

Abstract
Arginine (arg) methylation is a widespread posttranslational modification of proteins that impacts numerous cellular processes such as chromatin remodeling, RNA processing, DNA repair, and cell signaling. Known arg methylproteins arise mostly from yeast and mammals, and are almost exclusively nuclear and cytoplasmic. Trypanosoma brucei is an early branching eukaryote whose genome encodes five putative protein arg methyltransferases, and thus likely contains a plethora of arg methylproteins. Additionally, trypanosomes and related organisms possess a unique mitochondrion that undergoes dramatic developmental regulation and uses novel RNA editing and mitochondrial DNA replication mechanisms. Here, we performed a global mass spectrometric analysis of the T. brucei mitochondrion to identify new arg methylproteins in this medically relevant parasite. Enabling factors of this work are use of a combination digestion with two orthogonal enzymes, an efficient offline two dimensional chromatography separation, and high-resolution mass spectrometry analysis with two complementary activations. This approach led to the comprehensive, sensitive and confident identification and localization of methylarg at a proteome level. We identified 167 arg methylproteins with wide-ranging functions including metabolism, transport, chaperoning, RNA processing, translation, and DNA replication. Our data suggest that arg methylproteins in trypanosome mitochondria possess both trypanosome-specific and evolutionarily conserved modifications, depending on the protein targeted. This study is the first comprehensive analysis of mitochondrial arg methylation in any organism, and represents a significant advance in our knowledge of the range of arg methylproteins and their sites of modification. Moreover, these studies establish T. brucei as a model organism for the study of posttranslational modifications.
Arginine (arg)1 methylation is a widespread post-translational modification with roles in numerous cellular functions such as chromatin remodeling, RNA processing, DNA repair, and cell signaling (1). Methylation increases the hydrophobicity and bulkiness of arg residues, but does not alter their charge. This modification often results in dramatic positive and negative changes in protein-protein and protein-nucleic acid interactions, and it can also significantly affect nucleocytoplasmic localization. To date, it has not been definitively demonstrated that arg methylation is reversible; however, methylation can be antagonized by citrullination of arg residues (2). Arg methylation is catalyzed by a family of protein arg methyltransferases (PRMTs) that are categorized by their final products. Type I PRMTs produce both monomethylarg (MMA) and the final product asymmetric dimethylarg (ADMA), in which one terminal ω-nitrogen possesses both methyl groups. Type II PRMTs generate MMA and the final product symmetric dimethylarg (SDMA), where one methyl group is added to each terminal nitrogen. Type III PRMTs catalyze only MMA production. Arg methylproteins can be simultaneously decorated by more than one class of methylarg. Arg methylation often occurs within glycine/arg rich regions (1). However, reports of methylarg residues in non-canonical sequence contexts is becoming more common, suggesting a broader range of targets than originally believed (1, 3). Thus, PRMT substrates cannot be identified based on their sequences, and so must be empirically defined.
A subset of the known arg methylproteins were identified through targeted studies of specific pathways or through physical association with a PRMT (1, 4–6). Limited proteomic studies have also led to the identification of scores of arg methylproteins or putative arg methylproteins (7–9). The vast majority of arg methylproteins identified to date are cytoplasmic or nuclear. Strikingly, very little is known about arg methylation and its possible roles in organellar metabolism or gene expression. Only a single study exists in this regard, which identified 18 arg methylated proteins in the Golgi of human cells (9).
Kinetoplastid parasites are early branching eukaryotes with many intriguing biological features such as RNA polymerase I transcription of some protein-coding genes, polycistronic RNA polymerase II transcription, the apparent absence of RNA polymerase II regulation, and massive mitochondrial uridine insertion/deletion RNA editing (10–12). Trypanosoma brucei, a kinetoplastid that causes African sleeping sickness, is the only member this group in which arg methylation has been extensively studied (13). Of the five PRMTs in the T. brucei genome, four have been characterized: the Type I TbPRMT1 and TbPRMT6, the Type II TbPRMT5, and the Type III TbPRMT7 (14–17). Therefore, numerous targets of monomethylation, symmetric dimethylation, and asymmetric dimethylation presumably exist in T. brucei. Nevertheless, only a handful of in vivo targets or partners of trypanosome PRMTs have been identified (14–18). The mitochondria of kinetoplastids have been a subject of intensive study because of their unique mitochondrial DNA structure termed the kinetoplast, the extensive remodeling of mitochondrial RNAs by RNA editing, and the dramatic developmental regulation of mitochondrial gene expression and metabolism during the T. brucei life cycle (11, 12).
Here, we show that trypanosome mitochondria harbor numerous proteins that are modified by MMA, ADMA, and SDMA. Using a suite of technical advances, including a dual-enzyme proteolysis, an efficient two-dimensional chromatographic separation, and a sensitive and accurate mass spectrometry (MS) approach employing a dual activation strategy (CID and ETD alternatively), we were able to achieve an in-depth proteome-wide localization of methylarg sites and the identification of surrounding motifs with high confidence and accuracy. Overall, we identified 167 arg methylated mitochondrial proteins from diverse classes including metabolism, RNA processing, translation, and kinetoplast DNA (kDNA) replication, thereby significantly increasing the known range of arg methylproteins. These studies establish T. brucei as a model system for the study of posttranslational modifications. They further identify potential mechanisms of regulation unique to this deadly human pathogen.
EXPERIMENTAL PROCEDURES
Cells
Procyclic form (PF) T. brucei strain EATRO 164 Istar1 was cultured as described (16).
Antibodies
ASYM24, SYM11, and SYM10 antibodies were purchased from EMD Millipore; meRG antibodies were from CH3 Biosystems. Anti-Hsp70 was a kind gift from Jay Bangs. Antibodies against TbRGG2, TbRGG1, and TbRND were described previously (19, 20).
Western Blot and Immunoprecipitation
Mitochondrial extracts (equivalent of 1 × 1010 cells) were incubated with 200 μg normal rabbit serum, ASYM24, mRG, or SYM11 coupled to Protein A-Sepharose beads (GE Healthcare Life Sciences). After overnight incubation at 4 °C, unbound material was collected and the matrix was washed extensively with a 50-fold bed volume of PBST (phosphate buffered saline with 0.1% Nonidet P-40) containing a Complete protease inhibitor mixture tablet (Roche). The antibody-antigen complex was then disrupted and the antigen eluted from the column using five column volumes of 100 mm glycine (pH 2.5). The elution was neutralized using 1 m Tris (pH 8.0) and eluted proteins were analyzed by Western blotting with antibodies against specific mitochondrial proteins.
Protein Extraction, Sample Cleanup, and Digestion
Mitochondrial extracts were prepared as described from ∼1 × 1010 PF cells (21). Enriched mitochondria were homogenized in a detergent-containing extraction buffer (50 mm Tris, pH 8, 150 mm NaCl, 4% Nonidet P-40, 0.5% sodium deoxycholate, and 2% SDS). The protein concentration was determined with the BCA protein assay after an ultracentrifugation (140,000 × g, 40 min, 4 °C). To remove undesirable components while maintaining a high peptide recovery, we employed a precipitation/on-pellet-digestion protocol that we developed previously (22) for the digestion with either trypsin or GluC. For GluC digestion, the RapGest cleavable detergent was added to a concentration of 0.1% to facilitate pellet dissolution. The digest was fractionated immediately with SCX chromatography.
Two-dimensional Chromatography Coupled to CID/ETD Analysis
Offline two dimensional chromatography was used to resolve the highly complex mitochondrial proteome before MS analysis. A Waters 2796 Bioseparations HPLC system (Milford, MA) equipped with a Bio-Rad Biofrac fraction collector (Bio-Rad, Hercules, CA) was used for SCX fractionation. Separation was performed on a Thermo Scientific BioBasic SCX column (4.6 × 250 mm, 5-μm particle size) at a flow rate of 1 ml/min, with an injection volume of 600 μl, which contains tryptic or GluC peptides derived from 1 mg of mitochondrial proteins. The optimized mobile phases for SCX column were A: 3 mm ammonium formate in 25% acetonitrile, pH 3.0 and B: 400 mm ammonium formate in 25% acetonitrile, pH 4.5. After sample injection, 100% solvent A was held for 4 min to focus the peptides on the front end of the column, and thereby preventing band broadening during loading. The gradient steps for separation were: 0% to 4% B in 12 min, and 4–24% B in 34 min, 24–87% B in 22 min, then to 100% B in 2 min and held for another 10 min. A fraction was collected into tubes every 2 min after the start of the separation gradient. The wavelength for UV detection was set at 280 nm. A total of 40 fractions were collected for each sample, but these fractions with low-peptide contents (based on the UV chromatogram) were combined, which resulted in 25 total fractions. All fractions were lyophilized and reconstituted with 100 μl 2% acetonitrile containing 0.1% formic acid for Nano-RPLC-MS analysis.
The Nano-RPLC system consisted of a Spark Endurance autosampler (Emmen, Holland) and an ultra-high pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/nano-LC system. To achieve a comprehensive separation of the complex peptide mixture, a nano-LC/nanospray setup, which features low void volume and high chromatographic reproducibility (22), was employed. Mobile phase A and B were 0.1% formic acid in 2% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Samples containing 6 μg of peptides were loaded onto a large-ID trap (300 μm ID ×1 cm, packed with Zorbax 3-μm C18 material) with 1% B at a flow rate of 10 μl/min, and the trap was washed for 3 min. A series of nanoflow gradients (flow rate was 250 nL/min) was used to back-flush the trapped samples onto the nano-LC column (75 μm ID × 75 cm, packed with Pepmap 3-μm C18 material) for separation. The nano-LC column was heated at 52 °C to improve both chromatographic resolution and reproducibility. Depending on the complexity of the fraction as revealed by the SCX chromatogram, two different gradient elution profiles of different lengths were employed. For a less complex fraction, a shorter gradient was employed: (1) a linear increase from 3 to 8% B over 5 min; (2) an increase from 8 to 27% B over 65 min; (3) an increase from 27 to 45% B over 30 min; (4) an increase from 45 to 98% B over 20 min; and (5) isocratic at 98% B for 20 min. A shallower gradient was used to resolve a more complex sample: (1) 3 to 8% B over 5 min; (2) 8 to 24% B over 145 min; (3) 24 to 38% B over 95 min; (4) 38 to 63% B over 55 min; (5) 63 to 97% B in 35 min, and finally (6) isocratic at 97% B for 20 min.
An LTQ/Orbitrap-ETD hybrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) was used for protein identification. The instrument was operating under data-dependent product ion mode. One scan cycle included an MS1 scan (m/z 300–2000) at a resolution of 60,000 followed by either MS2 scans by alternating CID and ETD, to fragment the three most abundant precursors found in the MS1 spectrum. The target value for MS1 by Orbitrap was 8 × 106, under which the Orbitrap was calibrated carefully for mass accuracy and FT transmission. The use of high target value on the Orbitrap enabled a highly sensitive detection without compromising the mass accuracy and resolution. For CID, the activation time was 30 ms, the isolation width was 1.5 amu, the normalized activation energy was 35%, and the activation q was 0.25. Precursors with charge states >3 were rejected. As for ETD, a mixture of ultrapure helium and nitrogen (25% helium and 75% nitrogen, purity >99.995%) was used as the reaction gas. The ETD reaction time was set at 110 ms and the isolation width was 2 amu for the precursor and 10 amu for the fluoranthene anions; supplemental activation, which uses a short CID activation process to dissociate the charge-stripped precursors, was employed to enhance the fragmentation efficiency for doubly charged precursors. The AGC value of fluoranthene anions was set at 5 × 105. The singly charged precursors were rejected for ETD.
Database Searching
Bioworks 3.3.1 SP1 QF31448 (Thermofisher Scientific) was used to generate dta files. The dta filters of ZSA and Combolon were used for filtering CID spectra, whereas charger.exe (Thermofisher Scientific) was used to preprocess the ETD data. Filtered dta files were searched on Sequest Cluster 3.3.1 with a 64-CPU license (Thermofisher Scientific), against the TriTryp database containing 11425 gene entries (ver 10–20-2010). The search parameters are as following: 15 ppm tolerance for precursor ion masses and 1.0 Da for fragment ion masses to process all CID and ETD data. The fasta database was indexed for the tryptic and GluC peptides with the assumption of fully enzymatic cleavage at both ends, and two and five missed cleavages were permitted respectively for trypsin and GluC. In terms of modifications, carbamidomethylation of cysteines was specified as a static modification and variable modifications of methionine oxidation, di-methylation, and mono-methylation on arginine were allowed. The identification results were summarized by the Bioworks. A stringent set of criteria were employed to filter the data, including 15 ppm precursor mass tolerance and high Xcorr and delta-CN cut-off values (Xcorr>1.8 for 1+ charge, >2.1 for 2+ charge, >3 for 3+ charge and >4 for 4+ charge, and delta-CN>0.1) that resulted in a peptide FDR of 0.3%. The peptide FDR was estimated by searching a combined forward and reversed database. The sequence-informative ions (b and y ions for CID and c and z ions for ETD) of each identified methylarginine peptide were carefully inspected to evaluate the reliability of sequencing and localization of the methyl site(s). Suspected false-positives (including these with ambiguous localizations) were eliminated. For each methylated peptide identified by ETD, the corresponding CID spectrum was manually inspected to determine the symmetry of the methylation.
RESULTS
Mitochondria Contain a Distinct Set of Arg Methylproteins
We previously showed that arg methylation of a trypanosome mitochondrial RNA binding protein, RBP16, affects its macromolecular interactions and ability to stabilize specific RNAs (18, 23), suggesting that arg methylation could be more widespread in this organelle. To determine if trypanosome mitochondria contain a set of arg methylproteins distinct from that in other cellular compartments, we generated an enriched mitochondrial fraction from PF T. brucei (Fig. 1A) (21), and compared the profile of arg methylproteins in this fraction to that in whole cell lysate. We performed Western blots using ADMA specific antibodies ASYM24 and mRG (Fig. 1B) and SDMA specific antibodies SYM10 and SYM11 (Fig. 1C) (8, 24). Similar to what has been observed in yeast and mammals (4, 8), each of these antibodies detected a specific, but limited, set of methylproteins. Notably, all four antibodies reveal a pattern of methylproteins in mitochondria that is distinct from that detected by the same antibody in whole cell extract (arrowheads in Figs. 1B and 1C). We detected at least ten asymmetrically dimethylated and three symmetrically dimethylated proteins that are clearly enriched in mitochondria, likely a vast underestimate based on the published properties of these antibodies. Thus, numerous proteins containing both ADMA and SDMA are highly enriched in trypanosome mitochondria.
Fig. 1.

Specific arg methylproteins are enriched in mitochondria. A, Twenty micrograms of PF whole cell (WCL) and mitochondrial (Mito) lysates were analyzed by immunoblotting with antibodies against Hsp70 (cytoplasmic marker) and TbRGG2 (mitochondrial marker). Lysates in A were immunoblotted with ADMA specific antibodies ASYM24 and mRG (B) or SDMA specific antibodies SYM11 and SYM10 (C). Arrowheads, methylproteins enriched in mitochondria.
Two Dimensional LC Coupled to CID/ETD MS Reveals Abundant Mitochondrial Arg Methylproteins with Diverse Biological Functions
Next, we employed a suite of proteomics and analytical advances to perform an in-depth and nonbiased query of the mitochondrial proteome for arg methylproteins. A strong, detergent-containing buffer was used to obtain excellent recovery of proteins, especially those that are membrane-bound, as demonstrated in our previous works (22, 25). This was followed by efficient precipitation/on-pellet-digestion using two enzymes with orthogonal specificity (22), trypsin (which cuts at the C terminus of lysine and arginine) and GluC (which cuts at the C terminus of glutamic acid and aspartic acid), to digest the mitochondrial preparations in parallel. High-resolution, optimized strong cation exchange (SCX) chromatography was used to simplify the highly complex digestion mixture. Efficient fractionations were achieved for both tryptic and GluC digests, as indicated by the fact that >82% of identified peptides were only found in a single fraction (data not shown). Each fraction was further resolved with high chromatographic resolution by reversed-phase chromatography on a long, heated nano-scale column. Peptides were then sequenced by a high-resolution Orbitrap analyzer, using alternating CID and ETD activations, which when used with the dual-enzyme approach, markedly enhanced the proteomic coverage for analysis of the T. brucei mitochondrial preparation. Additionally, the use of ETD afforded more efficient sequencing of methylarginine peptides (26), for two main reasons. First, because of the internal arg residues, methylarginine peptides are often highly charged (e.g. ≥3+) under ESI, and thus they are sequenced more efficiently with ETD over CID. Second, because of the lability of the methylation moieties, CID promotes initial elimination of the methylation moiety, thus largely precluding further fragmentation of the peptide backbone. By comparison, ETD provides uniform cleavage of the peptide backbone and therefore yields sequence-informative fragments and provides abundant information on peptide sequencing and accurate methyl group localization. As a result, the majority of the methylarginine peptides identified in this study were identified by ETD (supplemental Table S1). Representative fragmentation spectra are shown in Fig. 2. Although ETD poses an overwhelming advantage for the identification of methylarginine peptides, the alternating CID analysis provided complimentary information on the type and symmetry of methylarginine, as illustrated in Figs. 2A and 2C. The symmetry-specific neutral losses from ADMA and SDMA have been well-characterized by our lab and others (25, 27, 28). Moreover, CID was helpful for some peptides that have lower charge states (supplemental Table S1). Aside from using the cutoff criteria, all CID and ETD spectra were manually inspected to rule out false-positive identification and PTM localization.
Fig. 2.

Representative CID and ETD fragmentation spectra of methylarginine peptides identified from enriched T. brucei mitochondrial preparation. The fragments of peptide R#LEFENGPLRDQVEAHTQR from Tb10.70.7760 by (A) CID and (B) ETD, and the fragments of peptide R#GVTSTPVSMQNARSGHFR from Tb09.211.3720 by (C) CID and (D) ETD. Symbols on peptide sequence: #, dimethylation on arginine residues; (a), asymmetric di-methylation; and (b), symmetric di-methylation. Neutral loss in CID: DMC, Dimethylcarbodiimide; DMA, Dimethylamine; and DMG, Dimethylguanidine.
In addition to mitochondrial proteins, arg methylproteins clearly originating from cytoplasm and nuclei were also present in our sample, and these will be reported elsewhere. To rigorously define mitochondrial proteins in the data set, we employed several criteria including classification as mitochondrial in the Trypanosoma proteome project (29), mitochondrial designation in TriTrypDB or GeneDB, GO annotations indicating a mitochondrial function, and published evidence of a mitochondrial function or presence in a mitochondrial protein complex. For hypothetical proteins with no identifying domains or other data, we assessed predicted mitochondrial localization using the MitoCarta (30) and PSORT II algorithms. Based on these criteria, we identified 256 unique methylpeptides, representing 167 mitochondrial proteins (supplemental Table S1).
Thirty-seven of the mitochondrial arg methylproteins (22% of total) were annotated as hypothetical and contained no discernible domains (Fig. 3; supplemental Table S1). Of the remaining 130 arg methylproteins, the largest category was proteins involved in metabolism, with a total of 44 such proteins (26% of total) containing one or more methylargs (Fig. 3 and Table I). This includes both conserved and kinetoplastid-specific components of the mitochondrial ATPase (31), and proteins involved in fatty acid metabolism and the citric acid cycle. The next most abundant class of mitochondrial arg methylproteins were those involved in translation, including numerous ribosomal proteins (32) and the translation initiation factor IF-2 (33) (Fig. 3 and Table S1). Also highly represented were chaperones and proteins involved in RNA processing, stability, and modification (Fig. 3 and supplemental Table S1). Regarding the latter, we identified the KREN2 and KREPB8 components of the editosome, several proteins reportedly associated with the MRB1 complex that functions in RNA editing and stabilization, and the KPAP1 poly(A) polymerase (19, 34–37). Interestingly, we also identified several proteins that function in replication of the novel kinetoplastid mitochondrial DNA structure, termed kDNA, including TbPol1B and TbPol1C, mitochondrial topoisomerase II, and the two mitochondrial DNA primases, Pri1 and Pri2 (38–41). Collectively, these data suggest that arg methylation has the potential to influence numerous mitochondrial processes including several aspects of metabolism, gene expression, and DNA replication.
Fig. 3.

Functional categories of mitochondrial arg methylproteins. The 167 identified mitochondrial arg methylproteins were categorized based on their known or likely functions. Proteins with no known domains and no annotations at GeneDB or TriTrypDB were categorized as hypothetical proteins. “Other” refers to proteins with known or expected functions that did not belong to a large general category.
Table I. Mitochondrial arginine methylproteins with functions in metabolism. Forty-four proteins that either have known functions in mitochondrial metabolism or that physically associate with the mitochondrial respiratome (31, 53) were identified as methylproteins.
| TriTryp DB number | Protein name | # methyl arginines |
|---|---|---|
| Tb11.47.0004 | 2-oxoglutarate dehydrogenase E1 component, putative | 4 |
| Tb11.01.3550 | 2-oxoglutarate dehydrogenase E2 component, putative | 1 |
| Tb927.8.2540 | 3-ketoacyl-CoA thiolase | 1 |
| Tb927.8.6970 | 3-methylcrotonyl-CoA carboxylase alpha subunit, putative | 3 |
| Tb927.8.7530 | 3,2-trans-enoyl-CoA isomerase | 1 |
| Tb927.8.2610 | 5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase, putative | 1 |
| Tb927.1.4490 | Acetyltransferase, putative Acyl-CoA N-acyltransferase | 1 |
| Tb10.70.5510 (Tb927.10.2230) | Adrenodoxin reductase, putative ferredoxin NADP reductase-like protein | 2 |
| Tb927.6.4210 | Aldehyde dehydrogenase, putative | 1 |
| Tb11.02.1340 | AMP deaminase, putative | 1 |
| Tb10.61.2130 (Tb927.10.14550)* | ATP-dependent DEAD/H RNA helicase, putative (TbDed1p) associated with complex V | 2 |
| Tb927.3.1380 | ATP synthase beta chain, mitochondrial precursor | 2 |
| Tb10.100.0070 (Tb927.10.180) | ATP synthase F1 subunit gamma protein, putative | 1 |
| Tb927.4.2560 | Cardiolipin synthetase, putative | 2 |
| Tb927.2.1560 | Cyclophilin type peptidyl-prolyl cis-trans isomerase precursor, putative associated with complex I | 1 |
| Tb927.2.1680† | Cyclophilin type peptidyl-prolyl cis-trans isomerase precursor, putative associated with complex I | 1 |
| Tb10.70.5510 | Ferredoxin NADP reductase-like protein | 2 |
| Tb09.160.4910 | Flavoprotein monooxygenase, putative | 1 |
| Tb11.02.2700 | Fumarate hydratase class I, putative | 1 |
| Tb11.02.0440 | Glycerolphosphate mutase, putative | 1 |
| Tb927.2.4380 | Hypothetical protein, conserved 30M24.390 (TbTob55) associated with complex I | 3 |
| Tb10.70.6930 (Tb927.10.1160) | Hypothetical protein, conserved associated with complex I | 1 |
| Tb927.5.2580 | Hypothetical protein, conserved associated with complex IV | 2 |
| Tb11.47.0022 | Hypothetical protein, conserved associated with complex V | 1 |
| Tb927.2.3610 | Hypothetical protein, conserved associated with complex V | 1 |
| Tb10.70.7760 (Tb927.10.520) | Hypothetical protein, conserved associated with complex V | 1 |
| Tb927.5.2930 | Hypothetical protein, conserved associated with complex V | 2 |
| Tb10.70.4980 (Tb927.10.2680) | Hypothetical protein, conserved FAD/NAD(P)-binding domain, oxidoreductase | 1 |
| Tb927.7.910 | Hypothetical protein, conserved NAD(P)-linked oxidoreductase | 2 |
| Tb11.0300† | Hypothetical protein 1 TM domain p27-like | 1 |
| Tb927.1.730 | Hypothetical protein, conserved PROKAR lipid attachment/P-loop containing nucleoside triphosphate hydrolases associated with complex I | 2 |
| Tb10.70.3940 (Tb927.10.3510) | Hypothetical protein, conserved Pseudouridine synthase, catalytic domain | 1 |
| Tb927.4.4300 | Hypothetical protein, conserved SET domain associated with complex I | 2 |
| Tb10.70.3640* | Hypothetical protein, conserved SET domain associated with respiratome | 1 |
| Tb927.4.2030* | Hypothetical protein, conserved (TbALBA4) associated with complex V | 7 |
| Tb11.02.2980 | Hypothetical protein, conserved ubiquinol-cytochrome-c reductase | 2 |
| Tb11.02.3130 | Malic enzyme, putative | 4 |
| Tb927.8.1060 | Malonyl-CoA decarboxylase, mitochondrial precursor, putative | 4 |
| Tb927.7.910 | NAD(P)-linked oxidoreductase | 2 |
| Tb09.211.4110 | NADPH–cytochrome p450 reductase, putative | 2 |
| Tb11.0400 | p27 protein, putative | 1 |
| Tb10.389.0890 (Tb927.10.12700) | Pyruvate dehydrogenase E1 alpha subunit, putative | 4 |
| Tb09.211.4700 | Reiske iron-sulfur protein, mitochondrial precursor | 1 |
| Tb10.61.0690 | tRNA pseudouridine synthase A-like protein | 4 |
| Tb11.02.2980 | Ubiquinol-cytochrome-c reductase | 2 |
| Tb10.61.0960 (Tb927.10.15420) | S-adenosyl-l-methionine-dependent methyltransferase (C20orf7) | 1 |
† Indicates proteins that exhibited the same methylpeptide and are likely repeat proteins in the genome.
* Proteins that have been shown to be cytoplasmic and/or nuclear, but are also reportedly associated with the mitochondrial respiratome.
Sites of Arg Methylation are Evolutionarily Conserved
Many of the arg methylated metabolic proteins identified here are conserved throughout the eukaryotic kingdom. To determine if specific sites of arg methylation are evolutionarily conserved, we aligned representative trypanosome proteins with their yeast and human homologues. Methylated arg positions in the homologs of the 3-methylcrotonyl-CoA carboxylase alpha subunit, pyruvate dehydrogenase E1 alpha subunit, and the beta subunit of ATP synthase are conserved between these three organisms, suggesting that these proteins may be arg methylated in higher eukaryotes (Fig. 4A). In other cases, the site of methylation is only conserved between the trypanosome and human (Fig. 4B) or trypanosome and yeast proteins (Fig. 4C). We also identified trypanosome proteins containing divergent sites of arg methylation. In the example of C20orf7, a complex I methyltransferase, neither the human nor the yeast protein contains the potential site for arg methylation, as it resides in a trypanosome specific C-terminal extension (Fig. 4D). Together, these data suggest that arg methylation in trypanosome mitochondria likely confers both trypanosome-specific and evolutionarily conserved regulatory properties, depending on the protein targeted.
Fig. 4.

Alignments of T. brucei arg methylproteins with yeast and human homologues. A, Examples in which the methylarg site is conserved in all three organisms. B, Conservation of a methyl site between T. brucei and human, but not yeast, homologues. C, Conservation of a methyl site between T. brucei and yeast, but not human, homologues. D, Presence of a methyl site in T. brucei that is absent from yeast and human homologues. A clear C20orf7 homolog was not evident in S. cerevisae; therefore, we used the homologue from the pathogenic yeast Candida albicans. Tb, T. brucei; Hs, H. sapiens; Sc, S. cerevisiae; Ca., C. albicans. Asterisks, methylargs in T. brucei proteins.
Co-immunoprecipitation Validates Arg Methylation of Two Mitochondrial RNA Binding Proteins
To validate biochemically a subset of the methylproteins identified by MS, we performed co-immunoprecipitations. We used antibodies against either ADMA or SDMA to precipitate arg methylproteins from mitochondrial lysates, and immunoblotted using protein specific antibodies. In some cases, MS revealed the identities of dimethylargs (DMA) as ADMA or SDMA, but often this distinction could not be made (supplemental Table S1). On TbRGG2, a protein essential for RNA editing (19), MS identified both ADMA and SDMA. TbRGG1, which functions in mitochondrial RNA stability and editing (35), harbored MMA, ADMA, and unclassified DMA. Both of these proteins were detected in ASYM24 and mRG immunoprecipitates, confirming that they contain ADMA (Fig. 5). Both proteins were also present in the SYM11 pulldowns to differing degrees, indicating the presence of SDMA. The specificity of the assay was confirmed by the absence of TbRGG1 and TbRGG2 from control immunoprecipitations using normal rabbit serum. Additionally, a mitochondrial protein for which 70% coverage by MS failed to reveal any methylargs, TbRND (20), was absent from any of the immunoprecipitates. These data confirm the identity of two methylproteins detected by MS, and further suggest that these proteins are substrates for both ADMA and SDMA as reported for some methylproteins in yeast and mammals (42–44).
Fig. 5.
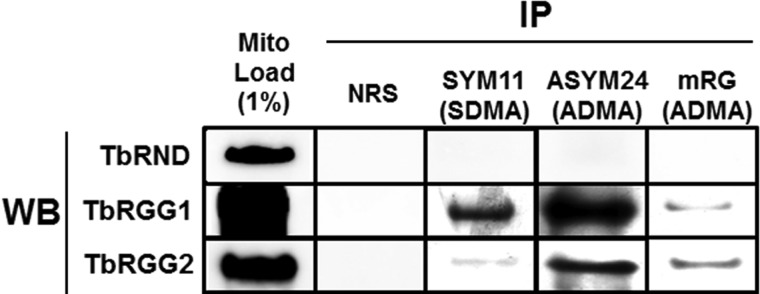
Immunoprecipitation of mitochondrial proteins using methyl-specific antibodies. Mitochondrial lysates were incubated with normal rabbit serum (NRS), the SDMA specific SYM11 antibody, or the ADMA specific ASYM24 or mRG antibodies. One percent of the mitochondrial lysate, or ten percent of immunoprecipitated material, was immunoblotted for TbRND, TbRGG1, and TbRGG2. IP, immunoprecipitation; WB, Western blot.
Amino Acid Motifs Surrounding Methylargs
We next wanted to utilize our MS data to identify common amino acids surrounding sites of DMA, and determine whether these differed between ADMA and SDMA. DMA is often found in RGG or RXR motifs, although numerous exceptions to this generalization have been reported (1, 3, 42, 45). We identified a total of 333 methylation sites (counting each instance of MMA or DMA separately). Of the 167 DMAs, 54 could be distinguished as ADMA and 13 as SDMA (supplemental Table S1). We also identified 166 sites of MMA, but because MMA could represent either a terminal PRMT7-catalyzed modification or an intermediate in the reaction catalyzed by any of the other TbPRMTs, we omitted MMA from our analysis. To define the amino acid motifs surrounding DMA residues, we analyzed the six amino acids immediately N-terminal and C-terminal of each of the 167 DMA-containing sites in our data set using WebLogo (Fig. 6). The most frequent amino acid surrounding DMA residues was glycine, with the DMA commonly positioned in an RGG motif (Fig. 6A). Arginine, alanine, and leucine, were also frequently present surrounding DMA. Amino acids surrounding conclusively identified ADMA residues were similar to the entire DMA population, suggesting that ADMA constitutes the majority of total DMA residues (Fig. 6B). In contrast, residues surrounding SDMA were enriched for glutamic and aspartic acid and for an arginine residue immediately upstream of the methylated arginine, indicating that different trypanosome PRMTs have distinct substrate specificities.
Fig. 6.

Amino acid motifs surrounding dimethylargs. WebLogo 3 (http://weblogo.threeplusone.com/create.cgi) was used to analyze the six amino acids N-terminal and C-terminal of DMA residues. A, Analysis of all 164 sites bearing DMA. B, Analysis of the 54 sites identified as bearing ADMA. C, Analysis of the 13 sites identified as bearing SDMA. Arginines are indicated in blue, glycines are in green, and charged amino acids are labeled in red.
DISCUSSION
Here, we report a survey of mitochondrial arg methylproteins in the early branching eukaryote, T. brucei. It is highly challenging to identify protein arg methylation in a complex mixture using conventional LC/MS techniques, because of the low stoichiometry of arg methylations and the difficulties in efficient sequencing and localization of the closely spaced, highly charged methylarg residues by CID techniques that are prevalently used for proteomic analysis. Previously, we discovered that ETD techniques are capable of sequencing methylarg-containing peptides and localizing the methylation with high accuracy, sensitivity and confidence (26). Although CID may not produce sequence-informative fragmentation spectra for some highly-charged methylated peptides, it reveals the forms of methylarg (e.g. ADMA versus SDMA) via neutral loss signatures. As a result, combining a high-resolution MS analyzer with alternating CID and ETD analyses enabled a comprehensive characterization of arg methylated peptides/proteins (26). More recently (20, 46), we demonstrated that the dual-activation method, in conjunction with proteolysis with orthogonal enzymes (e.g. trypsin and gluC) and efficient chromatographic separations, is capable of achieving a highly comprehensive analysis of proteins and post-translational modifications in very complex proteomes. Using the above strategy, we were able to achieve the first comprehensive analysis of arg methylproteins in the mitochondria of any organism, identifying 167 target proteins. The technique allowed us to precisely map the sites of mono- and dimethylation on these proteins, and in some cases distinguish between ADMA and SDMA. The most complete analysis of cellular arg methylproteins to date is the study by Boisvert et al., (8) who immunoprecipitated proteins from HeLa cells using four anti-methylarg antibodies and identified the recovered proteins by LC-MS/MS. These authors identified over 200 putative methylproteins, but they could not distinguish between bone fide methylproteins and those that precipitated because of interactions with methylproteins, and the sites of methylation were not identified. Ong et al. (7) used SILAC to identify 59 sites of arg methylation in 33 HeLa proteins, many of which were RNA binding proteins. The only study of arg methylation in an organelle prior this one identified 18 arg methylproteins in a Golgi-enriched fraction (9). Thus, the data reported here represent a very significant advance in our knowledge of the range of arg methylproteins and their sites of modification.
We identified methylarg proteins of wide-ranging function including metabolism, transport, chaperoning, RNA processing, translation, and DNA replication. Methylproteins were especially abundant in certain complexes. For example, proteins of the ATPase complex appeared overrepresented, with 8 ATPase components (31) containing methylarg. Interestingly, the ATPase complex of T. brucei has distinct functions at different phases of the life cycle, synthesizing ATP in the insect-borne life cycle stage (PF) and hydrolyzing ATP to maintain membrane potential in the mammalian-born stage (47, 48). It is tempting to speculate that changes in the methylation of ATPase components during the life cycle could impact differential ATPase function. Proteins of the mitochondrial small and large ribosomal subunits were also highly represented in our study. Arg methylation of ribosomal proteins impacts the assembly of cytosolic ribosomes in mammalian cells (49), and our results may indicate a similar role in mitochondrial ribosome biogenesis.
The sites of arg methylation in metabolic proteins were often conserved from trypanosomes to yeast or humans. Thus, many of the modifications reported for the first time here may reflect evolutionarily conserved mechanisms of mitochondrial protein regulation. On the other hand, kinetoplastid-specific processes are also likely to be impacted by arg methylation. Uridine insertion/deletion RNA editing involves both the catalytic editosome complex and numerous regulatory proteins (50), and several proteins in both categories contain methylarg. Thus, the dynamic association of accessory factors with the editosome and, potentially, dynamic association of endonuclease components (51) may be influenced by this modification. It was also striking that many proteins involved in kDNA replication are arg methylated. These proteins typically display discrete suborganellar localizations that presumably facilitate their functions (38, 40, 41), a parameter that could be affected by methylation status.
One question raised by this study is the identities of PRMTs that modify mitochondrial arg methylproteins. To date, PRMTs have not been reported to localize within the mitochondria of any organism, and we have not detected any of the four characterized T. brucei PRMTs (14–17) in mitochondrial preparations by Western blot. Knockdown of TbPRMT1 lead to hypomethylation and functional alteration of RBP16, indicating a role for this enzyme in methylation of mitochondrial proteins in trypanosomes (18). It is formally possible that all proteins get methylated before mitochondrial import, although this would be at odds with a regulatory function for methylation. It is also possible that a subset of TbPRMTs, potentially including TbPRMT1, are present in mitochondria below the level of detection. Interestingly, although most methylargs were surrounded by glycine residues, we did detect numerous modified args in other sequence contexts. The only known PRMT that preferentially modifies args in non-glycine-rich contexts is CARM1 (45), an enzyme for which T. brucei lacks a homologue. This suggests that a non-canonical PRMT could be acting within the mitochondrion. The T. brucei genome encodes over 100 proteins predicted to have methyltransferase activity, the majority of which have not been characterized. Thus, investigation of the PRMTs that modify mitochondrial proteins will be an exciting future direction.
In summary, the present study establishes T. brucei as a model system for analysis of protein modifications. The wealth of genetic techniques available in this organism (52) should permit rapid progress toward understanding the mechanisms by which the methylmarks identified here impact protein function.
Supplementary Material
Acknowledgments
We thank Amanda Pinak, Sarah McEvoy and Michel Pelletier for technical assistance.
Footnotes
* This work was supported by NIH grant RO1AI060260 to LKR, NIH grant U54HD071594 and AHA award 12SDG9450036 to JQ, and NIH NRSA postdoctoral fellowship F32AI07718501 to JCF.
 This article contains supplemental Table S1.
This article contains supplemental Table S1.
1 The abbreviations used are:
- ADMA
- asymmetric dimethyl arginine
- AGC
- automatic gain control
- Arg
- arginine
- CID
- collision induced dissociation
- DMA
- dimethyl arginine
- ESI
- electrospray ionization
- ETD
- electron-transfer dissociation
- FDR
- false discovery rate
- kDNA
- kinetoplast DNA
- MMA
- monomethyl arginine
- PF
- procyclic form T. brucei
- PRMT
- protein arginine methyltransferase
- PTM
- posttranslational modification
- SCX
- strong cation exchange
- SDMA
- symmetric dimethyl arginine
- SILAC
- stable isotope labeling by/with amino acids in cell culture.
REFERENCES
- 1. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cuthbert G. L., Daujat S., Snowden A. W., Erdjument-Bromage H., Hagiwara T., Yamada M., Schneider R., Gregory P. D., Tempst P., Bannister A. J., Kouzarides T. (2004) Histone deimination antagonizes arginine methylation. Cell 118, 545–553 [DOI] [PubMed] [Google Scholar]
- 3. Wooderchak W. L., Zang T., Zhou Z. S., Acuña M., Tahara S. M., Hevel J. M. (2008) Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the “RGG” paradigm. Biochemistry 47, 9456–9466 [DOI] [PubMed] [Google Scholar]
- 4. Bachand F., Silver P. A. (2004) PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J. 23, 2641–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olsson I., Berrez J. M., Leipus A., Ostlund C., Mutvei A. (2007) The arginine methyltransferase Rmt2 is enriched in the nucleus and co-purifies with the nuclear porins Nup49, Nup57 and Nup100. Exp. Cell Res. 313, 1778–1789 [DOI] [PubMed] [Google Scholar]
- 6. Perreault A., Lemieux C., Bachand F. (2007) Regulation of the nuclear poly(A)-binding protein by arginine methylation in fission yeast. J. Biol. Chem. 282, 7552–7562 [DOI] [PubMed] [Google Scholar]
- 7. Ong S.-E., Mann M. (2006) Identifying and quantifying sites of protein methylation by heavy methyl SILAC. Curr. Protoc. Protein Sci. Chapter 14, Unit 14.19 [DOI] [PubMed] [Google Scholar]
- 8. Boisvert F. M., Côté J., Boulanger M.-C., Richard S. (2003) A proteomic analysis of arginine-methylated protein complexes. Mol. Cell. Proteomics 2, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 9. Wu C. C., MacCoss M. J., Mardones G., Finnigan C., Mogelsvang S., Yates J. R., 3rd, Howell K. E. (2004) Organellar proteomics reveals Golgi arginine dimethylation. Mol. Biol. Cell 15, 2907–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clayton C., Shapira M. (2007) Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasit. 156, 93–101 [DOI] [PubMed] [Google Scholar]
- 11. Lukes J., Hashimi H., Ziková A. (2005) Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr. Genet. 48, 277–299 [DOI] [PubMed] [Google Scholar]
- 12. Stuart K. D., Schnaufer A., Ernst N. L., Panigrahi A. K. (2005) Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 30, 97–105 [DOI] [PubMed] [Google Scholar]
- 13. Fisk J. C., Read L. K. (2011) Protein arginine methylation in parasitic protozoa. Eukaryot Cell 10, 1013–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisk J. C., Zurita-Lopez C., Sayegh J., Tomasello D. L., Clarke S. G., Read L. K. (2010) TbPRMT6 Is a Type I Protein Arginine Methyltransferase That Contributes to Cytokinesis in Trypanosoma brucei. Eukaryot Cell 9, 866–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fisk J. C., Sayegh J., Zurita-Lopez C., Menon S., Presnyak V., Clarke S. G., Read L. K. (2009) A type III protein arginine methyltransferase from the protozoan parasite Trypanosoma brucei. J. Biol. Chem. 284, 11590–11600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pasternack D. A., Sayegh J., Clarke S., Read L. K. (2007) Evolutionarily divergent type II protein arginine methyltransferase in Trypanosoma brucei. Eukaryot Cell 6, 1665–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pelletier M., Pasternack D. A., Read L. K. (2005) In vitro and in vivo analysis of the major type I protein arginine methyltransferase from Trypanosoma brucei. Mol. Biochem. Parasitol. 144, 206–217 [DOI] [PubMed] [Google Scholar]
- 18. Goulah C. C., Read L. K. (2007) Differential effects of arginine methylation on RBP16 mRNA binding, guide RNA (gRNA) binding, and gRNA-containing ribonucleoprotein complex (gRNP) formation. J. Biol. Chem. 282, 7181–7190 [DOI] [PubMed] [Google Scholar]
- 19. Fisk J. C., Ammerman M. L., Presnyak V., Read L. K. (2008) TbRGG2, an essential RNA editing accessory factor in two Trypanosoma brucei life cycle stages. J. Biol. Chem. 283, 23016–23025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zimmer S. L., McEvoy S. M., Li J., Qu J., Read L. K. (2011) A novel member of the RNase D exoribonuclease family functions in mitochondrial guide RNA metabolism in Trypanosoma brucei. J. Biol. Chem. 286, 10329–10340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harris M. E., Moore D. R., Hajduk S. L. (1990) Addition of uridines to edited RNAs in trypanosome mitochondria occurs independently of transcription. J. Biol. Chem. 265, 11368–11376 [PubMed] [Google Scholar]
- 22. Duan X., Young R., Straubinger R. M., Page B., Cao J., Wang H., Yu H., Canty J. M., Qu J. (2009) A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J. Proteome Res. 8, 2838–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goulah C. C., Pelletier M., Read L. K. (2006) Arginine methylation regulates mitochondrial gene expression in Trypanosoma brucei through multiple effector proteins. RNA 12, 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duan P., Xu Y., Birkaya B., Myers J., Pelletier M., Read L. K., Guarnaccia C., Pongor S., Denman R. B., Aletta J. M. (2007) Generation of polyclonal antiserum for the detection of methylarginine proteins. J. Immunol. Methods 320, 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duan X., Abuqayyas L., Dai L., Balthasar J. P., Qu J. (2012) High-throughput method development for sensitive, accurate, and reproducible quantification of therapeutic monoclonal antibodies in tissues using orthogonal array optimization and nano liquid chromatography/selected reaction monitoring mass spectrometry. Anal. Chem. 84, 4373–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang H., Straubinger R. M., Aletta J. M., Cao J., Duan X., Yu H., Qu J. (2009) Accurate localization and relative quantification of arginine methylation using nanoflow liquid chromatography coupled to electron transfer dissociation and orbitrap mass spectrometry. J. Am. Soc. Mass Spectr. 20, 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiong L., Ping L., Yuan B., Wang Y. (2009) Methyl group migration during the fragmentation of singly charged ions of trimethyllysine-containing peptides: precaution of using MS/MS of singly charged ions for interrogating peptide methylation. J. Am. Soc. Mass Spectrom. 20, 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ong S. E., Mittler G., Mann M. (2004) Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 1, 119–126 [DOI] [PubMed] [Google Scholar]
- 29. Panigrahi A. K., Ogata Y., Ziková A., Anupama A., Dalley R. A., Acestor N., Myler P. J., Stuart K. D. (2009) A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 9, 434–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang X., Cui J., Nilsson D., Gunasekera K., Chanfon A., Song X., Wang H., Xu Y., Ochsenreiter T. (2010) The Trypanosoma brucei MitoCarta and its regulation and splicing pattern during development. Nucleic Acids Res. 38, 7378–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ziková A., Schnaufer A., Dalley R. A., Panigrahi A. K., Stuart K. D. (2009) The F(0)F(1)-ATP synthase complex contains novel subunits and is essential for procyclic Trypanosoma brucei. PLoS Pathog. 5, e1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziková A., Panigrahi A. K., Dalley R. A., Acestor N., Anupama A., Ogata Y., Myler P. J., Stuart K. (2008) Trypanosoma brucei mitochondrial ribosomes: affinity purification and component identification by mass spectrometry. Mol. Cell. Proteomics 7, 1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charrière F., Tan T. H. P., Schneider A. (2005) Mitochondrial initiation factor 2 of Trypanosoma brucei binds imported formylated elongator-type tRNA(Met). J. Biol. Chem. 280, 15659–15665 [DOI] [PubMed] [Google Scholar]
- 34. Etheridge R. D., Aphasizheva I., Gershon P. D., Aphasizhev R. (2008) 3′ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J. 27, 1596–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hashimi H., Ziková A., Panigrahi A. K., Stuart K. D., Lukes J. (2008) TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14, 970–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carnes J., Trotter J. R., Ernst N. L., Steinberg A., Stuart K. (2005) An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. 102, 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panigrahi A. K., Ernst N. L., Domingo G. J., Fleck M., Salavati R., Stuart K. D. (2006) Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12, 1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Melendy T., Sheline C., Ray D. S. (1988) Localization of a type II DNA topoisomerase to two sites at the periphery of the kinetoplast DNA of Crithidia fasciculata. Cell 55, 1083–1088 [DOI] [PubMed] [Google Scholar]
- 39. Hines J. C., Ray D. S. (2011) A second mitochondrial DNA primase is essential for cell growth and kinetoplast minicircle DNA replication in Trypanosoma brucei. Eukaryot Cell 10, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hines J. C., Ray D. S. (2010) A mitochondrial DNA primase is essential for cell growth and kinetoplast DNA replication in Trypanosoma brucei. Mol. Cell. Biol. 30, 1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klingbeil M. M., Motyka S. A., Englund P. T. (2002) Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol. Cell 10, 175–186 [DOI] [PubMed] [Google Scholar]
- 42. Pal S., Vishwanath S. N., Erdjument-Bromage H., Tempst P., Sif S. (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 24, 9630–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang H., Huang Z. Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B. D., Briggs S. D., Allis C. D., Wong J., Tempst P., Zhang Y. (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293, 853–857 [DOI] [PubMed] [Google Scholar]
- 44. Shire K., Kapoor P., Jiang K., Hing M. N., Sivachandran N., Nguyen T., Frappier L. (2006) Regulation of the EBNA1 Epstein-Barr virus protein by serine phosphorylation and arginine methylation. J. Virol. 80, 5261–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee J., Bedford M. T. (2002) PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO R 3, 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tu C., Li J., Young R., Page B. J., Engler F., Halfon M. S., Canty J. M., Jr., Qu J. (2011) Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome. Anal. Chem. 83, 4802–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brown S. V., Hosking P., Li J., Williams N. (2006) ATP synthase is responsible for maintaining mitochondrial membrane potential in bloodstream form Trypanosoma brucei. Euk. Cell 5, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schnaufer A., Clark-Walker G. D., Steinberg A. G., Stuart K. (2005) The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function.[erratum appears in EMBO J. 2006 Mar 8;25(5):1175–6]. EMBO J. 24, 4029–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ren J., Wang Y., Liang Y., Zhang Y., Bao S., Xu Z. (2010) Methylation of ribosomal protein S10 by protein-arginine methyltransferase 5 regulates ribosome biogenesis. J. Biol. Chem. 285, 12695–12705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aphasizhev R., Aphasizheva I. (2011) Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip. Rev. RNA 2, 669–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carnes J., Trotter J. R., Peltan A., Fleck M., Stuart K. (2008) RNA editing in Trypanosoma brucei requires three different editosomes. Mol. Cell. Biol. 28, 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alsford S., Turner D. J., Obado S. O., Sanchez-Flores A., Glover L., Berriman M., Hertz-Fowler C., Horn D. (2011) High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21, 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Acestor N., Ziková A., Dalley R. A., Anupama A., Panigrahi A. K., Stuart K. D. (2011) Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell. Proteomics 10, M110.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.