

Abstract
The hypothalamic paraventricular nucleus (PVN) plays an important role in the sympathoexcitatory response to elevated plasma angiotensin II (Ang II). However, the mechanism by which Ang II influences sympathetic activity is not fully understood. In this study, we tested the hypothesis that GABA(γ-aminobutyric acid)-ergic function in the PVN is reduced by peripheral infusion of Ang II. To accomplish this, rats received either intravenous Ang II (12 ng/kg per minute) or vehicle (D5W) for 7 days, and renal sympathetic nerve activity (SNA), mean arterial pressure (MAP), and heart rate (HR) responses were recorded after unilateral PVN microinjection of the GABA-A receptor antagonist bicuculline methiodide (BMI, 0.1 nmol). Results indicate that in contrast to a significant increase in renal SNA, MAP, and HR observed in vehicle-infused rats (P<0.05), BMI injection into the PVN of Ang II–infused animals was without effect on all recorded variables. In a separate groups of animals, ganglionic blockade produced a significantly greater fall in MAP (P<0.01) in Ang II–infused rats than in vehicle-infused control rats, indicating that the contribution of SNA to the maintenance of blood pressure was elevated in the Ang II–infused group. Overall, these data indicate that cardiovascular and sympathoexcitatory responses to acute GABA-A receptor antagonism in the PVN are significantly blunted in rats after 7 days of intravenous infusion of Ang II. We conclude that an Ang II–induced reduction in GABAergic inhibition within the PVN may contribute to elevated SNA observed in this study.
Keywords: angiotensin II, hypothalamus, sympathetic nervous system, hypertension, arterial
As the dominant inhibitory neurotransmitter in the mammalian brain, γ-aminobutyric acid (GABA)1 plays an important role in regulating cardiovascular function.2,3 In brain regions such as the nucleus tractus solitarius2 and rostral ventrolateral medulla2,3 that control sympathetic nerve activity (SNA), GABA tonically suppresses neuronal activity and excitability. In the hypothalamic paraventricular nucleus (PVN), neuronal activity is also regulated by GABA4–6 and by a number of excitatory neurotransmitters as well.7,8 Among the latter is the peptide angiotensin II (Ang II). Ang II–containing fibers arise from Ang II–sensitive regions of the forebrain known as circumventricular organs (CVO), which lack a complete blood-brain barrier.9,10 Neurons in two forebrain CVOs, the subfornical organ and organum vasculosum of the lamina terminalis, express Ang II AT1 receptors in high density11 and appear to sense and respond to circulating Ang II.9,12–15 Although Ang II increases SNA through these and other central actions,16,17 the mechanism by which circulating Ang II enhances PVN neuronal excitability is not fully understood.
On the basis of literature evidence, it is clear that GABA4–6 and Ang II13–15 can each act individually within the PVN to influence cardiovascular function. More recently, it has been shown that local GABA–Ang II interactions may be key to regulating PVN neuronal excitability and sympathetic outflow.4 Although mechanisms of GABA–Ang II interaction within the PVN have not been fully defined, in vitro electrophysiological studies indicate that GABA tonically opposes excitatory actions of Ang II on neurosecretory neurons,18 and excitation of spinally projecting neurons involves presynaptic actions of Ang II to reduce GABA release.19 In addition, functional studies in rats with sodium-dependent hypertension20 and heart failure6 have suggested that Ang II–induced sympathoexcitation involves reduced GABA-mediated inhibition within the PVN. This conclusion is consistent with evidence that both plasma Ang II and SNA are elevated whereas the sympathetic response to blockade of PVN GABA-A receptors is attenuated in these disease models.6,20
The aim of the present study was to determine more directly the effect of elevated plasma Ang II on cardiovascular and renal SNA responses to acute GABA-A receptor blockade in the PVN. This was accomplished by infusing rats intravenously with Ang II at a rate selected to leave plasma volume undisturbed. Results indicate that responses to blockade of GABA-A receptors in the PVN are blunted in Ang II–infused rats compared with vehicle-infused control rats and are consistent with the hypothesis that in heart failure6 and certain forms of hypertension,20 elevated peripheral Ang II could act to reduce GABAergic function within the PVN. Some of these results have been presented as an abstract.21
Methods
Experiments were performed in 40 male Sprague-Dawley rats (weight, 250 to 350 g; Charles River Laboratories) maintained in a 14:10-hour light-dark cycle and allowed free access to rat chow and water. All procedures were approved by the Institutional Animal Care and Use Committee.
Twelve-Day Infusion Protocol
Rats were anesthetized (pentobarbital, 50 mg/kg IP), and a telemetry probe was placed in the aorta to record blood pressure. The vena cava was catheterized to infuse Ang II or vehicle.17 After regaining consciousness, a solution of D5W and ampicillin (28 mg/24 h) was infused (5.7 mL/24 h) continuously for 5 days. Rats were then separated into two groups; one received fresh Ang II (12 ng/kg per minute) in the infusate daily and the other (control group) was maintained on the vehicle infusion for the final 7 days.
In a separate group of instrumented animals, the contribution of sympathetic activity to the maintenance of blood pressure was determined 3 days before and after completing the 7-day Ang II–infusion or vehicle-infusion period. While animals were conscious, the maximal fall in mean arterial pressure (MAP) produced by the ganglionic blocker chlorisondamine (2.5 mg/kg IV) was determined. Because of long-lasting drug effects, animals treated with chlorisondamine were not used in subsequent microinjection studies.
Experimental Procedures
On completing the infusion protocol, rats were anesthetized (α-chloralose, 80 mg/kg; urethane, 800 mg/kg IP) and a femoral vein and artery were catheterized to administer drugs and measure arterial pressure, respectively. Standard methods were used to record renal SNA.4,17 Renal SNA responses to intravenous sodium nitroprusside and phenylephrine were used to assess the signal-to-noise ratio of each recording. Hematocrit was measured from venous blood. Animals were paralyzed (gallamine triethiodide, 25 mg/kg IV) and artificially ventilated. End-tidal PCO2 and body temperature were maintained within normal limits.
Microinjection of Drugs
A 30-gauge injector was lowered stereotaxically into the PVN (coordinates: 1.7 to 1.9 mm caudal to bregma, 0.5 mm lateral to midline, and 7.4 to 7.6 mm ventral to the skull surface). The GABA-A receptor antagonist bicuculline methiodide (BMI) (or saline vehicle) was injected (0.1 nmol in 100 nL) unilaterally over a period of 60 seconds. In some experiments, BMI was purposely delivered outside the PVN (1.8 mm lateral to midline) to serve as an anatomical control.
Histology
After each experiment, Evans blue dye (2% in saline) was injected (100 nL) into the PVN (or adjacent control site). Brains were removed, placed in paraformaldehyde (4% for 24 hour), sectioned (40 μm), and viewed through a microscope to identify injections sites.
Analysis
During the infusion period, 24-hour averages of MAP and HR were calculated. Changes in MAP and HR were assessed by means of a 2-way repeated-measures ANOVA. A t test was used to compare hematocrit and responses to nitroprusside in vehicle-infused and Ang II–infused rats. Responses to BMI or vehicle microinjection were determined as the difference between a 60-second baseline average and a 60-second average of data centered on the maximum increase. BMI and vehicle effects were compared by means of a 2-way ANOVA. A Newman-Keuls test was used for pairwise comparisons. When group variances were unequal, data were transformed through the use of a logarithmic function. Significance was defined as P<0.05. Voltage caused by noise was subtracted from all SNA data before statistical analysis.4,17
Results
As shown in Figure 1, baseline MAP and HR were similar in rats before beginning the infusion protocol. In response to Ang II infusion (n=5), MAP increased significantly (P<0.05) within 24 hours and remained elevated throughout the 7-day infusion period. In contrast, MAP in vehicle-infused rats (n=5) remained unchanged. Although HR was significantly decreased (P<0.05) in Ang II–infused rats after 6 days of infusion (Figure 1), it did not significantly differ between groups at any point during the infusion period.
Figure 1.
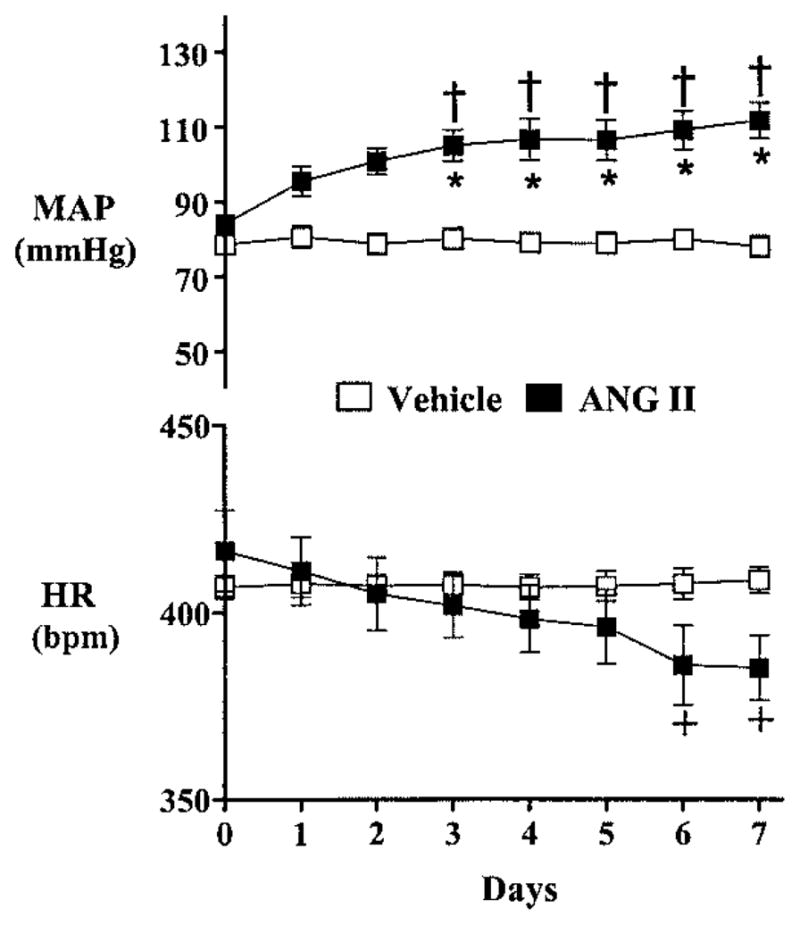
Twenty-four-hour average MAP (top) and HR (bottom) in animals receiving intravenous infusion of either Ang II (■, n=5) or vehicle (□, n=5) for 7 days. +P<0.05 compared with baseline; †P<0.01 compared with baseline; *P<0.001 between Ang II–infused and vehicle-infused rats.
As shown in Figure 2, 7 days of Ang II infusion significantly increased SNA, as indexed by the magnitude of the maximal depressor response to acute ganglionic blockade. Intravenous administration of chlorisondamine caused a significantly greater decrease (P<0.01) in MAP in Ang II–treated animals (117±3 to 42±1 mm Hg, n=5) than in vehicle-infused control rats (87±4 to 40±6 mm Hg, n=5). Likewise, there was a tendency for a greater fall in HR during ganglionic blockade in Ang II–infused animals, but differences did not reach statistical significance (Figure 2). Overall, these data indicate that the contribution of SNA in maintaining arterial pressure was significantly increased in animals infused with Ang II. Notably, this effect does not appear to result from differences in extracellular fluid volume, since hematocrit was not significantly different in animals infused with vehicle (43.2±2, n=5) or Ang II (45±1, n=5).
Figure 2.
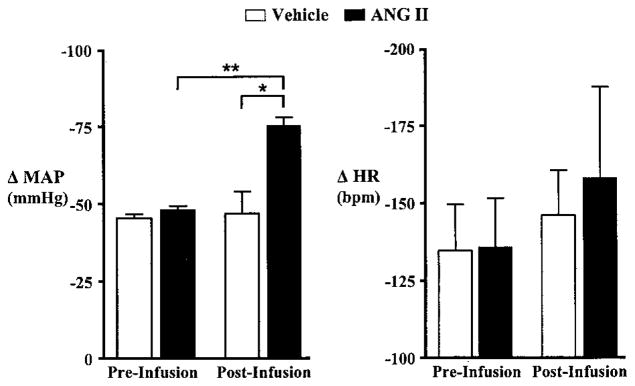
Effect of ganglionic blockade with chlorisondamine (2.5 mg/kg IV) on MAP (left) and HR (right) in rats before and after intravenous infusion of vehicle (n=5) or Ang II (n=5) for 7 days. The depressor response to ganglionic blockade was significantly greater in Ang II–infused animals compared with vehicle-infused control animals. Similarly, bradycardic responses tended to be larger in Ang II–infused than vehicle-infused animals, although differences did not reach statistical significance. *P<0.05, preinfusion vs postinfusion; **P<0.05, postinfusion vehicle-infused vs Ang II–infused.
Figure 3 shows the response to PVN microinjection of BMI (0.1 nmol) in a vehicle-infused (left) and an Ang II–infused rat (right). Note that in the vehicle-infused animal, renal SNA, ABP, and HR increased promptly. Conversely, in the Ang II–infused animal, PVN injection of BMI had little effect. Summary data in Figure 4 show that in vehicle-infused rats (n=6), PVN injection of BMI significantly increased (P<0.01) renal SNA by an average of 57±4%. In contrast, PVN injection of BMI had no effect on renal SNA (1±5.6%) in Ang II–infused rats (n=6). Similarly, MAP (P<0.01) and HR (P<0.05) were significantly increased after PVN injection of BMI in vehicle-infused rats (n=7), but again, no significant effect was observed in Ang II–infused animals (n=6) (Figure 4). Overall, these data indicate that Ang II infusion effectively attenuates renal sympathetic and cardiovascular responses to acute blockade of GABA-A receptors in the PVN.
Figure 3.
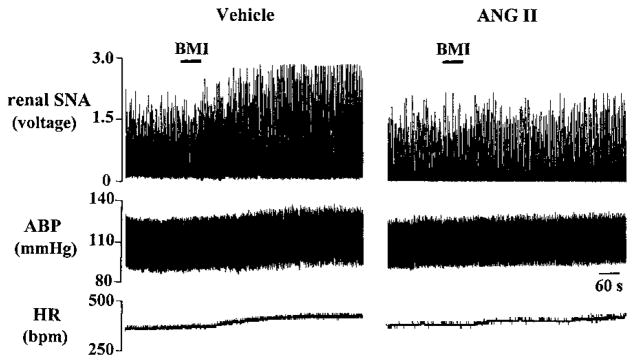
Individual records showing renal SNA, arterial blood pressure (ABP), and HR responses to unilateral PVN microinjection of BMI (0.1 nmol) in a vehicle-infused animal and an Ang II–infused animal. BMI microinjection in the PVN produced an increase in renal SNA in the vehicle-infused animal but was without effect in the Ang II–infused rat. Similarly, pressor and tachycardic responses to PVN-injected BMI were greater in the vehicle-infused than the Ang II–infused animal.
Figure 4.
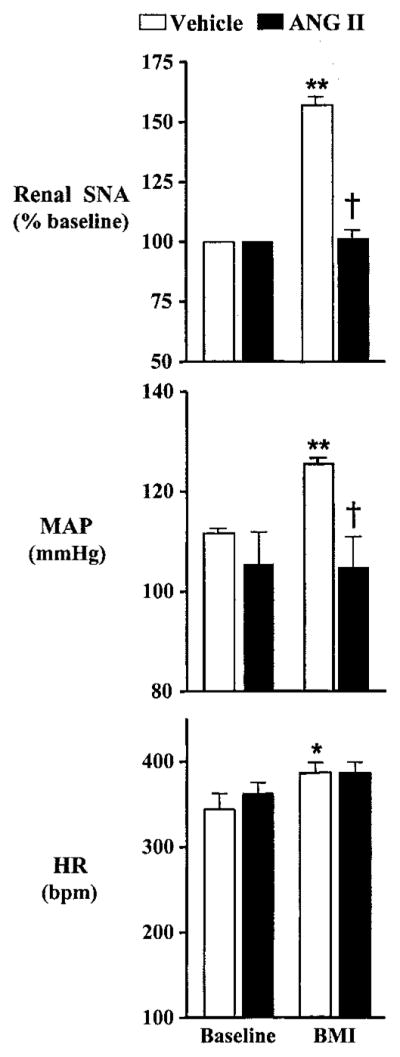
Group data showing renal SNA (top), MAP (middle), and HR (bottom) responses to unilateral PVN microinjection of BMI (0.1 nmol). Renal SNA (n=6 per group), MAP (n=7 vehicle; n=6 Ang II), and HR (n=7 per group) responses were significantly increased in response to PVN-microinjected BMI in vehicle-infused rats, whereas responses were significantly attenuated after 7 days of Ang II infusion. *P<0.05 compared with baseline; **P<0.01 compared with baseline; †P<0.01 compared with BMI in vehicle-infused animals.
It should be noted that differences in renal SNA, MAP, and HR responses to BMI in vehicle-infused and Ang II–infused animals did not depend on the level of ongoing MAP or HR, since values just before BMI injection were not statistically different between groups (Figure 4). Furthermore, intravenous administration of nitroprusside increased renal SNA similarly in Ang II–infused (169.7±3.2% baseline, n=6) and vehicle-infused (174±2.6% baseline, n=6) animals, indicating that Ang II treatment did not elevate basal SNA to a maximal level.
Given that microinjection of BMI outside the PVN and saline within the PVN were each without effect (P>0.1) on renal SNA, MAP, or HR in both vehicle-infused (n=6 per group) and Ang II–infused (n=6 per group) animals, it appears that actions of BMI were specific to the PVN. On the basis of dye diffusion patterns, microinjection sites appeared to be confined to superior portions of the PVN near the dorsal cap region without penetrating the ependyma of the third cerebral ventricle (Figure 5). Anatomic control injections were verified at ≈1.8 mm lateral to midline and did not significantly invade lateral portions of the PVN.
Figure 5.
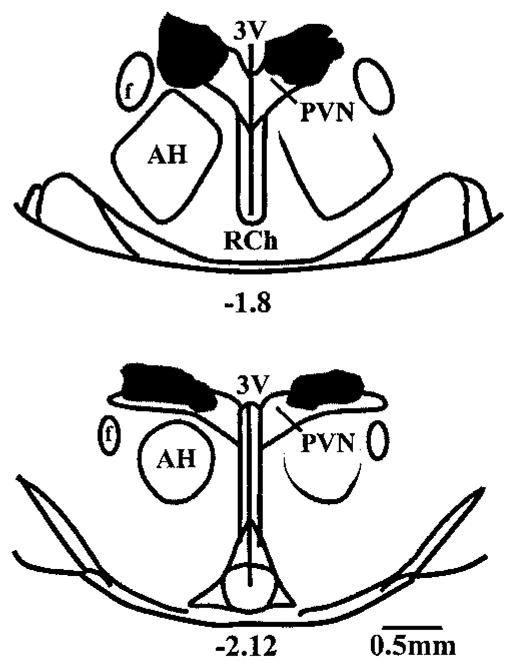
Schematic drawings of rat hypothalamus in coronal section. Shaded areas approximate regions of hypothalamus exposed to microinjected BMI in rats infused with vehicle (right) or Ang II (left). Shaded areas were determined by injecting 100 nL of Evans blue dye (2%) after each experiment and tracing the outline of the dye observed on each 40-μm-thick section through the PVN. Outermost distribution of dye was determined by overlaying areas from similar rostral-caudal sections taken from different brains. Hence, the areas shown are larger than the dye distribution observed in any single brain but represent the widest possible distribution of injectate for all BMI-treated animals. Note that all PVN injections were made unilaterally (left side) but are shown bilaterally to distinguish injection areas for each treatment group. Numbers below drawings indicate distance (in mm) posterior to bregma. AH indicates anterior hypothalamus; f, fornix; 3V, third cerebral ventricle; and RCh, retro-chiasmatic area.
Discussion
Similar to previous studies,4,5,6,22 our data show that BMI microinjected into the PVN in rats infused with vehicle for 7 days significantly increases renal SNA, MAP, and HR. In contrast, rats infused with Ang II had significantly blunted responses. In fact, continuous infusion of Ang II for 7 days effectively eliminated the renal SNA response to BMI injected into the PVN. Histological examination of injection sites revealed that the PVN was consistently targeted in both vehicle-infused and Ang II–infused animals.
What is becoming increasingly clear is that GABAergic control of SNA within the PVN is subject to modulation in diseases accompanied by sympathoexcitation. Indeed, it has been reported that GABA-mediated control of SNA within the PVN is reduced in rats with salt-sensitive hypertension20 and heart failure.6 One interpretation of these data is that elevated plasma Ang II present in both disease models may lead to reduced GABAergic function within the PVN. Results from the present study suggest that GABA-A receptor–mediated control of PVN neuronal excitability may indeed be reduced by direct intravenous infusion of Ang II.
It should be noted that these results appear to conflict with those of a recent study by DiBona and Jones,23 in which it was reported that the renal SNA response to BMI injected into the PVN is enhanced in rats fed a low versus normal sodium diet. Given that anesthesia has been reported to alter cardiovascular responses to PVN stimulation,24 differences could involve the type of anesthetic used. Additionally, it may be that the difference in methods used to increase circulating Ang II in their study23 and ours make the results not directly comparable. For example, different levels of sodium intake and associated differences in extracellular fluid volume could underlie these disparate responses. This could be the case because volume-sensitive inputs to the PVN have been reported to inhibit discharge of PVN autonomic neurons25 and chemical lesions of PVN parvocellular cells have been shown to reduce the renal SNA response to acute volume expansion.26 Given that hematocrit was unchanged by the Ang II infusion in our study, it does not appear that blood volume was altered and therefore effects of Ang II infusion do not appear to have involved a change in volume-sensitive input to the PVN. Moreover, effects of aldosterone do not appear to contribute significantly to the findings of the current study, since values of plasma aldosterone return toward baseline within 48 hours during continuous infusion of Ang II.27
Acute and long-term Ang II treatments are well known to differentially modulate SNA and arterial pressure through both peripheral28 and central17,29 actions. In the current study, 7-day peripheral Ang II infusion produced a significant and sustained increase in MAP. Furthermore, this Ang II infusion protocol consistently increased SNA, as indexed by the depressor response to ganglionic blockade. As a result, experiments were performed after at least 7 days of continuous infusion. Although MAP was unchanged throughout the infusion protocol in vehicle-treated control rats, it increased after urethane/α-chloralose anesthesia, as reported previously.17 Nevertheless, responses to BMI injected in the PVN in vehicle-infused animals were similar to those reported in previous studies.4–6 It is important to note that anesthesia did not affect the elevated MAP produced by continuous Ang II infusion. Overall, the present findings suggest that GABAergic function within the PVN may be reduced in response to Ang II infusion.
In addition to GABA, the PVN receives input from Ang II–sensitive regions of the forebrain,9,13,15 which supply the PVN with Ang II immunoreactive fibers.10,15 Furthermore, electrophysiological studies demonstrate that intravenous Ang II can increase the discharge of PVN neurons13,30 through activation of locally expressed AT1 receptors.11 Collectively, these data indicate that the PVN is an important central site where information concerning the level of circulating Ang II is processed. Although numerous studies describe the individual importance of PVN GABAergic and Ang II systems in the regulation of cardiovascular function, fewer investigations4 have examined interactions between GABA and Ang II in the control of SNA.
One possible explanation for the observed ability of Ang II infusion to effectively abolish the renal SNA response to PVN GABA-A receptor blockade is that basal SNA could have been increased to a level that did not allow for a further increase to occur. One argument against this is that renal SNA did increase in Ang II–infused animals when blood pressure was lowered with nitroprusside. Nevertheless, it remains possible that Ang II infusion could have maximally activated PVN neurons and thereby precluded any further increase in SNA during GABA-A receptor blockade. To address this possibility, additional studies will be needed to test whether PVN neuronal activity can be increased similarly in both treatment groups on exposure to an excitatory stimulus such as glutamate. Another possibility is that blunted responses to GABA-A receptor blockade in Ang II–infused rats could reflect an overall reduction in GABA-A receptor tone. This could result either from removal of excitatory input to local GABAergic neurons or it could be due to a reduction in the number and/or affinity of local GABA-A receptors. The latter appears to be the case in the spontaneously hypertensive rat (SHR), where GABA-A receptor expression31 and affinity32 in the hypothalamus are reduced compared with Wistar-Kyoto control rats, as are typical cardiovascular responses to hypothalamic injection of BMI.4–6 This does not appear to be a general mechanism, however, since rats with salt-sensitive hypertension showed no change in either GABA-A receptor expression or binding affinity compared with control rats.33 It is important to note that this discrepancy suggests that hypertension per se is not likely to explain or be explained by changes in GABA-A receptor function in the hypothalamus.
Despite differences noted in the aforementioned models, evidence indicates that Ang II can inhibit GABA release by acting presynaptically and can alter responses to GABA-A receptor activation by acting postsynaptically. That Ang II and GABA are colocalized in axon terminals in the subfornical organ34 supports both possibilities. Regarding presynaptic actions, Ang II has been reported to modulate GABA release in rat hippocampal neurons.35 Moreover, a recent in vitro patch clamp study by Li et al19 demonstrated that Ang II stimulates spinally projecting PVN neurons by acting presynaptically to reduce the frequency of inhibitory postsynaptic currents. In the same study, it was reported that AT1 receptors are expressed on putative synaptic terminals within the PVN. If such a process were activated under conditions of the present study, an Ang II–stimulated reduction in GABA release could explain the blunted renal SNA response observed during acute GABA-A receptor blockade.
In addition to presynaptic modulation of GABA release described above, Ang II can also act postsynaptically to modulate GABA-A receptor–mediated inhibition. For example, Cl− conductance through GABA-A receptor channels is reduced after phosphorylation by protein kinase C,36,37 a major signaling intermediate activated by stimulation of postsynaptic AT1 receptors.38 Furthermore, an in vitro patch-clamp study by Latchford and Ferguson18 recently reported that Ang II–induced excitation of PVN neurosecretory neurons is enhanced by blockade of GABA-A receptors, suggesting that GABAergic feedback restrains activation of these neurons by Ang II. Moreover, data from their study18 indicate that Ang II increases GABAergic input that buffers the Ang II–induced membrane depolarization. These findings describe a mechanism of postsynaptic interaction between Ang II and GABA in which Ang II input supplies at least a portion of the GABAergic tone present among nonautonomic neurons of the PVN. A recent in vivo study by Chen et al4 suggests that a similar process could occur among autonomic neurons, since prior blockade of AT1 receptors in the PVN nearly prevented the renal SNA response to subsequent blockade of PVN GABA-A receptors. It should be emphasized, however, that for feedback activation of local GABAergic neurons to contribute to the findings of the present study, it would seem necessary for this feedback restraint to be diminished after 7 days of Ang II infusion. Under this condition, GABA-mediated inhibition in the PVN could be reduced and therefore could result in attenuated cardiovascular and sympathetic responses to acute GABA-A receptor blockade. Regardless of the underlying mechanism, recent studies in both nonautonomic and sympathetic regulatory neurons of the PVN highlight the potential importance of local Ang II–GABA interactions in the control of cardiovascular and sympathetic function.
In conclusion, results from this study indicate that 7 days of intravenous infusion of Ang II significantly attenuates the increase in renal SNA, MAP, and HR induced by acute GABA-A receptor blockade in the PVN. These data as well as literature evidence6,20 raise the possibility that Ang II may act centrally to reduce GABAergic function in the PVN, thereby providing a potential mechanism by which sympathetic activity could become elevated in cardiovascular diseases such as congestive heart failure6 and arterial hypertension.20
Acknowledgments
This project was supported by National Heart, Lung, and Blood Institute grants HL-59326 (V.S.B.) and HL-56834 (G.M.T.). Dr LaGrange was supported by an National Institutes of Health-funded Institutional National Research Service Award (T32 HL-07446) during a portion of this study.
References
- 1.Decavel C, van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- 2.Schreihofer AM, Guyenet PG. The baroreflex and beyond: control of sympathetic vasomotor tone by GABAergic neurons in the ventrolateral medulla. Clin Exp Pharmacol Physiol. 2002;29:514–521. doi: 10.1046/j.1440-1681.2002.03665.x. [DOI] [PubMed] [Google Scholar]
- 3.Dampney RA. The subretrofacial vasomotor nucleus: anatomical, chemical and pharmacological properties and role in cardiovascular regulation. Prog Neurobiol. 1994;42:197–227. doi: 10.1016/0301-0082(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 4.Chen QH, Toney GM. Responses to GABA-A receptor blockade in the hypothalamic PVN are attenuated by local AT1 receptor antagonism. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1231–1239. doi: 10.1152/ajpregu.00028.2003. [DOI] [PubMed] [Google Scholar]
- 5.Kenney MJ, Weiss ML, Patel KP, Wang Y, Fels RJ. Paraventricular nucleus bicuculline alters frequency components of sympathetic nerve discharge bursts. Am J Physiol Heart Circ Physiol. 2001;281:1233–1241. doi: 10.1152/ajpheart.2001.281.3.H1233. [DOI] [PubMed] [Google Scholar]
- 6.Zhang K, Li YF, Patel KP. Reduced endogenous GABA-mediated inhibition in the PVN on renal nerve discharge in rats with heart failure. Am J Physiol Regul Integr Comp Physiol. 2002;282:1006–1015. doi: 10.1152/ajpregu.00241.2001. [DOI] [PubMed] [Google Scholar]
- 7.Moga MM, Saper CB. Neuropeptide-immunoreactive neurons projecting to the paraventricular hypothalamic nucleus in the rat. J Comp Neurol. 1994;346:137–150. doi: 10.1002/cne.903460110. [DOI] [PubMed] [Google Scholar]
- 8.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31:410–417. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 9.Johnson AK, Zardetto-Smith AM, Edwards GL. Integrative mechanisms and the maintenance of cardiovascular and body fluid homeostasis: the central processing of sensory input derived from the circumventricular organs of the lamina terminalis. Prog Brain Res. 1992;91:381–393. doi: 10.1016/s0079-6123(08)62357-2. [DOI] [PubMed] [Google Scholar]
- 10.Lind RW, Swanson LW, Ganten D. Organization of angiotensin II immunoreactive cells and fibers in the rat central nervous system: an immunohistochemical study. Neuroendocrinology. 1985;40:2–24. doi: 10.1159/000124046. [DOI] [PubMed] [Google Scholar]
- 11.Mendelsohn FA, Quirion R, Saavedra JM, Aguilera G, Catt KJ. Autoradiographic localization of angiotensin II receptors in rat brain. Proc Natl Acad Sci U S A. 1984;81:1575–1579. doi: 10.1073/pnas.81.5.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson AV. Systemic angiotensin acts at the subfornical organ to control the activity of paraventricular nucleus neurons with identified projections to the median eminence. Neuroendocrinology. 1988;47:489–497. doi: 10.1159/000124960. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Ferguson AV. Subfornical organ efferents to paraventricular nucleus utilize angiotensin as a neurotransmitter. Am J Physiol. 1993;265:302–309. doi: 10.1152/ajpregu.1993.265.2.R302. [DOI] [PubMed] [Google Scholar]
- 14.Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res. 1992;599:223–229. doi: 10.1016/0006-8993(92)90395-p. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka J, Kaba H, Saito H, Seto K. Subfornical organ neurons with efferent projections to the hypothalamic paraventricular nucleus: an electrophysiological study in the rat. Brain Res. 1985;346:151–154. doi: 10.1016/0006-8993(85)91106-0. [DOI] [PubMed] [Google Scholar]
- 16.Cox BF, Bishop VS. Neural and humoral mechanisms of angiotensin-dependent hypertension. Am J Physiol. 1991;261:1284–1291. doi: 10.1152/ajpheart.1991.261.4.H1284. [DOI] [PubMed] [Google Scholar]
- 17.LaGrange LP, Toney GM, Bishop VS. Chronic angiotensin II infusion attenuates the renal sympathoinhibitory response to acute volume expansion. Am J Physiol Regul Integr Comp Physiol. 2003;284:1098–1107. doi: 10.1152/ajpregu.00165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latchford KJ, Ferguson AV. Angiotensin II activates a nitric-oxide-driven inhibitory feedback in the rat paraventricular nucleus. J Neurophysiol. 2003;89:1238–1244. doi: 10.1152/jn.00914.2002. [DOI] [PubMed] [Google Scholar]
- 19.Li DP, Chen SR, Pan HL. Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci. 2003;23:5041–5049. doi: 10.1523/JNEUROSCI.23-12-05041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin DS, Haywood JR. Reduced GABA inhibition of sympathetic function in renal-wrapped hypertensive rats. Am J Physiol. 1998;275:1523–1529. doi: 10.1152/ajpregu.1998.275.5.R1523. [DOI] [PubMed] [Google Scholar]
- 21.LaGrange LP, Toney GM, Bishop VS. Chronic angiotensin II (AII) infusion abolishes GABA-A receptor blockade-induced renal sympathoexcitation. FASEB J. 2003;17:824.6. Abstract. [Google Scholar]
- 22.Tagawa T, Dampney RA. AT(1) receptors mediate excitatory inputs to rostral ventrolateral medulla pressor neurons from hypothalamus. Hypertension. 1999;34:1301–1307. doi: 10.1161/01.hyp.34.6.1301. [DOI] [PubMed] [Google Scholar]
- 23.DiBona GF, Jones SY. Effect of dietary sodium intake on the responses to bicuculline in the paraventricular nucleus of rats. Hypertension. 2001;38:192–197. doi: 10.1161/01.hyp.38.2.192. [DOI] [PubMed] [Google Scholar]
- 24.Kannan H, Hayashida Y, Yamashita H. Increase in sympathetic outflow by paraventricular nucleus stimulation in awake rats. Am J Physiol. 1989;256:R1325–R1330. doi: 10.1152/ajpregu.1989.256.6.R1325. [DOI] [PubMed] [Google Scholar]
- 25.Lovick TA, Coote JH. Effects of volume loading on paraventriculospinal neurones in the rat. J Auton Nerv Syst. 1988;25:135–140. doi: 10.1016/0165-1838(88)90018-5. [DOI] [PubMed] [Google Scholar]
- 26.Haselton JR, Goering J, Patel KP. Parvocellular neurons of the paraventricular nucleus are involved in the reduction in renal nerve discharge during isotonic volume expansion. J Auton Nerv Syst. 1994;50:1–11. doi: 10.1016/0165-1838(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 27.Cowley AC, McCaa RE. Acute and chronic dose-response relationships for angiotensin, aldosterone, and arterial pressure at varying levels of sodium intake. Circ Res. 1976;39:788–797. doi: 10.1161/01.res.39.6.788. [DOI] [PubMed] [Google Scholar]
- 28.Dendorfer A, Thornagel A, Raasch W, Grisk O, Tempel K, Dominiak P. Angiotensin II induces catecholamine release by direct ganglionic excitation. Hypertension. 2002;40:348–354. doi: 10.1161/01.hyp.0000028001.65341.aa. [DOI] [PubMed] [Google Scholar]
- 29.Brooks VL, Ell KR, Wright RM. Pressure-independent baroreflex resetting produced by chronic infusion of angiotensin II in rabbits. Am J Physiol. 1993;265:1275–1282. doi: 10.1152/ajpheart.1993.265.4.H1275. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka J, Kaba H, Saito H, Seto K. Electrophysiological evidence that circulating angiotensin II sensitive neurons in the subfornical organ alter the activity of hypothalamic paraventricular neurohypophyseal neurons in the rat. Brain Res. 1985;342:361–365. doi: 10.1016/0006-8993(85)91137-0. [DOI] [PubMed] [Google Scholar]
- 31.Czyzewska-Szafran H, Wutkiewicz M, Remiszewska M, Jastrzebski Z, Czarnecki A, Danysz A. Down-regulation of the GABA-ergic system in selected brain areas of spontaneously hypertensive rats (SHR) Pol J Pharmacol Pharm. 1989;41:619–627. [PubMed] [Google Scholar]
- 32.Kunkler PE, Hwang BH. Lower GABAA receptor binding in the amygdala and hypothalamus of spontaneously hypertensive rats. Brain Res Bull. 1995;36:57–61. doi: 10.1016/0361-9230(94)00164-v. [DOI] [PubMed] [Google Scholar]
- 33.Haywood JR, Mifflin SW, Craig T, Calderon A, Hensler JG, Hinojosa-Laborde C. Gamma-aminobutyric acid (GABA): a function and binding in the paraventricular nucleus of the hypothalamus in chronic renal-wrap hypertension. Hypertension. 2001;37:614–618. doi: 10.1161/01.hyp.37.2.614. [DOI] [PubMed] [Google Scholar]
- 34.Pickel VM, Chan J. Co-localization of angiotensin II and gamma-aminobutyric acid in axon terminals in the rat subfornical organ. Neurosci Lett. 1995;193:89–92. doi: 10.1016/0304-3940(95)11673-k. [DOI] [PubMed] [Google Scholar]
- 35.Hadjiivanova CH, Georgiev V. In vitro effect of angiotensin II on GABA release in rat hippocampus. Neuropeptides. 1998;32:431–434. doi: 10.1016/s0143-4179(98)90067-1. [DOI] [PubMed] [Google Scholar]
- 36.Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci. 1999;19:674–683. doi: 10.1523/JNEUROSCI.19-02-00674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sigel E. Functional modulation of ligand-gated GABAA and NMDA receptor channels by phosphorylation. J Recept Signal Transduct Res. 1995;15:325–332. doi: 10.3109/10799899509045224. [DOI] [PubMed] [Google Scholar]
- 38.Zhu M, Gelband CH, Posner P, Sumners C. Angiotensin II decreases neuronal delayed rectifier potassium current: role of calcium/calmodulin-dependent protein kinase II. J Neurophysiol. 1999;82:1560–1568. doi: 10.1152/jn.1999.82.3.1560. [DOI] [PubMed] [Google Scholar]