: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
References
- 1.Bertram L. Alzheimer’s disease genetics current status and future perspectives. Int Rev Neurobiol. 2009;84:167–84. doi: 10.1016/S0074-7742(09)00409-7. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–9. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 3.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashford JW. APOE genotype effects on Alzheimer’s disease onset and epidemiology. J Mol Neurosci. 2004;23:157–65. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- 5.Ertekin-Taner N. Genetics of Alzheimer disease in the pre- and post-GWAS era. Alzheimers Res Ther. 2010;2:3. doi: 10.1186/alzrt26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 8.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011 doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011 doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatz M, Pedersen NL, Berg S, et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52:M117–25. doi: 10.1093/gerona/52a.2.m117. [DOI] [PubMed] [Google Scholar]
- 11.Nee LE, Lippa CF. Alzheimer’s disease in 22 twin pairs--13-year follow-up: hormonal, infectious and traumatic factors. Dement Geriatr Cogn Disord. 1999;10:148–51. doi: 10.1159/000017115. [DOI] [PubMed] [Google Scholar]
- 12.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 13.Li YJ, Scott WK, Hedges DJ, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–93. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saczynski JS, Beiser A, Seshadri S, et al. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology. 2010;75:35–41. doi: 10.1212/WNL.0b013e3181e62138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Wang YJ, Zhang M, et al. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology. 2011 doi: 10.1212/WNL.0b013e318217e7a4. [DOI] [PubMed] [Google Scholar]
- 16.Sharp SI, Aarsland D, Day S, Sonnesyn H, Ballard C. Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry. 2011;26:661–9. doi: 10.1002/gps.2572. [DOI] [PubMed] [Google Scholar]
- 17.Savva GM, Stephan BC. Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke. 2010;41:e41–6. doi: 10.1161/STROKEAHA.109.559880. [DOI] [PubMed] [Google Scholar]
- 18.Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev. 2011;12:e426–37. doi: 10.1111/j.1467-789X.2010.00825.x. [DOI] [PubMed] [Google Scholar]
- 19.Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–66. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 20.Almeida OP, Hulse GK, Lawrence D, Flicker L. Smoking as a risk factor for Alzheimer’s disease: contrasting evidence from a systematic review of case-control and cohort studies. Addiction. 2002;97:15–28. doi: 10.1046/j.1360-0443.2002.00016.x. [DOI] [PubMed] [Google Scholar]
- 21.Rusanen M, Kivipelto M, Quesenberry CP, Jr, Zhou J, Whitmer RA. Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Arch Intern Med. 2011;171:333–9. doi: 10.1001/archinternmed.2010.393. [DOI] [PubMed] [Google Scholar]
- 22.Moceri VM, Kukull WA, Emanuel I, van Belle G, Larson EB. Early-life risk factors and the development of Alzheimer’s disease. Neurology. 2000;54:415–20. doi: 10.1212/wnl.54.2.415. [DOI] [PubMed] [Google Scholar]
- 23.Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol. 2002;156:445–53. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]
- 24.Laurin D, Verreault R, Lindsay J, MacPherson K, Rockwood K. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol. 2001;58:498–504. doi: 10.1001/archneur.58.3.498. [DOI] [PubMed] [Google Scholar]
- 25.Fratiglioni L, Wang HX. Brain reserve hypothesis in dementia. J Alzheimers Dis. 2007;12:11–22. doi: 10.3233/jad-2007-12103. [DOI] [PubMed] [Google Scholar]
- 26.Stern C, Munn Z. Cognitive leisure activities and their role in preventing dementia: a systematic review. Int J Evid Based Healthc. 2010;8:2–17. doi: 10.1111/j.1744-1609.2010.00150.x. [DOI] [PubMed] [Google Scholar]
- 27.Li G, Shofer JB, Rhew IC, et al. Age-varying association between statin use and incident Alzheimer’s disease. J Am Geriatr Soc. 2010;58:1311–7. doi: 10.1111/j.1532-5415.2010.02906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larrieu S, Letenneur L, Helmer C, Dartigues JF, Barberger-Gateau P. Nutritional factors and risk of incident dementia in the PAQUID longitudinal cohort. J Nutr Health Aging. 2004;8:150–4. [PubMed] [Google Scholar]
- 29.Verreault R, Laurin D, Lindsay J, De Serres G. Past exposure to vaccines and subsequent risk of Alzheimer’s disease. CMAJ. 2001;165:1495–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Morris MC. The role of nutrition in Alzheimer’s disease: epidemiological evidence. Eur J Neurol. 2009;16(Suppl 1):1–7. doi: 10.1111/j.1468-1331.2009.02735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helmer C, Peuchant E, Letenneur L, et al. Association between antioxidant nutritional indicators and the incidence of dementia: results from the PAQUID prospective cohort study. Eur J Clin Nutr. 2003;57:1555–61. doi: 10.1038/sj.ejcn.1601724. [DOI] [PubMed] [Google Scholar]
- 32.Mandel S, Amit T, Bar-Am O, Youdim MB. Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol. 2007;82:348–60. doi: 10.1016/j.pneurobio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Reznichenko L, Amit T, Zheng H, et al. Reduction of iron regulated amyloid precursor protein and amyloid peptide by (−) epigallocatechin 3 gallate in cell cultures: implications for iron chelation in Alzheimer’s disease. Journal of neurochemistry. 2006;97:527–36. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- 34.Rezai-Zadeh K, Shytle D, Sun N, et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. The Journal of neuroscience. 2005;25:8807. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 36.Jeong SK, Kim JM, Kweon SS, et al. Does arm length indicate cognitive and functional reserve? International journal of geriatric psychiatry. 2005;20:406–12. doi: 10.1002/gps.1295. [DOI] [PubMed] [Google Scholar]
- 37.Huang T, Carlson M, Fitzpatrick A, et al. Knee height and arm span: a reflection of early life environment and risk of dementia. Neurology. 2008;70:1818. doi: 10.1212/01.wnl.0000311444.20490.98. [DOI] [PubMed] [Google Scholar]
- 38.Shcherbatykh I, Carpenter DO. The role of metals in the etiology of Alzheimer’s disease. J Alzheimers Dis. 2007;11:191–205. doi: 10.3233/jad-2007-11207. [DOI] [PubMed] [Google Scholar]
- 39.Frisardi V, Solfrizzi V, Capurso C, et al. Aluminum in the diet and Alzheimer’s disease: from current epidemiology to possible disease-modifying treatment. J Alzheimers Dis. 2010;20:17–30. doi: 10.3233/JAD-2009-1340. [DOI] [PubMed] [Google Scholar]
- 40.Brown DR. Brain proteins that mind metals: a neurodegenerative perspective. Dalton Trans. 2009:4069–76. doi: 10.1039/b822135a. [DOI] [PubMed] [Google Scholar]
- 41.Gerhardsson L, Lundh T, Minthon L, Londos E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2008;25:508–15. doi: 10.1159/000129365. [DOI] [PubMed] [Google Scholar]
- 42.Baldi I, Lebailly P, Mohammed-Brahim B, et al. Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol. 2003;157:409–14. doi: 10.1093/aje/kwf216. [DOI] [PubMed] [Google Scholar]
- 43.Santibanez M, Bolumar F, Garcia AM. Occupational risk factors in Alzheimer’s disease: a review assessing the quality of published epidemiological studies. Occup Environ Med. 2007;64:723–32. doi: 10.1136/oem.2006.028209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kukull WA, Larson EB, Bowen JD, et al. Solvent exposure as a risk factor for Alzheimer’s disease: a case-control study. Am J Epidemiol. 1995;141:1059–71. doi: 10.1093/oxfordjournals.aje.a117370. discussion 72-9. [DOI] [PubMed] [Google Scholar]
- 45.Sobel E, Dunn M, Davanipour Z, Qian Z, Chui HC. Elevated risk of Alzheimer’s disease among workers with likely electromagnetic field exposure. Neurology. 1996;47:1477–81. doi: 10.1212/wnl.47.6.1477. [DOI] [PubMed] [Google Scholar]
- 46.Calderon-Garciduenas L, Reed W, Maronpot RR, et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol. 2004;32:650–8. doi: 10.1080/01926230490520232. [DOI] [PubMed] [Google Scholar]
- 47.Grosse SD, Matte TD, Schwartz J, Jackson RJ. Economic gains resulting from the reduction in children’s exposure to lead in the United States. Environ Health Perspect. 2002;110:563–9. doi: 10.1289/ehp.02110563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirkle JL, Brody DJ, Gunter EW, et al. The decline in blood lead levels in the United States. The National Health and Nutrition Examination Surveys (NHANES) JAMA. 1994;272:284–91. [PubMed] [Google Scholar]
- 49.Rabito FA, Shorter C, White LE. Lead levels among children who live in public housing. Epidemiology. 2003;14:263–8. [PubMed] [Google Scholar]
- 50.Lanphear BP, Byrd RS, Auinger P, Schaffer SJ. Community characteristics associated with elevated blood lead levels in children. Pediatrics. 1998;101:264–71. doi: 10.1542/peds.101.2.264. [DOI] [PubMed] [Google Scholar]
- 51.Edwards M, Triantafyllidou S, Best D. Elevated blood lead in young children due to lead-contaminated drinking water: Washington, DC, 2001-2004. Environ Sci Technol. 2009;43:1618–23. doi: 10.1021/es802789w. [DOI] [PubMed] [Google Scholar]
- 52.Jung CH, Matsuto T, Tanaka N, Okada T. Metal distribution in incineration residues of municipal solid waste (MSW) in Japan. Waste Management. 2004;24:381–91. doi: 10.1016/S0956-053X(03)00137-5. [DOI] [PubMed] [Google Scholar]
- 53.Nedwed T, Clifford DA. A survey of lead battery recycling sites and soil remediation processes. Waste Management. 1997;17:257–69. [Google Scholar]
- 54.Kaul B, Sandhu RS, Depratt C, Reyes F. Follow-up screening of lead-poisoned children near an auto battery recycling plant, Haina, Dominican Republic. Environ Health Perspect. 1999;107:917–20. doi: 10.1289/ehp.99107917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Lindern I, Spalinger S, Petroysan V, von Braun M. Assessing remedial effectiveness through the blood lead: soil/dust relationship at the Bunker Hill Superfund Site in the Silver Valley of Idaho. The Science of The Total Environment. 2003;303:139–70. doi: 10.1016/s0048-9697(02)00352-2. [DOI] [PubMed] [Google Scholar]
- 56.Lewin MD, Sarasua S, Jones PA. A multivariate linear regression model for predicting children’s blood lead levels based on soil lead levels: A study at four superfund sites. Environ Res. 1999;81:52–61. doi: 10.1006/enrs.1998.3952. [DOI] [PubMed] [Google Scholar]
- 57.Tong S, von Schirnding YE, Prapamontol T. Environmental lead exposure: a public health problem of global dimensions. Bull World Health Organ. 2000;78:1068–77. [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer PA, Brown MJ, Falk H. Global approach to reducing lead exposure and poisoning. Mutat Res. 2008;659:166–75. doi: 10.1016/j.mrrev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez-Avila M, Smith D, Meneses F, Sanin LH, Hu H. The influence of bone and blood lead on plasma lead levels in environmentally exposed adults. Environ Health Perspect. 1998;106:473–7. doi: 10.1289/ehp.106-1533211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rabinowitz MB, Wetherill GW, Kopple JD. Kinetic analysis of lead metabolism in healthy humans. J Clin Invest. 1976;58:260–70. doi: 10.1172/JCI108467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barry PSI, Mossman DB. Lead concentrations in human tissues. British Journal of Industrial Medicine. 1970;27:339–51. doi: 10.1136/oem.27.4.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rabinowitz MB. Toxicokinetics of bone lead. Environ Health Perspect. 1991;91:33–7. doi: 10.1289/ehp.919133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu H, Rabinowitz M, Smith D. Bone lead as a biological marker in epidemiologic studies of chronic toxicity: conceptual paradigms. Environ Health Perspect. 1998;106:1–8. doi: 10.1289/ehp.981061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbosa F, Jr, Tanus-Santos JE, Gerlach RF, Parsons PJ. A critical review of biomarkers used for monitoring human exposure to lead: advantages, limitations, and future needs. Environ Health Perspect. 2005;113:1669–74. doi: 10.1289/ehp.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pounds JG, Long GJ, Rosen JF. Cellular and molecular toxicity of lead in bone. Environ Health Perspect. 1991;91:17–32. doi: 10.1289/ehp.919117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silbergeld EK, Schwartz J, Mahaffey K. Lead and osteoporosis: mobilization of lead from bone in postmenopausal women. Environ Res. 1988;47:79–94. doi: 10.1016/s0013-9351(88)80023-9. [DOI] [PubMed] [Google Scholar]
- 67.Silbergeld EK. Lead in bone: implications for toxicology during pregnancy and lactation. Environ Health Perspect. 1991;91:63–70. doi: 10.1289/ehp.919163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu H, Shih R, Rothenberg S, Schwartz BS. The epidemiology of lead toxicity in adults: measuring dose and consideration of other methodologic issues. Environ Health Perspect. 2007;115:455–62. doi: 10.1289/ehp.9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu H, Aro A, Rotnitzky A. Bone lead measured by X-ray fluorescence: epidemiologic methods. Environ Health Perspect. 1995;103(Suppl 1):105–10. doi: 10.1289/ehp.95103s1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chettle DR. Three decades of in vivo x-ray fluorescence of lead in bone. X-Ray Spectrometry. 2005;34:446–50. [Google Scholar]
- 71.Wittmers LE, Jr, Aufderheide AC, Wallgren J, Rapp G, Jr, Alich A. Lead in bone. IV. Distribution of lead in the human skeleton. Arch Environ Health. 1988;43:381–91. doi: 10.1080/00039896.1988.9935855. [DOI] [PubMed] [Google Scholar]
- 72.Hu H, Milder FL, Burger DE. X-ray fluorescence: issues surrounding the application of a new tool for measuring burden of lead. Environ Res. 1989;49:295–317. doi: 10.1016/s0013-9351(89)80074-x. [DOI] [PubMed] [Google Scholar]
- 73.Garcia F, Ortega A, Domingo JL, Corbella J. Accumulation of metals in autopsy tissues of subjects living in Tarragona County, Spain. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2001;36:1767–86. doi: 10.1081/ese-100106258. [DOI] [PubMed] [Google Scholar]
- 74.Park SK, Mukherjee B, Xia X, et al. Bone lead level prediction models and their application to examine the relationship of lead exposure and hypertension in the Third National Health and Nutrition Examination Survey. J Occup Environ Med. 2009;51:1422–36. doi: 10.1097/JOM.0b013e3181bf6c8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coon S, Stark A, Peterson E, et al. Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ Health Perspect. 2006;114:1872–6. doi: 10.1289/ehp.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fewtrell LJ, Pruss-Ustun A, Landrigan P, Ayuso-Mateos JL. Estimating the global burden of disease of mild mental retardation and cardiovascular diseases from environmental lead exposure. Environ Res. 2004;94:120–33. doi: 10.1016/s0013-9351(03)00132-4. [DOI] [PubMed] [Google Scholar]
- 77.Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Brain Res Rev. 2005;49:529–54. doi: 10.1016/j.brainresrev.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Wright RO, Tsaih SW, Schwartz J, et al. Lead exposure biomarkers and mini-mental status exam scores in older men. Epidemiology. 2003;14:713–8. doi: 10.1097/01.EDE.0000081988.85964.db. [DOI] [PubMed] [Google Scholar]
- 79.Weisskopf MG, Wright RO, Schwartz J, et al. Cumulative lead exposure and prospective change in cognition among elderly men: the VA Normative Aging Study. Am J Epidemiol. 2004;160:1184–93. doi: 10.1093/aje/kwh333. [DOI] [PubMed] [Google Scholar]
- 80.Shih RA, Hu H, Weisskopf MG, Schwartz BS. Cumulative lead dose and cognitive function in adults: a review of studies that measured both blood lead and bone lead. Environ Health Perspect. 2007;115:483–92. doi: 10.1289/ehp.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Payton M, Riggs KM, Spiro A, 3rd, Weiss ST, Hu H. Relations of bone and blood lead to cognitive function: the VA Normative Aging Study. Neurotoxicol Teratol. 1998;20:19–27. doi: 10.1016/s0892-0362(97)00075-5. [DOI] [PubMed] [Google Scholar]
- 82.Schwartz BS, Stewart WF, Bolla KI, et al. Past adult lead exposure is associated with longitudinal decline in cognitive function. Neurology. 2000;55:1144–50. doi: 10.1212/wnl.55.8.1144. [DOI] [PubMed] [Google Scholar]
- 83.Weuve J, Kelsey KT, Schwartz J, et al. Delta-aminolevulinic acid dehydratase polymorphism and the relation between low level lead exposure and the Mini-Mental Status Examination in older men: the Normative Aging Study. Occupational and environmental medicine. 2006;63:746. doi: 10.1136/oem.2006.027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang FT, Hu H, Schwartz J, et al. Modifying effects of the HFE polymorphisms on the association between lead burden and cognitive decline. Environ Health Perspect. 2007;115:1210–5. doi: 10.1289/ehp.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weisskopf MG, Hu H, Sparrow D, Lenkinski RE, Wright RO. Proton magnetic resonance spectroscopic evidence of glial effects of cumulative lead exposure in the adult human hippocampus. Environ Health Perspect. 2007;115:519–23. doi: 10.1289/ehp.9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kamel F, Umbach DM, Hu H, et al. Lead exposure as a risk factor for amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:195–201. doi: 10.1159/000089625. [DOI] [PubMed] [Google Scholar]
- 87.Kamel F, Umbach DM, Munsat TL, et al. Lead exposure and amyotrophic lateral sclerosis. Epidemiology. 2002;13:311–9. doi: 10.1097/00001648-200205000-00012. [DOI] [PubMed] [Google Scholar]
- 88.Kamel F, Umbach DM, Lehman TA, et al. Amyotrophic lateral sclerosis, lead, and genetic susceptibility: polymorphisms in the delta-aminolevulinic acid dehydratase and vitamin D receptor genes. Environ Health Perspect. 2003;111:1335–9. doi: 10.1289/ehp.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weisskopf MG, Weuve J, Nie H, et al. Association of Cumulative Lead Exposure with Parkinson’s Disease. Environ Health Perspect. 2010 doi: 10.1289/ehp.1002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Basha MR, Murali M, Siddiqi HK, et al. Lead (Pb) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005;19:2083–4. doi: 10.1096/fj.05-4375fje. [DOI] [PubMed] [Google Scholar]
- 91.Basha MR, Wei W, Bakheet SA, et al. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu J, Basha MR, Brock B, et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morris KV. The emerging role of RNA in the regulation of gene transcription in human cells. Semin Cell Dev Biol. 2011 doi: 10.1016/j.semcdb.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 95.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1757–64. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–8. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 97.Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–83. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 99.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 100.DeBaun MR, Niemitz EL, McNeil DE, et al. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70:604–11. doi: 10.1086/338934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Faulk C, Dolinoy DC. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 6. 2011 doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barker DJ. The developmental origins of chronic adult disease. Acta Paediatr. 2004;(Suppl 93):26–33. doi: 10.1111/j.1651-2227.2004.tb00236.x. [DOI] [PubMed] [Google Scholar]
- 103.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–68. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hanson M, Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD. Developmental plasticity and developmental origins of non-communicable disease: Theoretical considerations and epigenetic mechanisms. Prog Biophys Mol Biol. 2011;106:272–80. doi: 10.1016/j.pbiomolbio.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 105.Jablonka E. Epigenetic epidemiology. Int J Epidemiol. 2004;33:929–35. doi: 10.1093/ije/dyh231. [DOI] [PubMed] [Google Scholar]
- 106.Fraga MF. Genetic and epigenetic regulation of aging. Curr Opin Immunol. 2009;21:446–53. doi: 10.1016/j.coi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 107.Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev. 2009;8:268–76. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 108.Sananbenesi F, Fischer A. The epigenetic bottleneck of neurodegenerative and psychiatric diseases. Biol Chem. 2009;390:1145–53. doi: 10.1515/BC.2009.131. [DOI] [PubMed] [Google Scholar]
- 109.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 110.Mill J. Toward an integrated genetic and epigenetic approach to Alzheimer’s disease. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 111.Chouliaras L, Rutten BP, Kenis G, et al. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog Neurobiol. 2010;90:498–510. doi: 10.1016/j.pneurobio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 112.Mastroeni D, Grover A, Delvaux E, et al. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mastroeni D, Grover A, Delvaux E, et al. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4:e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One. 2010;5:e8898. doi: 10.1371/journal.pone.0008898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ogawa O, Zhu X, Lee HG, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta neuropathologica. 2003;105:524–8. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- 117.Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 118.Belakavadi M, Dell J, Grover GJ, Fondell JD. Thyroid hormone suppression of beta-amyloid precursor protein gene expression in the brain involves multiple epigenetic regulatory events. Mol Cell Endocrinol. 2011;339:72–80. doi: 10.1016/j.mce.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 119.Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. 2009;68:880–91. doi: 10.1097/NEN.0b013e3181af2e46. [DOI] [PubMed] [Google Scholar]
- 120.Brohede J, Rinde M, Winblad B, Graff C. A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J Neurogenet. 2010;24:179–81. doi: 10.3109/01677063.2010.503978. [DOI] [PubMed] [Google Scholar]
- 121.Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70:288–92. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 122.Tohgi H, Utsugisawa K, Nagane Y, et al. The methylation status of cytosines in a tau gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci Lett. 1999;275:89–92. doi: 10.1016/s0304-3940(99)00731-4. [DOI] [PubMed] [Google Scholar]
- 123.Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21:28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marsit CJ, Karagas MR, Danaee H, et al. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006;27:112–6. doi: 10.1093/carcin/bgi172. [DOI] [PubMed] [Google Scholar]
- 127.Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27:1481–8. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- 128.Sun H, Zhou X, Chen H, Li Q, Costa M. Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol Appl Pharmacol. 2009;237:258–66. doi: 10.1016/j.taap.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286:355–65. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 130.Pilsner JR, Hu H, Ettinger A, et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect. 2009;117:1466–71. doi: 10.1289/ehp.0800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wright RO, Schwartz J, Wright RJ, et al. Biomarkers of Lead Exposure and DNA Methylation within Retrotransposons. Environ Health Perspect. 2010 doi: 10.1289/ehp.0901429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kovatsi L, Georgiou E, Ioannou A, et al. p16 promoter methylation in Pb2+ -exposed individuals. Clin Toxicol (Phila) 2010;48:124–8. doi: 10.3109/15563650903567091. [DOI] [PubMed] [Google Scholar]
- 133.Li YY, Chen T, Wan Y, Xu SQ. Lead exposure in pheochromocytoma cells induces persistent changes in amyloid precursor protein gene methylation patterns. Environ Toxicol. 2010 doi: 10.1002/tox.20666. [DOI] [PubMed] [Google Scholar]
- 134.Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006;20:43–9. doi: 10.1096/fj.05-4707com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Niculescu MD, Craciunescu CN, Zeisel SH. Gene expression profiling of choline-deprived neural precursor cells isolated from mouse brain. Brain Res Mol Brain Res. 2005;134:309–22. doi: 10.1016/j.molbrainres.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 136.Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–83. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 137.Clarke R, Smith AD, Jobst KA, et al. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55:1449–55. doi: 10.1001/archneur.55.11.1449. [DOI] [PubMed] [Google Scholar]
- 138.Bottiglieri T, Godfrey P, Flynn T, et al. Cerebrospinal fluid S-adenosylmethionine in depression and dementia: effects of treatment with parenteral and oral S-adenosylmethionine. J Neurol Neurosurg Psychiatry. 1990;53:1096–8. doi: 10.1136/jnnp.53.12.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cao XJ, Huang SH, Wang M, Chen JT, Ruan DY. S-adenosyl-L-methionine improves impaired hippocampal long-term potentiation and water maze performance induced by developmental lead exposure in rats. Eur J Pharmacol. 2008;595:30–4. doi: 10.1016/j.ejphar.2008.07.061. [DOI] [PubMed] [Google Scholar]
- 140.Chen T, Li YY, Zhang JL, et al. Protective effect of C(60) - methionine derivate on lead-exposed human SH-SY5Y neuroblastoma cells. J Appl Toxicol. 2011;31:255–61. doi: 10.1002/jat.1588. [DOI] [PubMed] [Google Scholar]
- 141.Schafer JH, Glass TA, Bressler J, Todd AC, Schwartz BS. Blood lead is a predictor of homocysteine levels in a population-based study of older adults. Environ Health Perspect. 2005;113:31–5. doi: 10.1289/ehp.7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guallar E, Silbergeld EK, Navas-Acien A, et al. Confounding of the relation between homocysteine and peripheral arterial disease by lead, cadmium, and renal function. Am J Epidemiol. 2006;163:700–8. doi: 10.1093/aje/kwj090. [DOI] [PubMed] [Google Scholar]
- 143.Bolin CM, Basha R, Cox D, et al. Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J. 2006;20:788–90. doi: 10.1096/fj.05-5091fje. [DOI] [PubMed] [Google Scholar]
- 144.Cerda S, Weitzman SA. Influence of oxygen radical injury on DNA methylation. Mutat Res. 1997;386:141–52. doi: 10.1016/s1383-5742(96)00050-6. [DOI] [PubMed] [Google Scholar]
- 145.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu J, Basha MR, Zawia NH. The environment, epigenetics and amyloidogenesis. J Mol Neurosci. 2008;34:1–7. doi: 10.1007/s12031-007-0009-4. [DOI] [PubMed] [Google Scholar]
- 147.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 148.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 149.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–56. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jack CR, Jr, Petersen RC, O’Brien PC, Tangalos EG. MR-based hippocampal volumetry in the diagnosis of Alzheimer’s disease. Neurology. 1992;42:183–8. doi: 10.1212/wnl.42.1.183. [DOI] [PubMed] [Google Scholar]
- 151.Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J Nucl Med. 1995;36:1238–48. [PubMed] [Google Scholar]
- 152.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 153.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 154.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–81. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 155.Ravelli AC, van der Meulen JH, Michels RP, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–7. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 156.Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ. 1990;301:259–62. doi: 10.1136/bmj.301.6746.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bergvall N, Iliadou A, Johansson S, et al. Genetic and shared environmental factors do not confound the association between birth weight and hypertension: a study among Swedish twins. Circulation. 2007;115:2931–8. doi: 10.1161/CIRCULATIONAHA.106.674812. [DOI] [PubMed] [Google Scholar]
- 158.Miller DB, O’Callaghan JP. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism. 2008;57(Suppl 2):S44–9. doi: 10.1016/j.metabol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 159.Borenstein AR, Copenhaver CI, Mortimer JA. Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:63–72. doi: 10.1097/01.wad.0000201854.62116.d7. [DOI] [PubMed] [Google Scholar]
- 160.Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Yaffe K. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005;330:1360. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rovio S, Kareholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. The Lancet Neurology. 2005;4:705–11. doi: 10.1016/S1474-4422(05)70198-8. [DOI] [PubMed] [Google Scholar]
- 162.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. American Journal of Epidemiology. 2001;154:635. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- 163.Ross MG, Desai M, Khorram O, et al. Gestational programming of offspring obesity: a potential contributor to Alzheimer’s disease. Curr Alzheimer Res. 2007;4:213–7. doi: 10.2174/156720507780362056. [DOI] [PubMed] [Google Scholar]
- 164.Lester-Coll N, Rivera EJ, Soscia SJ, et al. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 165.Landrigan PJ, Sonawane B, Butler RN, et al. Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect. 2005;113:1230–3. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hu H, Aro A, Payton M, et al. The relationship of bone and blood lead to hypertension. The Normative Aging Study. JAMA. 1996;275:1171–6. [PubMed] [Google Scholar]
- 167.Cheng Y, Schwartz J, Sparrow D, et al. Bone lead and blood lead levels in relation to baseline blood pressure and the prospective development of hypertension: the Normative Aging Study. Am J Epidemiol. 2001;153:164–71. doi: 10.1093/aje/153.2.164. [DOI] [PubMed] [Google Scholar]
- 168.Korrick SA, Hunter DJ, Rotnitzky A, Hu H, Speizer FE. Lead and hypertension in a sample of middle-aged women. Am J Public Health. 1999;89:330–5. doi: 10.2105/ajph.89.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jain NB, Potula V, Schwartz J, et al. Lead levels and ischemic heart disease in a prospective study of middle-aged and elderly men: the VA Normative Aging Study. Environ Health Perspect. 2007;115:871–5. doi: 10.1289/ehp.9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Weisskopf MG, Jain N, Nie H, et al. A prospective study of bone lead concentration and death from all causes, cardiovascular diseases, and cancer in the Department of Veterans Affairs Normative Aging Study. Circulation. 2009;120:1056–64. doi: 10.1161/CIRCULATIONAHA.108.827121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hu H, Tosteson T, Aufderheide AC, et al. Distribution of lead in human bone: I. Atomic absorption measurements. Basic Life Sci. 1990;55:267–74. doi: 10.1007/978-1-4613-1473-8_36. [DOI] [PubMed] [Google Scholar]
- 172.Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: Association with risk factors in epidemiologic studies. Epigenetics. 2011;6 doi: 10.4161/epi.6.7.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–9. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 175.Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr. 1999;69:179–97. doi: 10.1093/ajcn/69.2.179. [DOI] [PubMed] [Google Scholar]