

Abstract
Objectives
Increased sympathetic outflow, renin–angiotensin system (RAS) activity, and oxidative stress are critical mechanisms underlying the adverse cardiovascular effects of dietary salt excess. Nebivolol is a third-generation, highly selective β1-receptor blocker with RAS-reducing effects and additional antioxidant properties. This study evaluated the hypothesis that nebivolol reduces salt-induced cardiac remodeling and dysfunction in spontaneous hypertensive rats (SHRs) by suppressing cardiac RAS and oxidative stress.
Methods
Male SHRs (8 weeks of age) were given an 8% high salt diet (HSD; n = 22), whereas their age-matched controls (n = 10) received standard chow. In a subgroup of HSD rats (n = 11), nebivolol was given at a dose of 10 mg/kg per day by gastric gavage.
Results
After 5 weeks, HSD exacerbated hypertension as well as increased left-ventricular weight and collagen deposition while impairing left-ventricular relaxation. Salt-induced cardiac remodeling and dysfunction were associated with increased plasma renin concentration (PRC), cardiac angiotensin II immunostaining, and angiotensin-converting enzyme (ACE)/ACE2 mRNA and activity ratio. HSD also increased cardiac 3-nitrotyrosine staining indicating enhanced oxidative stress. Nebivolol treatment did not alter the salt-induced increase in arterial pressure, left-ventricular weight, and cardiac dysfunction but reduced PRC, cardiac angiotensin II immunostaining, ACE/ACE2 ratio, oxidative stress, and fibrosis.
Conclusions
Our data suggest that nebivolol, in a blood pressure-independent manner, ameliorated cardiac oxidative stress and associated fibrosis in salt-loaded SHRs. The beneficial effects of nebivolol may be attributed, at least in part, to the decreased ACE/ACE2 ratio and consequent reduction of cardiac angiotensin II levels.
Keywords: β1-adrenergic receptors, angiotensin II, blood pressure, cardiac fibrosis, nebivolol, salt
INTRODUCTION
Hypertensive left-ventricular hypertrophy associated with myocardial fibrosis and ischemia is one of the major risk factors underlying cardiovascular morbidity and mortality. A growing body of evidence derived from epidemiological, interventional, and experimental studies relates salt intake not only to blood pressure regulation but also to left-ventricular remodeling and dysfunction [1-5]. Various mechanisms have been suggested to explain the detrimental effects of dietary salt excess beyond its effect on hemodynamic load. Increased sympathetic outflow and renin–angiotensin system (RAS) activity as well as decreased nitric oxide bioavailability associated with increased oxidative stress have been increasingly appreciated as critical mechanisms underlying the adverse cardiovascular effects of dietary salt excess [6-10]. Therefore, nebivolol, a highly selective β1-adrenergic receptor blocker with antioxidant and vasodilator/antiproliferative potential related to an increased nitric oxide bioavailability [11], may be beneficial in the treatment of cardiovascular injury related to dietary salt excess. We recently showed that nebivolol treatment, in a blood pressure-independent manner, ameliorated renal injury and dysfunction caused by salt-induced renal oxidative stress in spontaneously hypertensive rats (SHRs), a well characterized experimental model of human essential hypertension [12]. Similarly, nebivolol improved diastolic dysfunction and myocardial remodeling associated with insulin resistance in the Zucker obese rats by reducing cardiac oxidative stress [13]. Although β1-adrenergic receptor stimulation up-regulates angiotensin-converting enzyme (ACE) in isolated cardio-myocyte as well as in the heart of normotensive rats [14,15], no data exist regarding the effects of nebivolol on the cardiac RAS. Thus, to further investigate the potential beneficial cardiac effects of nebivolol, we tested the hypothesis that nebivolol improves salt-induced cardiac dysfunction and remodeling in SHRs by suppressing cardiac RAS and oxidative stress.
METHODS
Animals
Eight week-old male SHRs (Charles River) were given 8% salt diet for 5 weeks (n = 22); their age and sex-matched controls received standard chow (control; 1% salt; n = 10). Animals from the salt-loaded group were randomized to receive nebivolol [high salt diet (HSD) and Neb; n = 11; 10 mg/kg per day] or the vehicle (HSD; n = 11; gum gastric 5% Arabica) by gavage. All rats were permitted free access to chow and tap water and were maintained in a temperature and humidity-controlled room with a 12-h light/dark cycle. The rats were handled in accordance with National Institute of Health guidelines; our Institutional Animal Care and Use Committee approved the study in advance.
Cardiac function
At the end of study, the rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.), and a catheter-tip transducer (2F; Millar Instruments, Houston, Texas, USA) was introduced into the left ventricle (LV) via the right carotid artery [6]. The catheter was connected to a pressure unit (MPVS 300; Millar Instruments) and the signal was then fed to a data acquisition system (Emka Technologies, Falls Church, Virginia, USA). Arterial pressure [systolic (SAP), diastolic (DAP), and mean arterial pressure (MAP)] records were obtained from a carotid artery before cardiac function measurements. Several indices of cardiac function including maximal developed pressure (Pmax), end-diastolic pressure (Ped), maximum rates of pressure rise +dP/dTmax, and time constant of isovolumic relaxation (tau; a load-independent index of the active phase of early diastolic relaxation) were obtained from ventricular pressure tracings. After the parameters of cardiac function were obtained the rats were then euthanized by an overdose of intravenously (i.v.) administered pentobarbital sodium, their hearts were quickly removed, and the ventricles were separated and weighed.
Biochemical analysis
A separate groups of rats [control (n = 6), HSD (n = 7), and HSD and Neb (n = 7)] the following same protocol were decapitated and trunk blood was collected for measurements of plasma renin concentration (PRC) as well as plasma 8-isoprostane as a measure of systemic oxidative stress. PRC was defined as the rate of Ang I generation from renin in the sample incubated at pH 6.5 for 90 min with excess exogenous substrate provided from nephrectomized rat plasma. Ang I generated in the sample was quantified by radioimmunoassay (Diosarin Corp, Stillwater, Minnesota, USA). Plasma isoprostane was determined by ELISA (Cayman Chemical).
Cardiac renin-angiotensin system, oxidative stress, and fibrosis
Hearts were fixed in 3% paraformaldehyde and a transverse section was used for histological examination of fibrosis as well as immunostaining for oxidative stress and RAS components. Fixed paraffin sections of the hearts were evaluated for 3-nitrotyrosine (3-NT) immunostaining as a marker for oxidative stress (primary rabbit polyclonal antinitrotyrosine antibody; 1: 200; Chemicon, Temecula, California, USA; ×400 original magnification) as described previously [16]. Sections were also examined for angiotensin (Ang) II (primary goat polyclonal antibody; 1: 100; Santa Cruz Biotechnology, Santa Cruz, California, USA), Ang-(1–12) (primary rabbit polyclonal Ang (1–12); 1: 50; Ana-Spec, San Jose, California, USA), and Ang II type 1 receptor (AT1) (primary rabbit polyclonal antibody; 1: 100; Santa Cruz Biotechnology) immunostaining as described previously [17]. Appropriate fluorescent secondary antibodies (Alexa fluor 647 donkey antigoat or antirabbit; 1: 400; Molecular Probes, Eugene, Oregon, USA) were used for visualization. Slides were examined under a biphoton confocal microscope (Zeiss LSM; 510 MLO, Thornwood, New York, USA) and captured images (×400; four fields) were analyzed and quantified with MetaView software (Boyce Scientific, Gary Summit, Missouri, USA). Fibrosis was evaluated on slides stained with picrosirius red as described earlier [1,6,18]. Eight images (×400 original magnification) per section were captured under bright light using Spot Advanced software 4.0.9 (Diagnostic Instruments, Inc., Sterling Heights, Michigan, USA) connected to an Olympus BX60 microscope (Olympus America, Inc., Center Valley, Pennsylvania, USA). The color images were converted to grayscale and analyzed by a blinded individual for interstitial collagen using Adobe Photoshop CS2 (Adobe Systems, San Jose, California, USA) and the results were expressed as percentages of the total area.
For cardiac ACE and ACE2 activity solubilized membranes were used as the source of peptidase activity [19]. 125I-Ang I and Ang II (2 × 106 counts/min, 2200 curies/mmol) were added with various inhibitors and the enzymatic products [(Ang II or Ang-(1–7)] were quantified on HPLC as previously described [19]. The following agents comprised the inhibitor cocktail in the assay: amastatin (10 μmol/l), bestatin (50 μmol/l), chymostatin (50 μmol/l), benzyl succinate (50 μmol/l), and parachloromercuribenzoic acid (250 μmol/l). MLN-4760 (50 μmol/l) and lisinopril (50 μmol/l) were added to inhibit ACE2 and ACE activity, respectively.
Nitric oxide metabolites (NOx) in tissue samples were determined using a Griess assay (nitrate/nitrite colorimetric assay kit; Alexis Biomedicals, California, USA) [12].
Gene expression analysis
Additional left-ventricular tissue samples from the apical portion were snap-frozen in liquid nitrogen and stored at −80°C for reverse transcriptase real-time PCR analysis of mRNA for ACE, ACE2 as well as p22phox and gp92phox, the critical components of NADPH oxidase [20]. RNA was isolated from tissue, using the TRIZOL reagent (GIBCO Invitrogen, Carlsbad, California, USA), as directed by the manufacturer. The RNA concentration and integrity were assessed using an Agilent 2100 Bioanalyzer with an RNA 6000 Nano LabChip (Agilent Technologies, Palo Alto, California, USA). Total RNA was reverse-transcribed using Avian Maloney Virus reverse transcriptase in a reaction mixture containing deoxyribonucleotides, random hexamers, and RNase inhibitor in reverse transcriptase buffer. Heating the reverse transcriptase reaction product at 95°C terminated the reaction. For real-time PCR, the resultant cDNA was added to TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, California, USA) with the appropriate gene-specific primer/probe set (Applied Biosystems) and amplification was performed on an ABI 7000 Sequence Detection System. The mixtures were heated at 50°C for 2 min, at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. All reactions were performed in triplicate and 18S ribosomal RNA, amplified using the TaqMan Ribosomal RNA Control Kit (Applied Biosystems), served as an internal control. The results were quantified as Ct values, where Ct is defined as the threshold cycle of PCR at which amplified product is first detected, and defined as relative gene expression (the ratio of target/control).
Statistics
All values are expressed as the mean ± 1 standard error of the mean (SEM). Data were analyzed by use of analysis of variance (ANOVA) followed by Newman–Keuls’ post-test. A value of P less than 0.05 was considered to be of statistical significance.
RESULTS
Direct measurements of blood pressure in anesthetized SHRs are presented in Table 1. As expected, high salt intake increased SBP, DBP, and MAP, whereas exposure to nebivolol had no effect on the salt-induced rise in arterial pressure. When compared to the control SHRs, SHRs fed a HSD had decreased heart rate. The rats treated with nebivolol had lower heart rates when compared to both untreated salt-loaded SHRs and the rats on the control diet. These data confirmed the effectiveness of nebivolol in blocking cardio-selective β1-receptors.
TABLE 1.
Body weight, arterial pressure, heart rate, isoprostane, PRC, cardiac NOx and fibrosis in nebivolol-treated SHRs fed HSD
| Control | High salt | High salt and nebivolol | |
|---|---|---|---|
| Body weight (g) | 304 ± 4 | 233 ± 9* | 264 ± 4*,# |
| SAP (mmHg) | 194 ± 6 | 234 ± 9* | 235 ± 8* |
| DAP (mmHg) | 145 ± 6 | 175 ± 5* | 170 ± 5* |
| MAP (mmHg) | 168 ± 6 | 201 ± 6* | 197 ± 6* |
| HR (beats/min) | 433 ± 11 | 391± 11* | 358 ± 9*,# |
| Plasma 8-isoprostane (pg/ml) | 133 ± 20 | 538±116* | 133±13* |
| PRC (ng/ml per h) | 2.08 ± 0.59 | 12.23 ± 3.24* | 1.21 ± 0.58* |
| Cardiac NOx (μg/mg protein) | 0.11 ± 0.01 | 0.14 ± 1.01 | 0.12 ± 0.01 |
| Fibrosis (% of total area) | 3.41 ± 0.48 | 13.05 ± 2.38* | 6.26 ± 0.97* |
Results are mean ± SEM. DAP, diastolic arterial pressure; HR, heart rate; MAP, mean arterial pressure; NOx, nitric oxide metabolites (nitrite and nitrate); PRC, plasma renin concentration; SAP, systolic arterial pressure.
P<0.05 vs. control.
P< 0.05 vs. untreated salt-loaded group.
Body weight was decreased in both salt-loaded groups of rats but nebivolol attenuated this effect (Table 1). Left-ventricular weight increased in both untreated and nebivolol-treated salt-loaded SHRs when compared to the rats on the control diet; however, nebivolol-treated rats had significantly higher left-ventricular weight when compared to the untreated salt-loaded SHRs (Fig. 1). In regards to right ventricle (RV), there was no significant difference in right-ventricular weight between untreated SHRs fed HSD and control SHRs; right-ventricular weight was increased in nebivolol-treated rats when compared to the untreated salt-loaded rats. Consequently, the LV/RV ratio, a marker for left-ventricular hypertrophy, was not different between the untreated and nebivolol-treated SHRs fed HSD, and was increased approximately to the same degree in both salt-loaded groups when compared to the control rats. Furthermore, high dietary salt intake increased cardiac fibrosis in SHRs (Table 1 and Fig. 2). Nebivolol treatment significantly attenuated the increased collagen deposition within the hypertrophied heart of salt-loaded rats (Table 1 and Fig. 2).
FIGURE 1.

Effects of high salt diet (HSD) diet and nebivolol (Neb) on left (LV) and right (RV) ventricular weight. *P < 0.05 vs. control. #P < 0.05 vs. untreated salt-loaded group.
FIGURE 2.
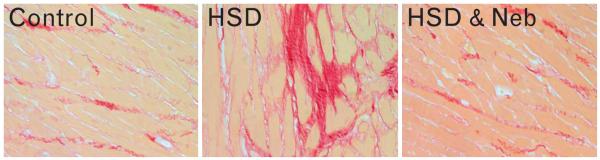
Interstitial fibrosis in the heart of control (left), salt-loaded (HSD; middle), and nebivolol-treated SHRs (HSD and Neb; right) (picrosirius red staining; ×400 original magnification). HSD, high salt diet; SHRs, spontaneous hypertensive rats.
Cardiac functional studies (Fig. 3) revealed impaired relaxation in untreated SHRs fed HSD as manifested by increased time constant of relaxation (tau). Nebivolol did not improve diastolic dysfunction in rats fed HSD. There were no differences in systolic function (dP/dTmax/Pmax) and Ped among the experimental groups.
FIGURE 3.

Effects of nebivolol on cardiac function of salt-loaded SHRs. Pmax = maximal developed left ventricular pressure, Ped end-diastolic pressure, +dP/dTmax = maximum rates of pressure rise, tau time constant of isovolumic relaxation; *P < 0.05 vs. control, #P < 0.05 vs. untreated salt-loaded group. HSD, high salt diet; Neb, nebivolol; SHRs, spontaneous hypertensive rats.
Plasma 8-isoprostane, a measure of systemic oxidative stress, was greater in untreated salt-loaded SHRs compared to the control rats, and nebivolol treatment significantly reduced the concentration of this biomarker (Table 1). Similarly, in untreated rats fed the HSD cardiac immunostaining for 3-NT was increased, suggestive of increased cardiac oxidative stress (Fig. 4). In contrast, nebivolol decreased 3-NT to the level observed in the control rats. Assessment of the gene expression of gp91phox and p22phox, the two critical components of NADPH oxidase, showed no significant change among the three experimental groups (Fig. 4). There was no difference in cardiac NOx between the three experimental groups (Table 1).
FIGURE 4.

Effects of nebivolol on cardiac oxidative stress in heart tissue of salt-loaded SHRs. 3-nitrotyrosine staining (3-NT; ×400 original magnification) and gene expression of gp91phox and g22phox. *P < 0.05 vs. control, #P < 0.05 vs. untreated salt-loaded group. HSD, high salt diet; Neb, nebivolol; SHRs, spontaneous hypertensive rats.
Consistent with an increased oxidative stress, increased PRC (Table 1) and cardiac Ang II immunostaining (Fig. 5) were found in untreated rats fed HSD. Nebivolol prevented the increase in both PRC and cardiac Ang II immunostaining (Table 1 and Fig. 5). Neither the salt diet nor nebivolol treatment affected the immunoreactive staining of AT1 receptors or Ang-(1–12) (Fig. 5), an extended form of Ang I found in other studies to form angiotensin peptides in both the heart and kidney [21]. The gene expression of two critical enzymes modulating Ang II formation and metabolism showed that salt loading was associated with increased cardiac ACE mRNA and no changes in ACE2 mRNA (Table 2). On the contrary, administration of nebivolol reversed the increase in ACE mRNA induced by the salt diet while now augmenting the expression of ACE2 mRNA (Table 2). The ACE activity corresponded to the changes in the gene expression, whereas ACE2 activity was nonsignificantly increased (24%) in nebivolol-treated rats when compared to the untreated SHRs (Table 2). Consequently, the elevated ACE/ACE2 mRNA and activity ratio found in untreated SHRs fed HSD was reduced by nebivolol treatment (Fig. 6).
FIGURE 5.

Cardiac immunostaining for Ang II (top), AT1 receptor (middle) and Ang-(1–12) (bottom) in the control and untreated and nebivolol-treated SHRs fed HSD (×400 original magnification). *P < 0.05 vs. control. #P < 0.05 vs. untreated salt-loaded group. HSD, high salt diet; Neb, nebivolol; SHRs, spontaneous hypertensive rats.
TABLE 2.
ACE and ACE2 in nebivolol-treated SHRs fed a HSD
| Control | High salt | High salt and nebivolol | |
|---|---|---|---|
| ACE mRNA (fold increase in arbitrary units) | 1.00 ± 0.06 | 2.00 ± 0.31* | 1.07 ± 0.11# |
| ACE2 mRNA (fold increase in arbitrary units) | 1.06 ± 0.06 | 0.78 ± 0.09 | 1.14 ± 0.12# |
| ACE activity (fmol/ml per min) | 4.39 ± 0.61 | 8.39 ± 1.00* | 4.37 ± 0.57# |
| ACE2 activity (fmol/ml per min) | 15.53 ± 2.26 | 14.63 ± 2.49 | 18.09 ± 1.56 |
Results are mean ±SEM. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; SHRs, spontaneous hypertensive rats.
p < 0.05 vs. control.
P< 0.05 vs. untreated salt-loaded group.
FIGURE 6.
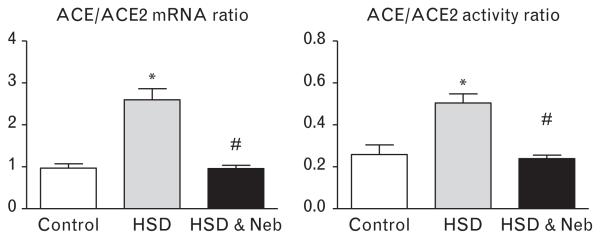
Cardiac ACE/ACE2 ratio in SHRs fed HSD and treated with nebivolol. *P < 0.05 vs. control, #P < 0.05 vs. untreated salt-loaded group. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; HSD, high salt diet; Neb, nebivolol; SHRs, spontaneous hypertensive rats.
DISCUSSION
Nebivolol, a potent cardioselective β1-receptor blocker with additional antioxidative and nitric oxide-releasing properties, reduced systemic and cardiac oxidative stress and associated cardiac fibrosis in SHRs fed a HSD. The decrease in oxidative stress was accompanied by reduced cardiac Ang II immunostaining potentially reflecting the effect of nebivolol on ACE/ACE2, the two critical enzymes involved in Ang II formation and metabolism, respectively. The beneficial effects of nebivolol on cardiac oxidative stress were not associated with changes in arterial pressure, cardiac function, and cardiac nitric oxide metabolites.
It is important to note that the current study confirmed our previous findings that nebivolol did not affect salt-related increase in arterial pressure [12]. These data underscore the view that dietary salt excess attenuates the anti-hypertensive properties of drug therapies [22-24]. In our previous study nebivolol did not facilitate systemic vascular nitric oxide availability in salt-loaded SHRs [12]. We extended these findings by now demonstrating that nebivolol, in concert with its antioxidative properties, reduced systemic oxidative stress. Thus, our results suggest that exacerbation of hypertension in SHRs fed HSD is not related to an increase in systemic oxidative stress or decreased nitric oxide availability due to increased nitric oxide scavenge by reactive oxygen species (ROS). Although increased systemic ROS production was coupled with development of hypertension [9,25], other studies suggest that vascular oxidative stress does not determine blood pressure regulation [26,27]. Further studies are necessary to elucidate the exact mechanisms underlying a salt-related blood pressure elevation in hypertensive patients. The data reported in this study do not negate a beneficial effect of this drug in hypertension, not aggravated by salt, since potent anti-hypertensive effects have been reported in SHRs even after treatment withdrawal [28]. Additional studies showed that nebivolol improved arterial structure coupled with improved endothelium-dependent relaxation [28,29]. Vasodilatory properties of nebivolol are mainly related to its ability to stimulate endothelial-dependent nitric oxide release shown not only in experimental animals [11] but also in healthy volunteers [30] and hypertensive patients [31].
Activation of β1-adrenergic receptors is one of the blood pressure-independent mechanisms contributing to cardiac hypertrophy and fibrosis [32,33]. To the best of our knowledge, this is the first study to show that nebivolol effectively reduced salt-related cardiac fibrosis independent of its effect on blood pressure. Nebivolol may have also exerted its antifibrotic effects by reducing salt-related cardiac oxidative stress reflected in pronounced 3-NT staining. It has been shown that gp91phox is critical for development of cardiac fibrosis in response to either pressure overload or RAS stimulation [34,35]. Moreover, left-ventricular failure in Dahl salt-sensitive rats was associated with increased ROS production and p22phox and gp91phox up-regulation that was corrected with both AT1 receptor antagonist and tempol [36]. We also previously found an increased gp91phox protein in the kidney of SHRs fed HSD [12]. However, in the present study, gene expression of the two critical components of NADPH oxidase, p22phox and gp91phox, was not altered with high dietary salt intake. Further studies are warranted to explore additional potential sources for ROS generation in response to dietary salt excess in SHRs such as xanthine oxidase, uncoupled nitric oxide synthase, mitochondria, and cytochrome P-460 enzymes [37]. Similarly, nebivolol did not affect the gene expression of these two NADPH oxidase subunits; however, its antioxidant properties could have arisen from its intrinsic direct superoxide scavenger properties [11].
β1-adrenergic receptor blockers have been used clinically for a long time in the treatment of different cardiovascular diseases, including hypertension as well as impaired renal and cardiac function [38-40]. Their beneficial effects are linked not only to their ability to decrease enhanced sympathetic drive but also to reduce renin release [41]. In the present study, we report that the beneficial antifibrotic and antioxidative effects of nebivolol in SHRs fed a HSD were associated with not only suppression of PRC but also with reduction in the cardiac expression of Ang II. It is well accepted that increased Ang II activity is linked to development of cardiac hypertrophy and fibrosis as well as dysfunction. Moreover, the reduction in cardiac Ang II content in nebivolol-treated SHRs fed HSD may be seen in the context of nebivolol’s ability to reduce the elevated ACE/ACE2 activity ratio observed in untreated rats. ACE has been involved not only in Ang II formation but also the metabolism of the antiproliferative and vasodilator actions of Ang-(1–7) [42-44], whereas ACE2 preferentially cleaves Ang II to form Ang-(1–7) [45]. Moreover, decreased cardiac Ang-(1–7) was associated with pronounced left-ventricular remodeling in SHRs fed HSD [18]. Thus, the imbalance between ACE/ACE2 may have favored Ang II over Ang-(1–7) in salt-loaded rats contributing to the specific phenotype observed in this group. Consistent with a multilevel interaction between sympathetic and RAS systems [46], we recently showed that nebivolol prevented the salt-induced reduction in Ang-(1–7) receptor mas along with improvement in renal blood flow, urinary protein excretion, and collagen deposition in salt-loaded SHRs [12]. On the contrary, Ang-(1–7)-producing fusion protein in the heart attenuated isoproterenol-induced cardiac fibrosis [47]. Future studies will explore in more details the contribution of counterbalancing Ang-(1–7) to the cardiac beneficial effects of nebivolol and the potential underlying mechanisms.
We also examined the expression of a novel angiotensin peptide precursor, Ang-(1–12) [21,48], in an attempt to further characterize the cardiac RAS under the condition of dietary salt excess and/or nebivolol treatment. Neither dietary salt excess nor nebivolol treatment altered the Ang-(1–12) level in the heart of SHRs. Since Ang II formation from Ang-(1–12) occurs via a nonrenin pathway in both the heart [49] and the systemic circulation [17], the current finding of unchanged Ang-(1–12) expression in the presence of PRC changes are in keeping with previous observations. Nagata et al. [50] recently reported that cardiac level of Ang-(1–12) did not change after exposure of normotensive rats to diets with different sodium content. They showed that increased plasma renin activity in response to low-salt diet was not accompanied with changes in Ang I, Ang II, and Ang-(1–12) in the heart. It is therefore possible that the absence of changes in the expression of cardiac Ang-(1–12) in response to either dietary sodium or the combination of dietary sodium and β1-receptor blockade reflects that this substrate is regulated by mechanisms that are independent of both renin and the deleterious effects of salt excess.
Finally, a growing body of evidence argues for favorable effects of nebivolol on left-ventricular dysfunction [13,51-53]. However, despite its beneficial effects on cardiac fibrosis and oxidative stress in SHRs fed a HSD in the present study, nebivolol did not improve impaired ventricular diastolic function nor did it reduce left-ventricular mass. Failure to improve cardiac function and left-ventricular hypertrophy may be related to the combined effects of salt-loading and hypertension since in the Zucker rat, a model of obesity and hypertension, nebivolol reduces myocardial structural maladaptive changes and improves diastolic relaxation in concert with improvements in insulin sensitivity and endothelial nitric oxide synthase activation [13]. Although increased left-ventricular wall thickness in both untreated and nebivolol-treated rats may have enhanced myocardial oxygen demand compromising coronary circulation and left-ventricular relaxation, decreased oxidative stress in rats treated with β1-adrenergic receptor blocker argues against compromised myocardial oxygen supply in these rats.
Consistent with this notion, decreased heart rate by nebivolol treatment would reduce oxygen consumption while prolonging diastolic time and in this way improve coronary hemodynamics since coronary flow occurs mainly through this phase of the cardiac cycle [54]. Thus, other mechanism of still impaired relaxation in nebivolol-treated rats should be explored. In addition to alteration in myofilament properties and cytoskeletal architecture, alteration in calcium handling has been linked to diastolic dysfunction in hypertrophied LV [55] and Ang II has an important contributory role [56]. Importantly, AT1 receptor antagonism under similar experimental conditions beneficially affected both left-ventricular hypertrophy and fibrosis improving left-ventricular dysfunction and coronary hemodynamic impairment, yet similarly to nebivolol, it did not prevent salt-induced rise in arterial pressure [6]. It is quite possible that, whereas nebivolol reduced Ang II in the heart of salt-loaded rats to the level comparable to the control rats, even that level of Ang II in response to an increase in salt intake might be inappropriately high.
In summary, our data suggest that nebivolol, in a blood pressure-independent manner, ameliorated cardiac oxidative stress and associated fibrosis but not left-ventricular dysfunction in salt-loaded SHRs. The beneficial effects of nebivolol may be attributed, at least in part, to the decreased ACE/ACE2 ratio and consequent reduction of cardiac Ang II levels.
ACKNOWLEDGEMENTS
J.V. and C.M.F. received investigator-initiated support from the Forest Research Institute.
The study was supported by the grants from Forest Research Institute (J.V. and C.M.F.), American Heart Association (2300114 to J.V.), NIH (2PO1 HL-051952 to C.M.F.). We also acknowledge partial support provided by the Farley-Hudson Foundation, Jacksonville, North Carolina, USA.
Abbreviations
- ACE
angiotensin-converting enzyme
- Ang
angiotensin
- dP/dTmax
maximum rate of pressure rise
- Ped
end-diastolic pressure
- PRC
plasma renin concentration
- RAS
renin–angiotensin system
- SHRs
spontaneously hypertensive rats
- tau
time constant of isovolumic relaxation
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Ahn J, Varagic J, Slama M, Susic D, Frohlich ED. Cardiac structural and functional responses to salt loading in SHR. Am J Physiol Heart Circ Physiol. 2004;287:H767–H772. doi: 10.1152/ajpheart.00047.2004. [DOI] [PubMed] [Google Scholar]
- 2.Du CG, Ribstein J, Grolleau R, Mimran A. Influence of sodium intake on left ventricular structure in untreated essential hypertensives. J Hypertens Suppl. 1989;7:S258–S259. doi: 10.1097/00004872-198900076-00125. [DOI] [PubMed] [Google Scholar]
- 3.Du CG, Ribstein J, Mimran A. Dietary sodium and target organ damage in essential hypertension. Am J Hypertens. 2002;15:222–229. doi: 10.1016/s0895-7061(01)02287-7. [DOI] [PubMed] [Google Scholar]
- 4.Frohlich ED, Varagic J. The role of sodium in hypertension is more complex than simply elevating arterial pressure. Nat Clin Pract Cardiovasc Med. 2004;1:24–30. doi: 10.1038/ncpcardio0025. [DOI] [PubMed] [Google Scholar]
- 5.Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, Johnston CI. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–2628. doi: 10.1161/01.cir.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 6.Varagic J, Frohlich ED, Susic D, Ahn J, Matavelli L, Lopez B, Diez J. AT1 receptor antagonism attenuates target organ effects of salt excess in SHRs without affecting pressure. Am J Physiol Heart Circ Physiol. 2008;294:H853–H858. doi: 10.1152/ajpheart.00737.2007. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension. 2001;38:606–611. doi: 10.1161/hy09t1.094005. [DOI] [PubMed] [Google Scholar]
- 8.Leenen FH, Yuan B. Dietary-sodium-induced cardiac remodeling in spontaneously hypertensive rat versus Wistar-Kyoto rat. J Hypertens. 1998;16:885–892. doi: 10.1097/00004872-199816060-00020. [DOI] [PubMed] [Google Scholar]
- 9.Welch WJ, Mendonca M, Blau J, Karber A, Dennehy K, Patel K, et al. Antihypertensive response to prolonged tempol in the spontaneously hypertensive rat. Kidney Int. 2005;68:179–187. doi: 10.1111/j.1523-1755.2005.00392.x. [DOI] [PubMed] [Google Scholar]
- 10.Susic D, Frohlich ED, Kobori H, Shao W, Seth D, Navar LG. Salt-induced renal injury in SHRs is mediated by AT1 receptor activation. J Hypertens. 2011;29:716–723. doi: 10.1097/HJH.0b013e3283440683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason RP, Kubant R, Jacob RF, Walter MF, Boychuk B, Malinski T. Effect of nebivolol on endothelial nitric oxide and peroxynitrite release in hypertensive animals: role of antioxidant activity. J Cardiovasc Pharmacol. 2006;48:862–869. doi: 10.1097/01.fjc.0000238593.67191.e2. [DOI] [PubMed] [Google Scholar]
- 12.Varagic J, Ahmad S, Brosnihan KB, Habibi J, Tilmon RD, Sowers JR, Ferrario CM. Salt-induced renal injury in spontaneously hypertensive rats: effects of nebivolol. Am J Nephrol. 2010;32:557–566. doi: 10.1159/000321471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Ma L, Habibi J, Whaley-Connell A, Hayden MR, Tilmon RD, et al. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the Zucker obese rat. Hypertension. 2010;55:880–888. doi: 10.1161/HYPERTENSIONAHA.109.145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busatto VC, Cunha V, Cicilini MA, Mill JG. Differential effects of isoproterenol on the activity of angiotensin-converting enzyme in the rat heart and aorta. Braz J Med Biol Res. 1999;32:355–360. doi: 10.1590/s0100-879x1999000300017. [DOI] [PubMed] [Google Scholar]
- 15.Ogiku N, Ishida R, Saeki K, Sugiura M. Induction of cardiac angiotensinogen mRNA and angiotensin converting enzyme (ACE) activity in isoproterenol-induced heart injury. Hypertens Res. 1996;19:179–187. doi: 10.1291/hypres.19.179. [DOI] [PubMed] [Google Scholar]
- 16.Whaley-Connell AT, Chowdhury NA, Hayden MR, Stump CS, Habibi J, Wiedmeyer CE, et al. Oxidative stress and glomerular filtration barrier injury: role of the renin-angiotensin system in the Ren2 transgenic rat. Am J Physiol Renal Physiol. 2006;291:F1308–F1314. doi: 10.1152/ajprenal.00167.2006. [DOI] [PubMed] [Google Scholar]
- 17.Ferrario CM, Varagic J, Habibi J, Nagata S, Kato J, Chappell MC, et al. Differential regulation of angiotensin-(1-12) in plasma and cardiac tissue in response to bilateral nephrectomy. Am J Physiol Heart Circ Physiol. 2009;296:H1184–H1192. doi: 10.1152/ajpheart.01114.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varagic J, Ahmad S, Brosnihan KB, Groban L, Chappell MC, Tallant EA, et al. Decreased cardiac Ang-(1-7) is associated with salt-induced cardiac remodeling and dysfunction. Ther Adv Cardiovasc Dis. 2010;4:17–25. doi: 10.1177/1753944709353337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaltout HA, Figueroa JP, Rose JC, Diz DI, Chappell MC. Alterations in circulatory and renal angiotensin-converting enzyme and angiotensin-converting enzyme 2 in fetal programmed hypertension. Hypertension. 2009;53:404–408. doi: 10.1161/HYPERTENSIONAHA.108.124339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 21.Jessup JA, Trask AJ, Chappell MC, Nagata S, Kato J, Kitamura K, Ferrario CM. Localization of the novel angiotensin peptide, angiotensin-(1-12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol Heart Circ Physiol. 2008;294 doi: 10.1152/ajpheart.91521.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayorh MA, Layas MF, Mann G, Feuerstein GZ, Eatman D. The effect of diet on simvastatin and losartan enhancement of endothelial function. Clin Exp Hypertens. 2007;29:311–325. doi: 10.1080/10641960701500463. [DOI] [PubMed] [Google Scholar]
- 23.Ekinci EI, Thomas G, Thomas D, Johnson C, Macisaac RJ, Houlihan CA, et al. Effects of salt supplementation on the albuminuric response to telmisartan with or without hydrochlorothiazide therapy in hypertensive patients with type 2 diabetes are modulated by habitual dietary salt intake. Diabetes Care. 2009;32:1398–1403. doi: 10.2337/dc08-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekinci EI, Thomas G, Macisaac RJ, Johnson C, Houlihan C, Panagiotopoulos S, et al. Salt supplementation blunts the blood pressure response to telmisartan with or without hydrochlorothiazide in hypertensive patients with type 2 diabetes. Diabetologia. 2010;53:1295–1303. doi: 10.1007/s00125-010-1711-2. [DOI] [PubMed] [Google Scholar]
- 25.Fortepiani LA, Zhang H, Racusen L, Roberts LJ, Reckelhoff JF. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. 2003;41:640–645. doi: 10.1161/01.HYP.0000046924.94886.EF. [DOI] [PubMed] [Google Scholar]
- 26.Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005;45:283–287. doi: 10.1161/01.HYP.0000153051.56460.6a. [DOI] [PubMed] [Google Scholar]
- 27.Sicard P, Oudot A, Guilland JC, Moreau D, Vergely C, Rochette L. Dissociation between vascular oxidative stress and cardiovascular function in Wistar Kyoto and spontaneously hypertensive rats. Vascul Pharmacol. 2006;45:112–121. doi: 10.1016/j.vph.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Guerrero EI, Ardanaz N, Sevilla MA, Arevalo MA, Montero MJ. Cardiovascular effects of nebivolol in spontaneously hypertensive rats persist after treatment withdrawal. J Hypertens. 2006;24:151–158. doi: 10.1097/01.hjh.0000198035.16634.c1. [DOI] [PubMed] [Google Scholar]
- 29.Guerrero E, Voces F, Ardanaz N, Montero MJ, Arevalo M, Sevilla MA. Long-term treatment with nebivolol improves arterial reactivity and reduces ventricular hypertrophy in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2003;42:348–355. doi: 10.1097/00005344-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Bowman AJ, Chen CP, Ford GA. Nitric oxide mediated venodilator effects of nebivolol. Br J Clin Pharmacol. 1994;38:199–204. doi: 10.1111/j.1365-2125.1994.tb04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tzemos N, Lim PO, MacDonald TM. Nebivolol reverses endothelial dysfunction in essential hypertension: a randomized, double-blind, crossover study. Circulation. 2001;104:511–514. doi: 10.1161/hc3001.094207. [DOI] [PubMed] [Google Scholar]
- 32.Boluyt MO, Long X, Eschenhagen T, Mende U, Schmitz W, Crow MT, Lakatta EG. Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. Am J Physiol. 1995;269:H638–H647. doi: 10.1152/ajpheart.1995.269.2.H638. [DOI] [PubMed] [Google Scholar]
- 33.Brouri F, Findji L, Mediani O, Mougenot N, Hanoun N, Le NG, et al. Toxic cardiac effects of catecholamines: role of beta-adrenoceptor downregulation. Eur J Pharmacol. 2002;456:69–75. doi: 10.1016/s0014-2999(02)02643-2. [DOI] [PubMed] [Google Scholar]
- 34.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 35.Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, Shah AM. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol. 2006;47:817–826. doi: 10.1016/j.jacc.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 36.Guo P, Nishiyama A, Rahman M, Nagai Y, Noma T, Namba T, et al. Contribution of reactive oxygen species to the pathogenesis of left ventricular failure in Dahl salt-sensitive hypertensive rats: effects of angiotensin II blockade. J Hypertens. 2006;24:1097–1104. doi: 10.1097/01.hjh.0000226200.73065.5d. [DOI] [PubMed] [Google Scholar]
- 37.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 38.Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med. 1998;339:489–497. doi: 10.1056/NEJM199808203390801. [DOI] [PubMed] [Google Scholar]
- 39.Hebert PR, Moser M, Mayer J, Glynn RJ, Hennekens CH. Recent evidence on drug therapy of mild to moderate hypertension and decreased risk of coronary heart disease. Arch Intern Med. 1993;153:578–581. [PubMed] [Google Scholar]
- 40.Staessen JA, Fagard R, Thijs L, Celis H, Arabidze GG, Birkenhager WH, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet. 1997;350:757–764. doi: 10.1016/s0140-6736(97)05381-6. [DOI] [PubMed] [Google Scholar]
- 41.Lopez-Sendon J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, et al. Expert consensus document on beta-adrenergic receptor blockers. Eur Heart J. 2004;25:1341–1362. doi: 10.1016/j.ehj.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Chappell MC, Allred AJ, Ferrario CM. Pathways of angiotensin-(1-7) metabolism in the kidney. Nephrol Dial Transplant. 2001;16(Suppl 1):22–26. doi: 10.1093/ndt/16.suppl_1.22. [DOI] [PubMed] [Google Scholar]
- 43.Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1-7): an evolving story in cardiovascular regulation. Hypertension. 2006;47:515–521. doi: 10.1161/01.HYP.0000196268.08909.fb. [DOI] [PubMed] [Google Scholar]
- 44.Ferrario CM. ACE2: more of Ang-(1-7) or less Ang II? Curr Opin Nephrol Hypertens. 2011;20:1–6. doi: 10.1097/MNH.0b013e3283406f57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 46.Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation. 2003;108:1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira AJ, Castro CH, Guatimosim S, Almeida PW, Gomes ER, Dias-Peixoto MF, et al. Attenuation of isoproterenol-induced cardiac fibrosis in transgenic rats harboring an angiotensin-(1-7)-producing fusion protein in the heart. Ther Adv Cardiovasc Dis. 2010;4:83–96. doi: 10.1177/1753944709353426. [DOI] [PubMed] [Google Scholar]
- 48.Ahmad S, Varagic J, Westwood BM, Chappell MC, Ferrario CM. Uptake and metabolism of the novel peptide angiotensin-(1-12) by neonatal cardiac myocytes. PLoS One. 2011;6:e15759. doi: 10.1371/journal.pone.0015759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trask AJ, Jessup JA, Chappell MC, Ferrario CM. Angiotensin-(1-12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol Heart Circ Physiol. 2008;294:H2242–H2247. doi: 10.1152/ajpheart.00175.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagata S, Kato J, Kuwasako K, Kitamura K. Plasma and tissue levels of proangiotensin-12 and components of the renin-angiotensin system (RAS) following low- or high-salt feeding in rats. Peptides. 2010;31:889–892. doi: 10.1016/j.peptides.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Fang Y, Nicol L, Harouki N, Monteil C, Wecker D, Debunne M, et al. Improvement of left ventricular diastolic function induced by beta-blockade. A comparison between nebivolol and metoprolol. J Mol Cell Cardiol. 2011;51:168–176. doi: 10.1016/j.yjmcc.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Sorrentino SA, Doerries C, Manes C, Speer T, Dessy C, Lobysheva I, et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional beta1-blockade. J Am Coll Cardiol. 2011;57:601–611. doi: 10.1016/j.jacc.2010.09.037. [DOI] [PubMed] [Google Scholar]
- 53.Vinereanu D, Gherghinescu C, Ciobanu AO, Magda S, Niculescu N, Dulgheru R, et al. Reversal of subclinical left ventricular dysfunction by antihypertensive treatment: a prospective trial of nebivolol against metoprolol. J Hypertens. 2011;29:809–817. doi: 10.1097/HJH.0b013e3283442f37. [DOI] [PubMed] [Google Scholar]
- 54.Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153:1589–1601. doi: 10.1038/sj.bjp.0707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Periasamy M, Janssen PM. Molecular basis of diastolic dysfunction. Heart Fail Clin. 2008;4:13–21. doi: 10.1016/j.hfc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jessup JA, Westwood BM, Chappell MC, Groban L. Dual ACE-inhibition and AT1 receptor antagonism improves ventricular lusitropy without affecting cardiac fibrosis in the congenic mRen2.Lewis rat. Ther Adv Cardiovasc Dis. 2009;3:245–257. doi: 10.1177/1753944709338489. [DOI] [PMC free article] [PubMed] [Google Scholar]