

Abstract
Hydrogen-deuterium exchange mass spectrometry is an important method for protein structure-function analysis. The bottom-up approach uses protein digestion to localize deuteration to higher resolution, and the essential measurement involves centroid mass determinations on a very large set of peptides. In the course of evaluating systems for various projects, we established two (HDX-MS) platforms that consisted of a FT-MS and a high-resolution QTOF mass spectrometer, each with matched front-end fluidic systems. Digests of proteins spanning a 20–110 kDa range were deuterated to equilibrium, and figures-of-merit for a typical bottom-up (HDX-MS) experiment were compared for each platform. The Orbitrap Velos identified 64% more peptides than the 5600 QTOF, with a 42% overlap between the two systems, independent of protein size. Precision in deuterium measurements using the Orbitrap marginally exceeded that of the QTOF, depending on the Orbitrap resolution setting. However, the unique nature of FT-MS data generates situations where deuteration measurements can be inaccurate, because of destructive interference arising from mismatches in elemental mass defects. This is shown through the analysis of the peptides common to both platforms, where deuteration values can be as low as 35% of the expected values, depending on FT-MS resolution, peptide length and charge state. These findings are supported by simulations of Orbitrap transients, and highlight that caution should be exercised in deriving centroid mass values from FT transients that do not support baseline separation of the full isotopic composition.
Hydrogen-deuterium exchange mass spectrometry (HDX-MS)1 provides a powerful means to study the link between protein structure and function (1). The method involves a chemical process in which labile hydrogens within a protein are exchanged with hydrogen from bulk water. When D2O is used in place of H2O, a mass shift results at every point of exchange, but it is the backbone amide hydrogens that offer exchange rates on a measurable timescale (2, 3). Measuring an amide hydrogen exchange rate can provide access to conformational dynamics, stability, and the interaction characteristics in that location of structure (4, 5). H/D exchange rates have be used to explore mechanisms of protein folding (6), determine the allosteric impact of post-translational modifications and ligand binding (7, 8), define truncation points for enhancing crystallization success (9), and they have also found a role in mapping interactions between proteins (10). Applications have stepped outside of primary research to include the characterization of protein drugs for stability and similarity testing (11–13). The capacity to provide such information has attracted increased attention from regulatory bodies and is generating a push for standardizing HDX methods.
Mass spectrometers are very effective tools for measuring exchange rates, from whole proteins down to the individual amide levels. Classical methods of rate measurement have used NMR (3), but mass spectrometry offers all the advantages of speed, sensitivity and scale that have made the tool so useful in proteomics. Measurements at the peptide level provide an important intermediate resolution. As with bottom-up proteomics, rendering deuterated proteins into smaller peptides through digestion provides opportunities to analyze protein systems of considerable complexity, and at the same time support analysis at higher structural resolution through MS/MS methods (14, 15). A considerable amount of effort has been applied by the research community and instrument manufacturers to produce instrument configurations that are suitable for managing the many processing steps required for labeling, digesting, separating and introducing deuterated peptides into the mass spectrometer (16). This has been supported by parallel efforts to develop software tools for the detection of deuterated peptides and the extraction of deuteration data (17–19).
A successful application of the bottom-up HDX-MS method requires a full peptide sequence map of the protein, so that deuteration rates at every point in protein structure can be quantified and related back to structure. Once the peptide is identified, the primary measurement is the peptide centroid mass of the deuterated state, relative to the unlabeled state. It requires intensity measurements for a minimum of two peaks in the isotopic cluster to determine when the centroid mass changes (20), although most often the full distribution is quantified in HDX-MS applications. As the range of applications continues to grow, particularly in the regulatory area, it is important to better understand how various elements of the HDX platform deliver the essential data (21, 22). In the current study, we are interested in the contribution of the mass spectrometer alone. Most users of the HDX-MS method are migrating from low resolution to high resolution systems, operated in a single-stage MS mode. This includes FT-MS and higher-resolution QTOF platforms, therefore in this study, we explore how an LTQ Orbitrap Velos (Thermo) and a 5600 TripleTOF (AB Sciex) influence the measurement of deuteration data for proteins of increasing size. Identical front-end fluidic systems and protein digests, as well as back-end analysis procedures, allow us to perform a direct comparison of performance in areas of sequence mapping, centroid measurement precision and centroid mass accuracy. We demonstrate that the Orbitrap system returns greater sequencing depth and marginally better precision than the 5600, however the measurement accuracy is strongly influenced by destructive interference arising from unequal mass defects between 13C and 2H. This has implications for any application that involves centroid mass determinations, beyond HDX-MS.
EXPERIMENTAL PROCEDURES
Sample Preparation
Three proteins were processed for the analyses described in this study. The DNA repair protein “x-ray repair cross-complementing protein 4” (XRCC4, amino acids 1–200) with a molecular weight of 20 kDa, polynucleotide kinase (PNK) with a molecular weight of 55 kDa, and α/β-tubulin dimer (TUB) with a combined molecular weight of 110 kDa were predigested with immobilized pepsin (Pierce) at room temperature for 4 min (pH 2.3) at a concentration of 10 μm for each protein. Each digest was then split into two pools. Aliquots of the first pool were stored at –80 °C and destined for replicative sequencing on each platform. The second pool was deuterated to equilibrium in 15% D2O for 24 h and aliquots were also stored at –80 °C for eventual deuteration analysis on the two liquid chromatography (LC)-MS platforms. Proteins were sourced from collaborators (XRCC4 and PNK, gifts from Dr. S.P. Lees-Miller, University of Calgary) or purchased (TUB from Cytoskeleton Inc., Denver, CO).
Sequence Mapping
Digest samples (5 pmol) were injected using a HTX PAL autosampler (Leap Technologies, Carrboro, NC) and peptides were trapped on a 5 cm, 200 μm i.d. Onyx C18 monolithic column (Phenomenex, Torrance, CA) held at 4 °C in the cooled chamber of the autosampler. Peptides were eluted by a 10 min acetonitrile gradient (3–40%) using either a Dionex Ultimate 3000 coupled to an LTQ Orbitrap Velos (Thermo) or an Eksigent nanoLC Ultra2D coupled to a 5600 Triple TOF (AB Sciex). The same flow rate was used in both configurations (4 μl/min). Transfer lines were matched so that peptide arrival times to the mass spectrometers were equivalent. Each digest was analyzed eight times on each platform: one full scan from m/z 300–1250 and six gas-phase fractionation scans (m/z 300–500, 495–600, 595–700, 695–700, 685–800, 795–1000, 995–1250) (23).
Sequencing with the LTQ Orbitrap Velos used a survey scan in the Orbitrap at 30 k resolution and data-dependent acquisition (DDA) for MS/MS in the ion trap using the top 20 most intense ions, for a total cycle time not exceeding 5.4 s. The collision-induced dissociation (CID) state was configured with a minimal peak selection threshold of 500 counts, an isolation width of 2 mass units, a normalized collision energy of 35, an activation q value of 0.25 and an activation time of 10 msec. Dynamic exclusion was configured with a repeat count of one for 10 s, before exclusion for 15 s. Known background ions were added to an exclusion list for the entire run. Source conditions were set to 3.1 kV for ionization and a sheath gas flow of 1. No auxiliary gas flow was used. For ion transmission the S-lens was set to an RF level at 44% and a capillary temperature of 275°C.
Sequencing with the 5600 TripleTOF used an optimized set of parameters as previously described (24). Briefly, information-dependent acquisition (IDA) employed a 250 msec survey scan and up to 20 product ion scans, on ions exceeding 500 counts, for a total cycle time of 2.3 s. A swept collision energy setting of 35 ± 15 eV was applied for CID. Dynamic exclusion was enabled and set for 15 s. Source conditions were set to 5.2 kV for ionization, 12 for gas1 and 10 for gas2. The curtain gas was set at 20.
Deuteration Measurement by LC-MS
Deuterated digests (5 pmol) were injected using the same autosampler and pump configurations as described above, with the mass spectrometers operated in MS mode only. On the Orbitrap, ten replicates for each digest were collected at nominal resolution settings of 30 k, 60 k and 100 k. Ten replicates for each digest were also collected on the 5600 TripleTOF for a total of 120 runs. For acquisition on the Orbitrap, the 30 k resolution was collected first, with 60 k and 100 k resolutions collected on subsequent days. Fresh protein aliquots were analyzed each day. Instrument calibration was performed before each set of acquisitions. Blanks were inserted between each run, and protein digests were analyzed in an alternating fashion. For acquisition on the 5600 TripleTOF, proteins were analyzed in the same alternating fashion, with blanks. After each of the three protein digests were analyzed, the instrument was calibrated to minimize mass drift.
Data Analysis - Sequence Identification
A merged .mgf file was created from the output of each instrument (Analyst TF v1.51 and Xcalibur v2.1) using Mass Matrix File Conversion (v3.9) containing the full-scan and all six gas-phase fractionation data files for each protein. Mascot Version 1.01 was used to identify peptide hits for each protein, from a limited database containing only the proteins in this study, including multiple isoforms of TUB, as described previously (25). Data were mapped to sequence using the following search terms: a mass tolerance of 10 ppm on precursor ions and 0.6 Da on fragment ions, no modifications, and no enzyme specificity. A standard probability cutoff of p = 0.05 was implemented and matches near the cutoff manually verified. The results were exported and formatted for import into Mass Spec Studio.
Data Analysis - Deuteration Measurement
Centroid mass measurements and deuterium content determinations were made using Mass Spec Studio, which incorporates a rebuild of Hydra software previously described for such purposes (18). Briefly, the rebuild centered on producing a generalized software framework to provide common infrastructure for analysis of any mass spectrometry data set. The framework provides a pluggable architecture allowing rapid development of new analysis packages for any purpose, offering the extensibility to support custom data providers, experiment types, user interface modules and algorithms. The current iteration provides some common modules for each of these categories. For example, a default data provider has been included, which uses the ProteoWizard library for reading vendor-specific mass spectrometry data files. Using this new infrastructure, we refactored Hydra into a processing package that plugs into the framework. For the current study, the only upgrade affecting Hydra functionality involves the inclusion of a custom data decompression algorithm to support the handling of high-resolution data. The accuracy of this algorithm was verified by comparing intensity data from within the Studio to intensities read from the vendor-supplied peak-viewing software (Xcalibur and PeakView). The Mass Spec Studio is available on request, and periodic new releases are planned, of Hydra and the framework.
As input to the Hydra process, Mass Spec Studio requires a .csv file containing a list of peptides, charge states, and retention times (generated from the Mascot output) and the set of LC-MS files containing the replicate deuteration measurements. The output of peptide centroid values was culled to ensure that only high-quality peptide isotopic clusters with nonoverlapping, high signal-to-noise ratios were used in measurements of deuteration accuracy and precision. We achieved this by (a) requiring that plots of mass versus number of isotopic peaks in mass calculation for each peptide followed a monotonically increasing function and (b) that a minimum of eight replicates contributed to the final average. When these conditions were met, the full isotopic cluster was used for centroid mass measurements. These selection criteria were manually validated to represent clean, single peptide selections.
Simulations of FT-MS Spectra for Deuterated Peptides
To determine the effect of deuterium addition on centroids measured by FT-MS, we generated transients computationally in Igor Pro (v. 4.09A, Wavemetrics), consisting of the superposition of a set of waveforms representing the isotopic cluster of a given peptide. We used the output of MS Isotope (prospector.ucsf.edu/prospector/mshome.htm) for each unlabeled peptide and assumed the isotopic cluster was represented by 13C/12C content only, except where noted. Deuteration was represented as a binomial expansion of this distribution, assuming N-2 exchangeable amide hydrogens, where N is the number of residues in the peptide. The waveform was therefore represented as follows (equation 1):
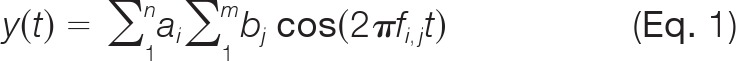 |
where ai represents the binomial coefficients for a given deuteration level, bj the coefficients drawn from the intensity values for each isotopic peak in the native peptide cluster and fi,j the frequency of the respective peptide isotopic composition. Transients were generated with a “sampling” rate of 1 × 106 points/s, for 0.5 s or more. Frequencies were based on values reported for earlier Orbitrap prototypes, that is, 711 kHz for 56Fe+ (26), and calculated through the basic relationship (27):
 |
Transients generated in this fashion were windowed with a Hanning function in Igor before Fourier transformation with a standard FFT algorithm. Frequency-domain spectra were generated in magnitude mode, and the centroid mass determined using peak intensities in the usual way. Deuterium values were obtained by subtraction of the centroid mass for the unlabeled peptide.
RESULTS AND DISCUSSION
System and Samples
A comparison of data from different MS platforms requires the analysis of identical samples, front-end fluidic systems and data processing routines to minimize any bias in the analysis (Fig. 1). For this purpose, we chose to define a common stock of protein sample digests, in both undeuterated and deuterated forms. The deuterated samples were equilibrated before analysis and storage, to ensure no drift in measurements that might result from slow kinetics of deuteration. We tested aliquots of the 15% deuterated X4 stock digest on successive days using the 5600 platform. There were no detectable changes in deuterium content for a random sampling of peptides, confirming equilibration (data not shown). Although we used different LC systems to simplify instrument communications, the autosampler, columns, and mobile phases were identical, and the gradients and total analysis times were closely matched.
Fig. 1.

Configuration of HDX-MS platforms for comparing MS performance in peptide sequence mapping, deuteration precision, and accuracy. Sample digests (deuterated or undeuterated) were introduced into either an Orbtrap Velos or a 5600 TripleTOF using matched gradient microLC systems, to ensure that peptide retention times were equivalent. Mass Spec Studio was used for deuteration analysis.
Sequencing
A bottom-up H/DX-MS analysis requires indexing the protein sequence using MS/MS methods, such that each amino acid residue is represented by a minimum of one digest peptide. Higher redundancy supports higher structural resolution and validates the deuteration measurements, so exhaustive peptide identification is usually performed. One procedure involves extending the gradient to increase time for MS/MS sequencing, but we chose an iterative method instead, to simulate a greater degree of spectral complexity as might be found with very large protein systems. For each of three proteins, we injected replicate samples in the “gas-phase fractionation” strategy, preserving the gradient and temperature conditions of the actual HDX-MS experiment, but without deuteration. This allowed us to generate a master list of peptides with their associated retention times, which could be parsed from the Mascot search files and used directly in Mass Spec Studio, for extracting deuteration data from the subsequent HDX-MS runs. The indexing statistics for three proteins are shown in Table I.
Table I. Peptide identification statistics for the Orbitrap and the 5600 TripleTOF for three different protein substrates.
| XRCC4 |
PNK |
α/β-TUB |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 5600 | Orbitrap | Overlap | 5600 | Orbitrap | Overlap | 5600 | Orbitrap | Overlap | |
| Counta | 241 | 409 | 200 | 294 | 450 | 223 | 446 | 750 | 341 |
| Seq. Cov. (%) | 100 | 100 | 100 | 97 | 98.1 | 94.5 | 93.3 | 95.8 | 92.1 |
| Redundancyb | 13 | 19 | 10 | 7 | 12 | 5 | 6 | 9 | 4 |
| Average scorec | 34 (20) | 28 (14) | 31 (14) | 32 (20) | 26 (14) | 30 (15) | 37 (20) | 32 (15) | 34 (16) |
| Average chargec | 2.1 (1.0) | 2.1 (0.9) | 2.0 (0.8) | 2.3 (1.0) | 2.4 (1.1) | 2.3 (0.9) | 2.1 (0.8) | 1.9 (0.8) | 2.1 (0.8) |
| Average lengthc | 12 (5) | 11 (4) | 11 (4) | 14 (7) | 14 (8) | 14 (7) | 12 (5) | 12 (5) | 12 (5) |
a Unique pairs of retention time and m/z.
b Average number of times a given location is represented by a peptide.
c Value (std. dev.).
The fractional overlap between peptide sets is modest and remarkably consistent between the two analytical platforms: ∼42% of all identified peptides are found in both data sets, with no obvious dependence on protein size. The high selection rate and narrow m/z range per sequencing replicate provided excess capacity for identification, as the table shows the number of identified peptides increases from XRCC4 to TUB. This suggests that other reasons are behind the modest overlap in peptide identifications. The Mascot scores for the common and unique peptide subsets do not show a significant difference. Substantially lower scores for the unique subsets might have implied lower intensities but this appears not to be the case. When the data sets were inspected visually, most of the peptides identified uniquely by one or the other platform actually can be found in both LC-MS sets, at least based on an observation of common retention time and accurate mass, and at intensities sufficient to trigger a sequencing event. It suggests that at least some of the unique identifications are because of differences between beam-type and trap fragmentations, which is not surprising (28). In this case, ions would be selected but would generate weak fragmentation patterns in one or the other instrument, which translate into scores below the identification thresholds. We also note that peak detection algorithms have moderately high false-negative rates and thus some of the unique identifications could be because of missed peak selections (29). The high false negative rate has been noted by others as well in the context of HDX-MS experiments (30). As the numbers of peptides identified are different between platforms, it also makes sense that the redundancy (or fold-coverage) is different. The Orbitrap outperformed the 5600 in numbers of peptides by an average of 64%, which translated into a 62% increase in redundancy. This did not affect the sequence coverage to any appreciable degree, as each data set supported coverage approaching 100%. There were no significant differences detected in average peptide charge or length.
Although the number of identified peptides did increase with protein molecular weight, the correlation is weak. There are likely three reasons. In the first place, the sequence composition can bias detection; for example TUB has post-translational modifications and extensive acidic tails that can reduce depth of coverage. Second, although the substrate size increases, the digestion conditions are fixed because of the time constraints in HDX experiments. This can lead to a decrease in overall digestion efficiency, which would influence the map. Third, the LC conditions are also fixed, and as complexity increases, ion suppression and/or chimeric MS/MS spectra may reduce the rate of identification (31, 32). These could be improved by extending the run-time for sequencing purposes but it might not benefit the HDX runs, which require short gradients. The implications of reduced coverage in the TOF platform may not be especially troublesome, as there remains a large number of un-sequenced peptides that could be “tapped” on an as-needed basis by targeted sequencing efforts.
Precision of deuteration measurements
HDX applications that monitor the effect of a change in protein state require high measurement precision, to identify perturbations of the mass shift with high confidence. For example, a region of stable secondary structure in a protein or ligand binding-site may have a low exchange rate, which on binding reduces even further. These events can be more difficult to detect than structural transitions in flexible loops, and thus requires both sensitive and precise measures of deuteration. We explored precision on each platform by selecting a subset of data from the analysis of 15% deuterated XRCC4 and PNK digests, and compared the high resolution TOF data to three different resolution settings on the Orbitrap (Fig. 2).
Fig. 2.

Cumulative distribution functions highlighting the precision of deuteration measurements from a large set of technical replicates. A, 5600 TripleTOF data from 165 peptides, (B) Orbitrap Velos 30k resolution data from 144 peptides (C) Orbitrap Velos 60k resolution data from 210 peptides and (D) Orbitrap Velos 100k resolution data from 190 peptides. Data shown as percent relative standard deviation (%RSD), fit with a sigmoidal function (black trace). Insets show the mean and median values for the respective instrument configuration.
Peptides without spectral overlap and with moderate or high signal-to-noise ratios were selected for this comparison, and precision was expressed as % relative standard deviation (%RSD). The plots show a cumulative distribution function, describing the probability at which a certain %RSD (or better) can be expected. The data is well-fit with a sigmoid, as expected for data with a normal error distribution. It is clear that high precision can be achieved with each platform, although the Orbitrap delivers marginally better performance regardless of resolution setting. The Orbitrap performance erodes slightly at the highest instrument resolution, however. Because the transient length is greater at the higher resolution, the sampling rate is reduced so less points define the chromatographic peaks, which could impact precision. Regardless, in all cases analytical precision is high and in typical HDX-MS experiments, error arising from other sources, such as reagent dispensing and protein heterogeneity, would likely dominate.
Accuracy of Deuteration Measurements
Accurate deuterium levels are required when measuring the kinetics of label uptake. Kinetics measurements are used to extract rate constants, which in turn provide access to important parameters such as protection factors that are related to protein stability. When investigating protein interactions, accurate deuteration levels are also necessary for quantifying Kd values and binding kinetics (33). To explore the influence of the mass spectrometry platform on accuracy, we first determined if both systems generated the same values for a common set of deuterated peptides. The centroid masses for the subset of common peptides described in Table I were measured, using the XRCC4 and PNK digests, and the deuteration level determined by subtracting the corresponding unlabeled peptide centroid masses. These are plotted in Fig. 3.
Fig. 3.

Comparing deuteration values between the Orbitrap Velos and the 5600 TripleTOF for a common set of peptides. (A, D) A comparison of the Orbitrap 30k resolution setting with the 5600. (B, E) A comparison of the Orbitrap 60k resolution setting with the 5600. (C, F) A comparison of the Orbitrap 100k resolution setting with the 5600. Plots A–C show the mass shifts calculated by subtracting the unlabeled centroid mass from the labeled centroid mass. Plots D–F show the data as a ratio of mass shifts, as a function of the neutral peptide mass. Dashed lines in all figures represent values expected if measurements were equivalent. All masses as unified atomic mass units, u.
The deuteration values as measured by the Orbitrap, at any resolution, were consistently lower than the deuteration values measured by the 5600. The deviation worsens considerably with increasing resolution on the Orbitrap, and Figs. 3D–3F indicates that the bias appears strongest at low peptide mass. Interestingly, when the charge state is overlaid on the data, the bias is strongest for singly charged and weakest for multiply charged peptides. We see no discrepancy between the theoretical and expected mass for a singly charged peptide infused into the 5600 for a range of deuteration values (supplemental Fig. S1), thus the anomalous values appear to arise from some aspect of the FT-MS measurement.
Isotope Beating
FT-MS measurements in the Orbitrap rely on the generation of image current for ion packets oscillating in the axial direction, moving as concentric rings along the central spindle electrode (34). The waveform that results from the simultaneous detection of ions with varied masses is a complex time-domain transient, which is the superposition of the correspondingly varied frequencies. These are deconvolved with the aid of Fourier transformation and the spectrum generated through the application of Equation 3.
 |
However, individual frequencies are correlated in the Orbitrap experiment and as a result, generate both constructive and destructive interference patterns at the detector. This leads to classical time-domain signals that show beat patterns, with beat frequencies that are simply a function of the difference between the two interfering signals (35), equation 4:
In an FT-MS experiment, beat patterns observable in the timeframe of a typical transient could arise from the slightly different frequencies found within an isotopic cluster. Studies with mid-resolution FT-ICR instruments have shown that isotopic peaks nominally one mass unit apart in multiply charged protein ions beat at a frequency of 8.8 Hz for a 6+ ion of bovine ubiquitin, for example (35). The time between beats does not contribute to the measurement as the signal is essentially absent, which imparts a stepwise relationship between resolution and the acquisition time. In this case, resolution only increases when additional beats are recorded. Isotopic beat patterns of this nature also affect intensity, as was shown in the measurement of polymer distributions (36). It was demonstrated that transients must be sampled for at least two beats in order generate polymer spectra that faithfully returned peak intensity data. A similar situation could occur with deuterated peptides (Fig. 4).
Fig. 4.

Potential for isotope beating between an unlabeled peptide and the peptide mass-shifted with deuterium. Black trace shows an isotopic cluster of a peptide with 50 carbons, showing only the 13C contribution to the cluster. Red trace shows the same peptide shifted in mass through the exchange of one 1H for a 2H. Inset shows an expansion around the nominal M+1 peak, and the expected mass difference.
To determine how signal interference could affect measurements of deuteration, we simulated transients for two different peptides at various deuteration levels, AEGFSAI (1+ and 2+ charge states) and VVEKLGVPFQVL (1+ charge state). These simulations used frequencies based on values reported for earlier Orbitrap instrument configurations. Conventional routines for windowing and transforming the transients were applied to generate frequency-domain spectra, and peak intensities were measured from this domain. The results are shown in Fig. 5, describing the deviation between simulated and expected deuteration level in two ways. In Fig. 5A, the normalized deuteration values are plotted for each of the three peptide forms, over the full range of possible amide bond deuteration levels. In Fig. 5B, the same data is plotted as a percentage of the expected deuteration level, over the full deuteration range. Deviations between the simulated and expected levels are very pronounced at the lower deuteration levels, but remain observable even at 80% deuteration. As with the experimental data (Fig. 3), there is a charge state bias, with singly charged AEGFSAI showing a higher deviation than the doubly charged form. Similarly, as the peptides increase in size the deviation diminishes. In these simulations, peptide deuteration was modeled as a binomial expansion of the native peptide isotopic cluster, which we produced from the 12C/13C contribution only. This expansion can be viewed as a collection of native distributions shifted by the mass difference between D and H, a value of 1.00628u, for each additional amide deuteration event. The difference between peaks in the native distribution is 1.00335u (13C-12C), and although the mass difference between these two values is small (0.00292u), it represents a beat frequency of 0.5 Hz for singly charged AEGFSAI (see equations 3 and 4).
Fig. 5.
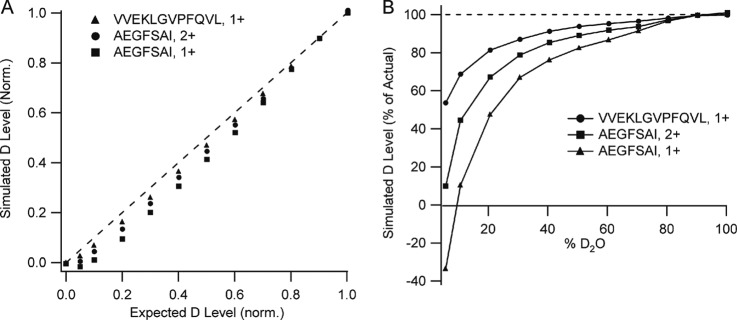
Simulation of mass shift error arising from deuteration. A, Plot of deuterium content for the indicated peptides returned after transient generation and transformation versus the deuteration content used as input to the simulation. B, Alternative plot of the same data, showing the data as a percentage of the expected, or actual, deuterium content. Simulations based on a 2 s transient.
The full isotopic cluster represents multiple small offsets from various combinations of 2H and 13C that complicate the beat pattern, but the 0.00292u mass difference represents the closest overlap and it dominates the beat pattern (Fig. 6A). Our simulations do not include any physical factors for erosion of resolution with acquisition time, so to determine the effect of increased resolution on the biased centroid measurements, we simply increased the duration of the transients before processing, and then recalculated centroid masses (Fig. 6B). The bias was observed to be greatest at one beat period (2 s.) and it required approximately four beat periods to return a centroid mass that approached the expected value. At this transient length, all isotopic forms could be distinguished from each other in the transformed signal. At shorter acquisition times, for example 0.25 of a beat period, centroid accuracy is better than an acquisition 4 times longer in duration. It reflects the fact that destructive interference underlies this phenomenon, as short acquisitions relative to the beat frequency effectively renders the two signal components equal.
Fig. 6.

Effect of transient sampling time on the mass error generated by isotope beating. A, An unprocessed transient simulating the signal for a 10% deuterated AEGFSAI. B, The mass shift resulting from transforming the transient after the indicated sampling times.
The experimental observations in Fig. 3 are consistent with this outcome of the simulation as well. We observed the centroid mass bias to worsen with increasing Orbitrap resolution (from 30 k to 100 k). The transient length in the Orbitrap Velos at a nominal resolution of 60k is 768 msec (37), which suggests our measurements fall somewhere to the left of the maximum centroid bias observed in Fig. 6B. For the larger singly charged VVEKLGVPFQVL, the primary beat frequency is 0.2 Hz with a beat period of 5 s. The destructive interference should not be as great for the reasons described above, which our simulations support (Fig. 5), but what about the doubly charged AEGFSAI? Simulations show that the centroid bias is not as high as the singly charged version. The primary beat frequency for this ion is 0.75 Hz, thus the simulations have sampled more than one beat period, suggesting that the measurement falls to the right of its maximum centroid bias. Without access to the raw transients and the vendor's data processing routine, it is somewhat difficult to make a rigorous comparison between our model and the experimental data. However, we compared our interference-based model with the measured deuteration data for the doubly charged peptides in our data set, and found that the simulations of the 60k resolution set fit the data well (supplemental Fig. S2). When taken together, these simulations are consistent with the experimental observations of bias, and point to a problem that may best be described as a moving target. Unless sufficient resolution is available to separate all isotopic forms of a peptide, there will be a subset of the peptides that meet the criteria for maximum destructive interference when standard transient lengths are used.
The studies by Hofstadler et al. (35) and Easterling et al. (36) indicate that this is not a new problem in FT-MS. With respect to Orbitraps specifically, isotope beating has been proposed recently to explain deviations in relative ion abundances for applications involving small molecule identification and quantification (38, 39). The effects observed were smaller than shown here (and potentially correctable), for the simple reason that the compounds were defined by their native isotopic compositions, with a lower % abundance of the isotopic interference. Variable deuteration clearly increases the severity of the beating phenomenon. Physical effects arising in FT-MS may also influence the measurement of isotopic abundances by an Orbitrap. These can include space charging and peak coalescence phenomena (40, 41). Such effects are usually discussed in the context of mass accuracy, but they may also exert an influence on observed peak intensities when clouds of ions have near-equivalent m/z values. Assessing the relative impact of these other influences is not part of the current study. We propose that isotope beating is a major influence, but perhaps not the only one, based on a strong correlation with the experimental data.
Implications for HDX
It may be argued that most applications of bottom-up measurements are used in a comparative fashion, where measurement precision may ultimately be more important than accuracy. This may be the case when the technique is applied to large-scale screening activities, or coarse-mapping of binding interfaces. However, segments of otherwise useful data may be poorly represented in these studies as a result of isotope beating. The effect of signal interference is strongest for short, singly charged peptides. These represent a rich segment of HDX data because they have the greatest structural resolution. A reduction in the apparent deuterium content will restrict the sensitivity for measuring changes, and could cause changes to go unnoticed. The bias is weak at the highest deuteration levels, which suggests that measurements should be made using high percentages of D2O. This is usually how experiments are conducted. In practice, peptides actually span a wide deuteration range, as deuteration levels depend on structure, dynamics and back exchange, which are all sequence dependent. Subtracting deuteration values for nested peptides, in an attempt to localize deuteration changes to higher resolution (30), may not be valid in the face of the variable destructive interference we observe.
Bottom-up measurements are also used to determine the kinetics of labeling in regions of a protein, and these involve measuring the rate of change in deuterium levels over time. Accurate protection factors obviously require accurate deuterium values, which the Orbitrap underrepresents depending on the resolution setting. Kinetics plots will reflect the bias initially by a delayed onset of label uptake, followed by a faster uptake rate. Kinetics constants are not yet utilized quantitatively, but if recent studies relating kinetics to protein structure are any indication (42), they will find application in structure refinement and accuracy will be important.
Conclusions and Further Implications
Both platforms return rich data sets sufficient for in-depth HDX-MS analysis of large protein systems, when using typical low-pmol amounts of sample. Sequence coverage in the Orbitrap is more extensive due primarily to higher rates of peptide identification, but there is no particular bias in the TOF data suggesting that a protein will experience markedly lower sequence coverage. Both support high levels of redundancy. Measurement precision offered by both platforms is high, and it will be sample handling that ultimately limits precision in most cases. With regards to the accuracy of deuteration measurements, we show that the mass difference between the 2H shift and the 13C shift, if not fully resolved, may generate destructive interference using conventional transient lengths available on the Orbitrap platform.
It is worth considering the impact of the isotope beating phenomenon in other applications as well. Any centroid determination or measurement of an isotopic cluster may be prone to this effect. Un-deuterated versions of the same peptide set used in this study also demonstrate a progressive negative bias, although the effect is not quite as large (supplemental Fig. S3). Contributors to the bias in the unlabeled set mainly arise from significant levels of 15N and 18O in peptides, and it extends the concerns described by Kaufmann (39) in the area of compound identification and drug quantitation. With conventional peptide sequences, the effect will always lead to a lower centroid mass measurement, as the monoisotopic ion is exactly that, isotopically pure and thus not prone to destructive interference at available resolutions. Proteomics applications involving identification and label-free quantitation methods will not likely be influenced by isotope beating, as isotopic clusters are not invoked for these applications, but isotopically labeled quantitative methods could be to a minor degree, for example 18O/16O labeling (43) or stable isotope enrichment methods if enrichment levels are not high (44, 45). Here, clusters could partially overlap and conditions may be created for destructive interference. In SILAC, the clusters are well separated and the effect is not likely to be a problem. In all cases, the bias may be reduced by either increasing resolution sufficiently to resolve all isotopic contributors, or eroding resolution so that the elemental mass defects are all effectively equal.
Supplementary Material
Footnotes
* Acknowledgments: The work was supported by an NSERC Discovery Grant 298351–2010 (DCS). DCS acknowledges the additional support of the Canada Research Chair program, Alberta Ingenuity - Health Solutions and the Canada Foundation for Innovation.
 This article contains supplemental Figs. S1 to S3.
This article contains supplemental Figs. S1 to S3.
1 The abbreviations used are:
- HDX-MS
- hydrogen/deuterium exchange mass spectrometry
- PNK
- polynucleotide kinase
- XRCC4
- Xray repair cross-complementing protein 4
- TUB
- α/β-tubulin
- DDA
- data-dependent acquisition
- IDA
- information-dependent acquisition
- %RSD
- percent relative standard deviation
- Hz
- hertz.
REFERENCES
- 1. Zhang Z., Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linderstøm-Lang K. (1955) Deuterium exchange between peptides and water. Chem. Soc. 2, 1–20 [Google Scholar]
- 3. Dempsey C. E. (2001) Hydrogen exchange in peptides and proteins using NMR-spectroscopy. Prog. Nucl. Magnetic Resonance Spectrosc. 39, 135–170 [Google Scholar]
- 4. Wales T. E., Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 5. Konermann L., Pan J., Liu Y. H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 6. Konermann L., Simmons D. A. (2003) Protein-folding kinetics and mechanisms studied by pulse-labeling and mass spectrometry. Mass Spectrom. Rev. 22, 1–26 [DOI] [PubMed] [Google Scholar]
- 7. Hamuro Y., Coales S. J., Morrow J. A., Molnar K. S., Tuske S. J., Southern M. R., Griffin P. R. (2006) Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 15, 1883–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Betts G. N., van der Geer P., Komives E. A. (2008) Structural and functional consequences of tyrosine phosphorylation in the LRP1 cytoplasmic domain. J. Biol. Chem. 283, 15656–15664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pantazatos D., Kim J. S., Klock H. E., Stevens R. C., Wilson I. A., Lesley S. A., Woods V. L., Jr. (2004) Rapid refinement of crystallographic protein construct definition employing enhanced hydrogen/deuterium exchange MS. Proceedings of the National Academy of Sciences of the United States of America 101, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ling J. M. L., Shima C. H., Schriemer D. C., Schryvers A. B. (2010) Delineating the regions of human transferrin involved in interactions with transferrin binding protein B from Neisseria meningitidis. Mol. Microbiol. 77, 1301–1314 [DOI] [PubMed] [Google Scholar]
- 11. Bobst C. E., Kaltashov I. A. (2011) Advanced mass spectrometry-based methods for the analysis of conformational integrity of biopharmaceutical products. Curr. Pharm .Biotechnol. 12, 1517–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houde D., Berkowitz S. A., Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen G., Warrack B. M., Goodenough A. K., Wei H., Wang-Iverson D. B., Tymiak A. A. (2011) Characterization of protein therapeutics by mass spectrometry: recent developments and future directions. Drug Discov. Today 16, 58–64 [DOI] [PubMed] [Google Scholar]
- 14. Zehl M., Rand K. D., Jensen O. N., Jorgensen T. J. (2008) Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc. 130, 17453–17459 [DOI] [PubMed] [Google Scholar]
- 15. Pan J., Han J., Borchers C. H., Konermann L. (2008) Electron capture dissociation of electrosprayed protein ions for spatially resolved hydrogen exchange measurements. J. Am. Chem. Soc. 130, 11574–11575 [DOI] [PubMed] [Google Scholar]
- 16. Iacob R. E., Engen J. R. (2012) Hydrogen exchange mass spectrometry: are we out of the quicksand? J. Am. Soc. Mass Spectrom. 23, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pascal B. D., Chalmers M. J., Busby S. A., Griffin P. R. (2009) HD desktop: an integrated platform for the analysis and visualization of H/D exchange data. J. Am. Soc. Mass Spectrom. 20, 601–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slysz G. W., Baker C. A., Bozsa B. M., Dang A., Percy A. J., Bennett M., Schriemer D. C. (2009) Hydra: software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinformatics 10, 162–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kazazic S., Zhang H. M., Schaub T. M., Emmett M. R., Hendrickson C. L., Blakney G. T., Marshall A. G. (2010) Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Slysz G. W., Percy A. J., Schriemer D. C. (2008) Restraining expansion of the peak envelope in H/D exchange-MS and its application in detecting perturbations of protein structure/dynamics. Anal. Chem. 80, 7004–7011 [DOI] [PubMed] [Google Scholar]
- 21. Burkitt W., O'Connor G. (2008) Assessment of the repeatability and reproducibility of hydrogen/deuterium exchange mass spectrometry measurements. Rapid Commun. Mass Spectrom. 22, 3893–3901 [DOI] [PubMed] [Google Scholar]
- 22. Chalmers M. J., Pascal B. D., Willis S., Zhang J., Iturria S. J., Dodge J. A., Griffin P. R. (2011) Methods for the analysis of high precision differential hydrogen-deuterium exchange data. Int. J. Mass Spectrom. 302, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yi E. C., Marelli M., Lee H., Purvine S. O., Aebersold R., Aitchison J. D., Goodlett D. R. (2002) Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis 23, 3205–3216 [DOI] [PubMed] [Google Scholar]
- 24. Andrews G. L., Simons B. L., Young J. B., Hawkridge A. M., Muddiman D. C. (2011) Performance Characteristics of a New Hybrid Quadrupole Time-of-Flight Tandem Mass Spectrometer (TripleTOF 5600). Anal. Chem. 83, 5442–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bennett M. J., Chik J. K., Slysz G. W., Luchko T., Tuszynski J., Sackett D. L., Schriemer D. C. (2009) Structural mass spectrometry of the alpha beta-tubulin dimer supports a revised model of microtubule assembly. Biochemistry 48, 4858–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Makarov A. (2000) Electrostatic axially harmonic orbital trapping: A high-performance technique of mass analysis. Anal. Chem. 72, 1156–1162 [DOI] [PubMed] [Google Scholar]
- 27. Scigelova M., Hornshaw M., Giannakopulos A., Makarov A. (2011) Fourier transform mass spectrometry. Mol Cell Proteomics 10, M111.009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jedrychowski M. P., Huttlin E. L., Haas W., Sowa M. E., Rad R., Gygi S. P. (2011) Evaluation of HCD- and CID-type Fragmentation Within Their Respective Detection Platforms For Murine Phosphoproteomics. Mol. Cell. Proteomics 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Renard B. Y., Kirchner M., Steen H., Steen J. A., Hamprecht F. A. (2008) NITPICK: peak identification for mass spectrometry data. BMC Bioinformatics 9, 355–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mayne L., Kan Z. Y., Chetty P. S., Ricciuti A., Walters B. T., Englander S. W. (2011) Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass Spectrom. 22, 1898–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang K., Page J. S., Smith R. D. (2004) Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1416–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Houel S., Abernathy R., Renganathan K., Meyer-Arendt K., Ahn N. G., Old W. M. (2010) Quantifying the Impact of Chimera MS/MS Spectra on Peptide Identification in Large-Scale Proteomics Studies. J. Proteome Res. 9, 4152–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Percy A. J., Rey M., Burns K. M., Schriemer D. C. (2012) Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry-A review. Anal. Chim. Acta 721, 7–21 [DOI] [PubMed] [Google Scholar]
- 34. Hu Q., Noll R. J., Li H., Makarov A., Hardman M., Graham, Cooks R. (2005) The Orbitrap: a new mass spectrometer. J. Mass Spectrom. 40, 430–443 [DOI] [PubMed] [Google Scholar]
- 35. Hofstadler S. A., Bruce J. E., Rockwood A. L., Anderson G. A., Winger B. E., Smith R. D. (1994) Isotopic beat patterns in fourier-transform ion-cyclotron resonance mass-spectrometry - implications for high-resolution mass measurements of large biopolymers. Int. J. Mass Spectrom. Ion Processes 132, 109–127 [Google Scholar]
- 36. Easterling M. L., Amster I. J., van Rooij G. J., Heeren R. M. A. (1999) Isotope beating effects in the analysis of polymer distributions by Fourier transform mass spectrometry. J. Am. Soc. Mass Spectrom. 10, 1074–1082 [Google Scholar]
- 37. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Muller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., Makarov A. (2012) Ultra High Resolution Linear Ion Trap Orbitrap Mass Spectrometer (Orbitrap Elite) Facilitates Top Down LC MS/MS and Versatile Peptide Fragmentation Modes. Mol. Cell. Proteomics 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Erve J. C., Gu M., Wang Y., DeMaio W., Talaat R. E. (2009) Spectral Accuracy of Molecular Ions in an LTQ/Orbitrap Mass Spectrometer and Implications for Elemental Composition Determination. J. Am. Soc. Mass Spectrom. 20, 2058–2069 [DOI] [PubMed] [Google Scholar]
- 39. Kaufmann A., Walker S. (2012) Accuracy of relative isotopic abundance and mass measurements in a single-stage orbitrap mass spectrometer. Rapid Commun. Mass Spectrom. 26, 1081–1090 [DOI] [PubMed] [Google Scholar]
- 40. Aizikov K., Mathur R., O'Connor P. B. (2009) The spontaneous loss of coherence catastrophe in Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nikolaev E. N., Jertz R., Grigoryev A., Baykut G. (2012) Fine Structure in Isotopic Peak Distributions Measured Using a Dynamically Harmonized Fourier Transform Ion Cyclotron Resonance Cell at 7 T. Anal. Chem. 84, 2275–2283 [DOI] [PubMed] [Google Scholar]
- 42. Liu T., Pantazatos D., Li S., Hamuro Y., Hilser V. J., Woods V. L., Jr. (2012) Quantitative Assessment of Protein Structural Models by Comparison of H/D Exchange MS Data with Exchange Behavior Accurately Predicted by DXCOREX. J. Am. Soc. Mass Spectrom. 23, 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramos-Fernandez A., Lopez-Ferrer D., Vazquez J. (2007) Improved method for differential expression proteomics using trypsin-catalyzed O-18 labeling with a correction for labeling efficiency. Mol. Cell. Proteomics 6, 1274–1286 [DOI] [PubMed] [Google Scholar]
- 44. Whitelegge J. P., Katz J. E., Pihakari K. A., Hale R., Aguilera R., Gómez S. M., Faull K. F., Vavilin D., Vermaas W. (2004) Subtle modification of isotope ratio proteomics; an integrated strategy for expression proteomics. Phytochemistry 65, 1507–1515 [DOI] [PubMed] [Google Scholar]
- 45. Zhao Y., Lee W. N., Lim S., Go V. L., Xiao J., Cao R., Zhang H., Recker R. R., Xiao G. G. (2009) Quantitative Proteomics: Measuring Protein Synthesis Using N-15 Amino Acid Labeling in Pancreatic Cancer Cells. Anal. Chem. 81, 764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.