

Abstract
Background
Bacillus subtilis is a very important Gram-positive model organism of high biotechnological relevance, which is widely used as a host for the production of both secreted and cytoplasmic proteins. We developed a novel and efficient expression system, based on the liaI promoter (PliaI) from B. subtilis, which is under control of the LiaRS antibiotic-inducible two-component system. In the absence of a stimulus, this promoter is kept tightly inactive. Upon induction by cell wall antibiotics, it shows an over 100-fold increase in activity within 10 min.
Results
Based on these traits of PliaI, we developed a novel LiaRS-controlled gene expression system for B. subtilis (the “LIKE" system). Two expression vectors, the integrative pLIKE-int and the replicative pLIKE-rep, were constructed. To enhance the performance of the PliaI-derived system, site-directed mutagenesis was employed to optimize the ribosome binding site and alter its spacing to the initiation codon used for the translational fusion. The impact of these genetic modifications on protein production yield was measured using GFP as a model protein. Moreover, a number of tailored B. subtilis expression strains containing different markerless chromosomal deletions of the liaIH region were constructed to circumvent undesired protein production, enhance the positive autoregulation of the LiaRS system and thereby increase target gene expression strength from the PliaI promoter.
Conclusions
The LIKE protein expression system is a novel protein expression system, which offers a number of advantages over existing systems. Its major advantages are (i) a tightly switched-off promoter during exponential growth in the absence of a stimulus, (ii) a concentration-dependent activation of PliaI in the presence of suitable inducers, (iii) a very fast but transient response with a very high dynamic range of over 100-fold (up to 1,000-fold) induction, (iv) a choice from a range of well-defined, commercially available, and affordable inducers and (v) the convenient conversion of LIKE-derived inducible expression strains into strong constitutive protein production factories.
Keywords: two-component system, liaIH operon, antibiotic-inducible promoter, cell envelope stress response, protein expression, Bacillus subtilis, bacitracin
Background
Bacillus subtilis is a widely exploited bacterium for basic research, but also industrial and biotechnological applications [1] owing to the ease of genetic manipulation, a systems level understanding of its genome and physiology [2-4], its efficient protein secretion systems [5], non-pathogenic GRAS-status [6] and well-characterized mechanisms for gene expression [7]. Over the years, numerous genetic devices and expression systems have been developed for this organism to facilitate the production of homologous or heterologous proteins [7-14], usually based on strong inducible promoters. Such systems can either be integrated into the chromosome or located on replicative plasmids to increase the gene copy number under the control of the inducible promoter.
A number of new expression systems based on induction by peptide antibiotics were described for Gram-positive bacteria [9,15,16]. The nisin-controlled gene expression (NICE) system was developed for different species of Lactococcus and Lactobacillus and allows the production of the desired proteins in high amounts (comparable to other expression systems), reaching a maximum 3 h after nisin induction [15,16]. A very similar subtilin-regulated expression system (SURE) was recently constructed for B. subtilis[9]. Both systems enable the controlled overexpression of a variety of homologous and heterologous proteins and enzymes and show a number of advantages to other control elements, such as the strict control of gene expression, no leakage of the promoter regulation under non-inducing conditions, high levels of expression upon induction and almost no limitations in the choice of sugar-containing media [9,15]. For the use in B. subtilis, the SURE system has several advantages over the NICE system: (i) The SURE system only requires a single plasmid, thereby ensuring a stable expression platform; (ii) the expression levels achieved by the SURE system are significantly higher; and (iii) it also requires lower concentrations of the inducer molecule [9,17].
Despite significant progress in the field, no exisiting system works equally well for all proteins and none of the existing expression systems for B. subtilis is without pitfalls or limitations. While the SURE system represents a major improvement, its inducer, the lantibiotic subtilin, is not commercially available. Instead, culture supernatant of the lantibiotic producer must be used, which introduces a source of variation and requires testing the potency each time a new supernatant is used for induction. Therefore, novel tightly controllable gene expression systems are still in demand to expand and complement the existing repertoire in order to find the optimal solution for a given protein to be produced in B. subtilis.
Here, such an addition to the existing bioengineering toolbox for B. subtilis will be described. The LIKE (from the German “LIa-Kontrollierte Expression”) system is based on the cell envelope stress-responsive liaI promoter. This promoter was initially identified in the course of studies on the response of B. subtilis to the presence of harmful concentrations of various cell wall antibiotics [18]. The underlying regulatory network of the cell envelope stress response in this organism is rather complex and consists of at least four extracytoplasmic function (ECF) σ factors and a similar number of two-component systems (TCS) and has been extensively studied [19,20]. One such TCS, LiaRS, is a central player in the envelope stress response network of B. subtilis. It strongly responds to antibiotics that interfere with the lipid II cycle, such as bacitracin. Activation of the LiaRS system of B. subtilis specifically leads to the strong induction of a single target promoter, PliaI, which drives the expression of the liaIH operon. This promoter is basically shut off in the absence of inducing condition during logarithmic growth and shows an impressive dynamic range of over 100- up to 1,000-fold in the presence of suitable stimuli [21-23].
Because of its specificity and sensitivity, PliaI has already been developed as a powerful screening tool for mechanism-of-action studies of novel peptide antibiotics interfering with envelope integrity [22,24,25]. But its tightly regulated, concentration-dependent and highly dynamic behavior also makes this promoter a very promising candidate for the development of a novel gene expression system. This prospect is further supported by transcriptome studies of mutants that are constitutively switched Lia-ON or Lia-OFF, which revealed a very specific response with only very few genes being indirectly affected [23]. Moreover, B. subtilis is highly resistant to bacitracin, a commercially available compound, which can be used as an ideal inducer to activate PliaI-driven gene expression in growing cultures of B. subtilis. Moreover, a simple gene deletion can convert the inducible into a high-level constitutive promoter activity. Based on these traits of PliaI, we developed vectors and strains to apply this promoter as a powerful protein expression system in B. subtilis.
Results and discussion
Features of the native liaI promoter (PliaI)
Previously, we have characterized the cell envelope stress-inducible promoter PliaI, which controls the expression of the liaIH operon in B. subtilis. During normal logarithmic growth, this promoter is virtually switched off and hence does not show any significant basal activity. In the presence of suitable inducers such as the cell wall antibiotic bacitracin, it strongly responds in a concentration-dependent manner, resulting in a more than 100-fold increased activity already 5–10 min after the addition of bacitracin. This activity strictly depends on the activity of the response regulator LiaR [21-23] (Figure 1A). This tight regulation and the impressive strength of PliaI under inducing conditions are illustrated by the protein gel shown in Figure 1B, which demonstrates that even from the native PliaI, present in single copy on the chromosome, LiaH is the predominant protein produced under inducing conditions, as has already been indicated previously by 2D gelectrophoresis [23]. These features make PliaI a very promising candidate for developing a novel protein-expression system for the Gram-positive model organism B. subtilis, which is widely used in the biotechnological industry as a protein production host [1]. To achieve this, the liaI promoter was first sequence-optimized and then integrated into two expression vectors. Moreover, a set of suitable expression strains was developed and evaluated to further improve the promoter strength while simultaneously avoiding the metabolic burden of overexpressing the native target proteins of LiaR-dependent gene regulation, LiaI and LiaH [23], as indicated in Figure 1B.
Figure 1.
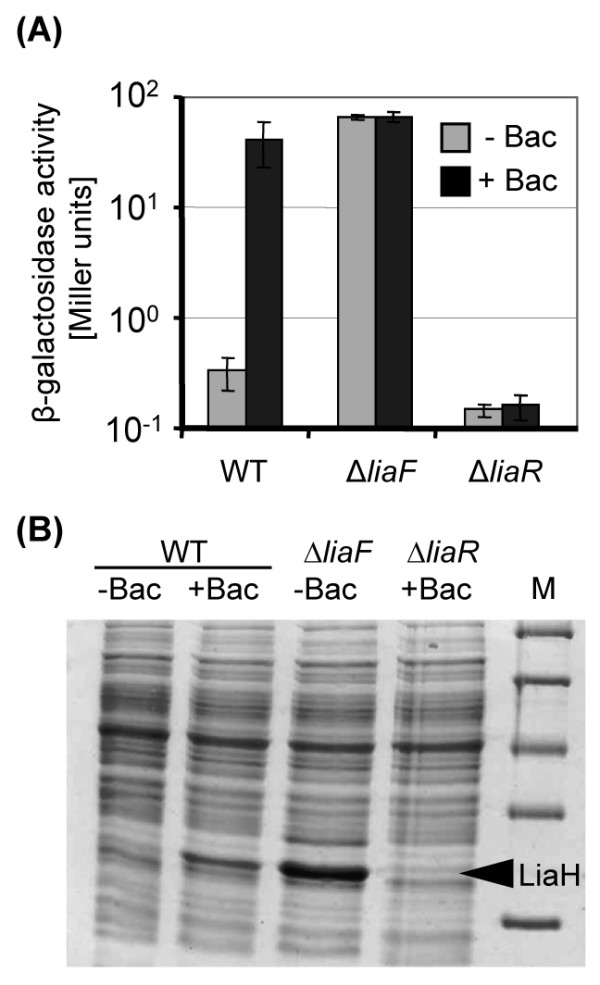
Activity of the native liaI promoter (PliaI) as monitored by (A) promoter-reporter gene fusions and (B) SDS-PAGE. (A) β-Galactosidase reporter assays of a PliaI-lacZ fusion in the wild type W168 (TMB016) and the corresponding liaF and liaR mutants (TMB331 and TMB020, respectively) in the presence and absence of bacitracin (Bac; final concentration 50 μg/ml). The assay was performed as previously described [21], the promoter activity is expressed as Miller units (β-galactosidase activity normalized against cell density). (B) SDS-PAGE of the soluble protein fraction (15 μg/lane) of the wild type (WT) and isogenic liaF and liaR mutants, challenge for 30 min with bacitracin as described above. The position of the band corresponding to the LiaH protein is marked. The identity of LiaH was verified by mass spectroscopy. M, molecular weight markers.
Design and construction of PliaI-based expression vectors and B. subtilis protein production strains for the LIKE system
A closer inspection of the liaI promoter sequence revealed a poorly conserved Shine-Dalgarno sequence (SD) with a suboptimal spacing to the liaI start codon (data not shown). As a first step in developing a PliaI-derived bacitracin-inducible expression system, we therefore optimized its SD sequence by introducing a strong B. subtilis ribosome binding site (TAAGGAGG) with an optimal spacing of seven nucleotides upstream of the start codon, which was used for all subsequent constructions and will be referred to as PliaI(opt) from now on (Figure 2). This optimized SD sequence is well established for B. subtilis[26,27], and provides optimal complementarity to the 3’-end of the 16S rRNA, thereby increasing the ribosome’s affinity to the mRNA and enhancing the translation initiation efficiency.
Figure 2.
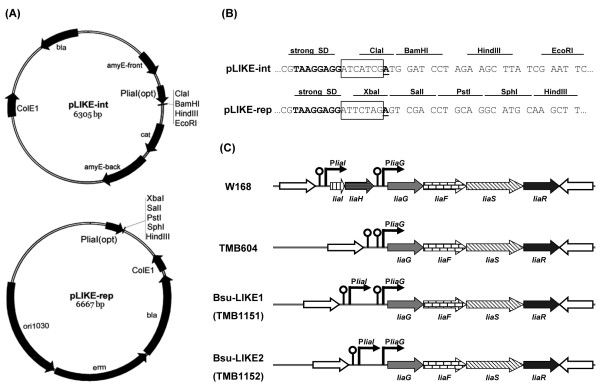
Vectors and strains of the LIKE system. (A) Vector maps of integrative plasmid pLIKE-int and E. coli/B. subtilis shuttle vector pLIKE-rep. Abbreviations: PliaI(opt), liaI promoter with optimized SD sequence; bla, ampicilin resistance; erm, erythromycin resistance; cat, chloramphenicol resistance; ColE1, origin of replication for E. coli; ori1030, origin of replication for B. subtilis. (B) Sequence of the multiple cloning sites for each plasmid. The optimized SD sequence is indicated in bold, the 7 bp ‘spacer’ is boxed, the first nucleotide of the coding sequence is underlined. For pLIKE-int, ClaI can be used as restriction enzyme to create an ATG start codon. For pLIKE-rep, XbaI must be used to reconstruct the ATG start codon. (C) Schematic representation of the liaI operon and genotype of deletion strains constructed in this study. Open reading frames are depicted as solid arrows. Promoters of the genes indicated by thin arrows, terminators by hairpin symbols.
For the construction of new PliaI-derived bacitracin-inducible gene expression systems, we chose two vectors as backbones: the E. coli/B. subtilis shuttle vector pGP380, and pDG1662 for ectopic integration at the amyE locus of B. subtilis[28,29], thus enabling both expression from a multi-copy replicative vector, as well as the stable chromosomal integration at single copy. The optimized regulatory element PliaI(opt) was amplified by PCR and cloned into the two vectors (see Materials and Methods for details) resulting in the expression vectors pLIKE-rep and pLIKE-int, respectively (Figure 2A/B).
Previous work has demonstrated that the liaIH operon is the only relevant target of LiaFSR-dependent gene expression, and that activation of PliaI results in a strong accumulation of LiaH in the cytosol (Figure 1B) [23]. Based on the organization and expression of genes in the liaIH-liaGFSR locus, activation of PliaI also leads to an increased expression of liaGFSR, due to read-through transcription [22]. Such positive autoregulatory feedback loops often have beneficial effects on the activity of their target genes [30]. Hence, it might be desirable to maintain this feedback loop. On the other hand, the observed very strong production of the native LiaFSR-target proteins LiaIH is not desired in a protein production host, since it depletes the cells of energy, amino acids and ribosomes required for heterologous protein production.
To account for these two opposing goals, we constructed a number of clean deletion mutants as potential hosts of the LIKE-system. The features of the resulting strains are summarized in Figure 2C. Strain TMB604 lacks both the liaIH operon including the native liaI promoter. Hence, no autoregulation can occur under inducing conditions. In contrast, strains TMB1151/TMB1152, which are also deleted for the liaIH operon, still maintain PliaI and therefore autoregulation. They differ in the presence or absence of the weak terminator located downstream of liaH (Figure 2C).
As a measure for PliaI(opt)-dependent protein production in the two expression plasmids, gfpmut1 gene was used as a reporter gene [31]. Translational fusions of PliaI(opt) with gfpmut1 were constructed in both pLIKE-int and pLIKE-rep and subsequently introduced into the aforementioned B. subtilis strains.
Evaluation of the LIKE-system, based on the bacitracin-induced GFP production
The range of inducers for the envelope-stress responsive LiaFSR three-component system is well-defined and includes, amongst others, the cell wall antibiotic bacitracin [22,25]. As an inducer for protein production in B. subtilis, this compound has a number of advantages: (i) It is one of the strongest inducers for the Lia-system and is easily commercially available. (ii) B. subtilis is highly resistant against bacitracin, and even above inhibitory antibiotic concentrations, cellular damage occurs only very slowly [18,32]. (iii) The maximum PliaI activity occurs well below the inhibitory concentration, thereby avoiding any damage to the producing cultures. (iv) In addition to its major inhibitory activity on cell wall biosynthesis, bacitracin also acts as a weak protease inhibitor [33], which can be viewed as a beneficial side effect of using this inducer. For all of these reasons, bacitracin will be used as the model inducer for the subsequently described evaluation of the LIKE expression system.
Nevertheless, it should be pointed out that a number of other compounds and conditions can also be considered as suitable alternative inducers, including antibiotics such as vancomycin or nisin, as well as non-antibiotic conditions such as alkaline shock [34], making the LIKE-system highly variable even in cases where bacitracin is not suitable for a given application (for example for heterologous protease production).
Initially, we compared the promoter activity of PliaI(opt) between pLIKE-int and pLIKE-rep in all four different host strains described above (Figure 2C). For this purpose, the dynamics of expression of recombinant GFP was determined after bacitracin addition (30 μg mL-1) over the course of 4 h in growing populations. In all strains, a swift and strong accumulation of fluorescence was detected already 30 min after bacitracin induction (Figure 3A). As expected, gfp expression was significantly higher in strains harboring the replicative pLIKE-rep derivative (multiple copies) compared to strains with chromosomally integrated pLIKE-int derivatives (Figure 3A). In the wild type background of W168, the fluorescence intensity of the expression strain TMB1172, harboring the integrated expression plasmid, reached less than 10% of the activity measured for the otherwise identical strain TMB1176 with the replicative construct (Figure 3A).
Figure 3.

Growth, absolute fluorescence (A) and promoter activity (B) of strains carrying translational fusion of PliaI-gfpmut1 and PliaI(opt)-gfpmut1. Growth profiles are shown without symbols and expression by symbols: (○) (W168), (□) (TMB604), (◊) (TMB1151), (∆) (TMB1152) and (●) (TMB408). Vertical dotted line indicates time point of bacitracin addition (final conc. 30 μg mL-1; OD600~0.4-0.5). Fluorescence is expressed in arbitrary units (AU) (C) Western blot analysis of the cytoplasmic fractions of cells expressing the same fusions probed with LiaH or GFP antisera. Lanes 1–2, protein expression with native modified PliaI; 3–10, with optimized PliaI(opt) in the absence (−) and presence (+) of bacitracin (final conc. 30 μg mL-1).
The benefit of improving the ribosome binding site in PliaI(opt) compared to the original SD sequence could also be demonstrated by comparing GFP production in two strains, TMB1172 and TMB408, respectively, harboring integrated expression systems. Our analysis revealed that the level of GFP expression from the wild type PliaI promoter in strain TMB408 was significantly lower compared to PliaI(opt)-mediated expression (Figure 3A and Figure 3C, lanes 2 and 4). A deletion of the native PliaI upstream of the liaGFSR operon in strain TMB604 resulted in an approx. two-fold decreased promoter activity compared to the wild type background, indicating that the presence of the autoregulatory feedback loop is important for full PliaI activity (Figure 3A and Table 1). On the other hand, deletion of liaH while maintaining the native PliaI upstream of the liaGFSR operon (strains TMB1151/1152) resulted in only a small increase of PliaI(opt) activity in case of the pLIKE-int derived expression strain. This effect was more pronounced in case of the pLIKE-rep derived strains, where the promoter activity even surpassed that of the wild type (Figure 3A and Table 1). Taken together, these results demonstrate both the important role of the autoregulatory feedback and of improving the SD sequence for achieving a maximal level of GFP production.
Table 1.
Effect of mutations in the liaIH operon on the expression of translational PliaI(opt)-gfp fusions
| Strain |
Relevant genotypea |
Promoter activity (fluorescence)b | |
|---|---|---|---|
| Expression plasmid | Strain background | ||
| TMB408 |
amyE:: pSJ5101 (PliaI-gfp) |
(WT168) PliaIliaIHTerm+ |
264 |
| TMB1172 |
amyE:: pLIKE-int+gfp |
(WT168) PliaIliaIHTerm+ |
1440 |
| TMB1174 |
(TMB604) ∆PliaIliaIH |
958 |
|
| TMB1153 |
(TMB1151) ∆liaIH |
1080 |
|
| TMB1318 |
(TMB1152) ∆liaIHTerm |
1416 |
|
| TMB1176 |
pLIKE-rep+gfp | (WT168) PliaIliaIHTerm+ |
9570 |
| TMB1178 |
(TMB604) ∆PliaIliaIH |
3372 |
|
| TMB1342 |
(TMB1151) ∆liaIH |
10607 |
|
| TMB1343 | (TMB1152) ∆liaIHTerm | 10870 | |
a The terminator downstream of liaH is abbreviated “Term”, its presence is indicated by a “+”. b Promoter activities were calculated taking the derivative of the fluorescence divided by the OD600 (dGFP/dt/OD600) at each time point.
Determination of the PliaI activity revealed that the window of promoter activity was narrower in case of the integrated promoter, both for activation and shut-off, relative to the replicative derivatives (Figure 3B). For the pLIKE-int derivatives, maximum promoter activity was reached already 20–30 min after addition of bacitracin and the total window of activity was less than 60 min. In contrast, pLIKE-rep derivatives required almost 60 min to reach maximum promoter activity and the total window of activity was about 120 min. But in light of the overall 10-times higher promoter activity in case of the latter, this result is maybe not too surprising.
All major conclusions drawn above were verified at the protein level by Western analysis, using antibodies against GFP and LiaH. Both proteins were not detectable in uninduced cultures, supporting the previously demonstrated tight control of PliaI and the absence of any significant promoter activity under non-inducing conditions. Upon addition of bacitracin, both proteins accumulated to different level, depending on strain background. These studies demonstrate both the positive effect of improving the ribosome binding site and the negative effect of deleting the autoregulatory feedback loop at the level of protein production (Figure 3C).
Taken together, both pLIKE-int and pLIKE-rep were successfully established as vectors for bacitracin-dependent protein production in strains that maintain the positive autoregulatory feedback loop. While expression based on the replicative vector yields higher protein amounts, the integrative system has the advantage of genetic stability and does not require any selection.
Effect of the inducer (bacitracin) concentration on the activity of PliaI(opt)
Next, we wanted to investigate the dynamics of PliaI activity and the resulting GFP production as a function of the inducer concentration. It is already well established that PliaI-mediated gene expression occurs in a dosage-dependent manner, at least in case of the wild type promoter [22,23,32]. Here, we performed similar experiments, using the pLIKE-int and pLIKE-rep derivatives pAT6203 and pAT3803, respectively, in the W168 (wild type) background. The resulting strains TMB1172 and TMB1176 were inoculated in microtiter plates and challenged in the mid-exponential growth phase with increasing concentrations of bacitracin (Figure 4). The results are in very good agreement with the previous observations. The promoter activity increases as a function of the bacitracin concentration, reaching a maximum at bacitracin concentrations of about 30 μg mL-1 (Figure 4A/B). At higher concentrations (above 50 μg mL-1), the ongoing promoter activity after 250 mins indicates an ongoing bacitracin stress. Especially at the highest bacitracin concentration, 100 μg mL-1, the GFP yield is clearly reduced concomitant with a reduced final cell density, at least in case of the pLIKE-rep derived strain TMB1176 (Figure 4A). This result was also confirmed by Western blot analysis (Figure 4C). To ensure optimal protein production without causing severe antibiotic stress, our data suggests the use of a bacitracin concentration of no more than 30 μg mL-1, although this concentration may have to be optimized for individual target proteins, especially if toxicity is a problem.
Figure 4.

Concentration-dependent induction of the PliaI(opt)in B. subtilis W168 cultures. (A) Growth, absolute fluorescence and (B) promoter activity of strains carrying translational fusion of PliaI(opt)-gfpmut1 on plasmids pLIKE-int and pLIKE-rep treated with different concentration of bacitracin. Growth profiles are shown without symbols and expression by symbols: (◊) 1 μg mL-1, (□) 3 μg mL-1, (■) 10 μg mL-1, (○) 30 μg mL-1, (●) 50 μg mL-1, (∆) 100 μg mL-1. Vertical dotted line indicates the point of mid-log phase (OD600~0.4-0.5) when bacitracin was added. Fluorescence is expressed in arbitrary units (AU) (C) Western blot analysis revealed the amount of GFP produced by cells harboring pAT6203 (pLIKE-int) and pAT3803 (pLIKE-rep) 90 min post induction.
Overproduction of YdfG using the LIKE system
To demonstrate the suitability of the LIKE system for the overexpression of a heterologous protein, we performed an expression experiment using the protein YdfG of Bacillus licheniformis. This protein is a putative carboxymuconolactone decarboxylase. We could recently demonstrate that its gene represents the only target of the extracytoplasmic function σ factor ECF41Bli[35]. It consists of 148 amino acids and an estimated molecular weight of 16,6 kDa.
Based on the results shown in Figures 3 and 4, we used strains TMB1151 and TMB1152 as expression hosts for the pLIKE-rep+His6-ydfG (pKSLIKEr01) and pLIKE-int+His6-ydfG (pKSLIKEi01) derivative, respectively. YdfG production was induced in mid-log growing cultures by addition of 30 μg ml-1 bacitracin. The cells were harvested 30 min post-induction and disrupted by sonication. For each sample, 10 μg of total protein was separated on a 14% tricine gel and subsequently stained by colloidale Coomassie staining solution. The result is shown in Figure 5. For both derivatives, a clear additional band can be observed in the induced fractions at ~17 kDa. As expected, the YdfG yield received from the pLIKE-rep derivative is much higher compared to the integrative one. To be sure that this band is not a bacitracin effect, control samples of the expression host TMB1151 were treated equally and were also loaded on the gel. Here, no distinct band can be observed in the bacitracin-induced sample (Figure 5). By using the pLIKE-rep derivative, it was possible to achieve a protein yield comparable to the one shown for LiaH in bacitracin-induced B. subtilis wild type cells (Figure 1B).
Figure 5.
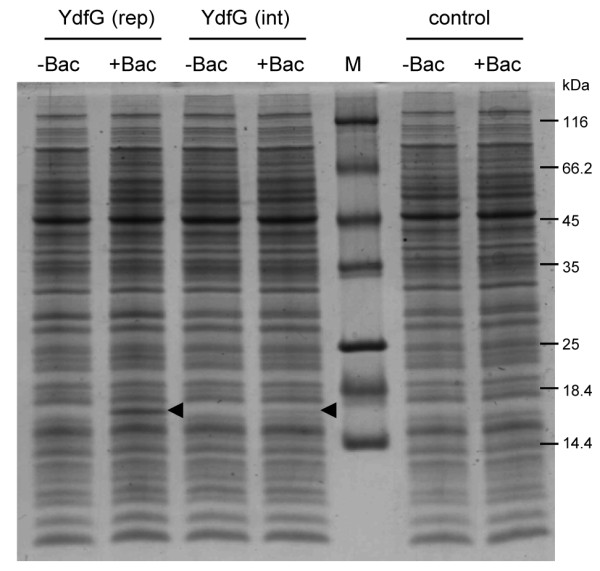
Overproduction of YdfG using the LIKE system. 14% tricine SDS-PAGE of total proteins (10 μg/lane) of strains TMB1566 (YdfG-rep), TMB1570 (YdfG-int), and TMB1151 (control) treated with and without 30 μg ml-1 bacitracin for 30 min. The position of the band corresponding to the protein YdfG is marked. M, molecular weight marker (in kDa).
Conclusions
We have developed a novel and efficient LiaFSR-dependent gene expression system, which places target proteins under the control of an optimized bacitracin-responsive PliaI promoter. The LIKE system offers first a single-copy, integrative option (pLIKE-int in strain Bsu-LIKE2), which is genetically stable without selective pressure, but reaches lower protein production levels. The second option consists of the replicative vector pLIKE-rep in combination with strain Bsu-LIKE1 to obtain a maximum gene expression. The LIKE-system has a number of important features: (i) There is no detectable background expression in the non-induced state. (ii) Using bacitracin as inducer, the promoter has an impressive dynamic range of up to 1,000-fold above background level that can be titrated as a function of inducer concentration. (iii) The described range of inducers is wide, including cell wall antibiotics that function as strong (bacitracin, nisin, daptomycin) or intermediately strong (vancomycin) inducers, as well as non-antibiotic conditions that act as intermediate to weak inducers of the Lia-system, including pH-upshift, organic solvents, some detergents, or ethanol [22,25,36-38]. All of these inducers are well-defined and readily available at low prices. Moreover, a recent study demonstrated that the Lia-system can also be induced by the overexpression of certain heterologous and secreted proteins, especially the universal shock protein USP45 from Lactococcus lactis and the TEM-1 β-lactamase from E. coli[39]. (iv) Lastly, an antibiotic-inducible LIKE-expression strain can easily be converted into a strong constitutive expression platform by the simple deletion of liaF, encoding the LiaRS-specific inhibitor protein [21-23]. The effect of such a deletion is shown in Figure 1, which demonstrates that a liaF deletion results in a protein production that even surpasses that of the fully induced strains, even in the absence of an inducer.
This flexibility distinguishes the LIKE system from other available expression systems. Taken together, the expression vectors and strains described in this report expand the genetic toolbox already available for protein production, based on the tight and highly dynamic bacitracin-inducible promoter PliaI. We hope and believe that the vectors and strains described in this report will provide valuable tools for protein expression in B. subtilis. The LIKE system, consisting of both expression vectors as well as the host strains Bsu-LIKE1 and Bsu-LIKE2, is available for the scientific community through the Bacillus Genetic Stock Center (http://www.bgsc.org; accession numbers ECE255, ECE256 for the two vectors and 1A1070, 1A1071 for the two B. subtilis expression strains).
Methods
Growth conditions
All bacterial strains (Table 2) were grown in Luria–Bertani (LB) medium at 37°C with aeration. The cell density was determined by measuring the OD600 with the Ultrospec™ 2100 pro UV/visible spectrophotometer (GE Healthcare). When appropriate, the growth media were supplemented with chloramphenicol (5 μg mL-1), erythromycin (1 μg mL-1) plus lincomycin (25 μg mL-1) for macrolide-lincosamide-streptogramin (MLS) resistance (B. subtilis), or ampicillin (100 μg mL-1; E. coli). Protein expression was induced by using zinc bacitracin (Sigma).
Table 2.
Bacterial strains used in this study
| Strain | Relevant genotype | Source and/or reference |
|---|---|---|
|
E. coli DH5α |
recA1 endA1 gyrA96 thi hsdR17(rK- mK+) relA1 supE44 ϕ80ΔlacZΔM15 Δ(lacZYA-argF)U169 |
Laboratory stock |
|
Bacillus subtilis |
|
|
| W168 |
Wild type, trpC2 |
Laboratory stock |
| HB0933 |
W168 attSPβ2∆2 trpC2, liaR::kan |
[18] |
| TMB016 |
W168 amyE::(cat PliaI-lacZ) |
[21] |
| TMB020 |
HB0933 amyE::(cat PliaI-lacZ) |
[21] |
| TMB329 |
W168 ∆liaF (clean deletion) |
[23] |
| TMB331 |
TMB329 amyE::(cat PliaI-lacZ) |
This work |
| TMB408 |
W168 amyE::pSJ5101 (PliaI-gfp) |
S. Jordan |
| TMB604 |
W168 ΔPliaI-liaIH (clean deletion) |
[23] |
| Bsu-LIKE1 (TMB1151) |
W168 ∆liaIH (clean deletion) |
This work |
| Bsu-LIKE2 (TMB1152) |
W168 ∆liaIH-terminator (clean deletion) |
This work |
| TMB1172 |
W168 amyE::pAT6203 (pLIKE-int PliaI(opt)-gfp) |
This work |
| TMB1176 |
W168 pAT3803 (pLIKE-rep PliaI(opt)-gfp) |
This work |
| TMB1174 |
TMB604 amyE::pAT6203 (pLIKE-int PliaI(opt)-gfp) |
This work |
| TMB1178 |
TMB604 pAT3803 (pLIKE-rep PliaI(opt)-gfp) |
This work |
| TMB1153 |
TMB1151 amyE::pAT6203 (pLIKE-int PliaI(opt)-gfp) |
This work |
| TMB1342 |
TMB1151 pAT3803 (pLIKE-rep PliaI(opt)-gfp) |
This work |
| TMB1318 |
TMB1152 amyE::pAT6203 (pLIKE-int PliaI(opt)-gfp) |
This work |
| TMB1343 |
TMB1152 pAT3803 (pLIKE-rep PliaI(opt)-gfp) |
This work |
| TMB1566 |
TMB1151 pKSLIKEr01 (pLIKE-rep PliaI(opt)-His6-ydfG) |
This work |
| TMB1570 | TMB1152 pKSLIKEi01 (pLIKE-int PliaI(opt)-His6-ydfG) | This work |
DNA manipulations, transformation and PCR
All plasmid constructions were done in E. coli and isolated by alkaline lysis method [40], then used to transform B. subtilis[41]. Procedures for DNA manipulation and transformation of E. coli were carried out as described [42]. The primers used in this study are listed in Table 3. For all PCR reactions the Phusion DNA Polymerase (Finnzymes) was used according to the manufacturer’s instructions. Sequencing was performed in-house by the Sequencing Facility of the LMU Biocenter.
Table 3.
Oligonucleotides used in this study
| Primers | Sequence (5' to 3')a | Description/position |
|---|---|---|
| Plasmid construction |
|
|
| TM2064 |
CATGGTCTCAGATCTTTAAAACGCCATGCCTCG |
BsaI; 5' end of PliaI |
| TM1980 |
CTTGTTGGATCCATCGATGATCCTCCTTACGTTTTCCTTGTCTTC |
Strong SD region; BamHI, ClaI; 3' end of PliaI |
| TM1991 |
ATCTGAATTCGGTTTTAAAACGCCATGCC |
EcoRI; 5' end of PliaI |
| TM1992 |
ATTTTCTCTAGAATCCTCCTTACGTTTTCCTTGTCTTC |
Strong SD region; XbaI; 3' end of PliaI |
| TM1981 |
TCCTATCGATGAGTAAAGGAGAAGAACTTTTCACTGG |
ATG start codon; ClaI; 5' end of gfpmut1 |
| TM1982 |
GGCCAAGCTTGAACTAGTTTCATTTATTTGTAGAGC |
HindIII; 3' end of gfpmut1 |
| TM1993 |
TTCCTCTAGATGAGTAAAGGAGAAGAACTTTTC |
ATG start codon; XbaI; 5' end of gfpmut1 |
| TM1994 |
GGCCGTCGACGAACTAGTTTCATTTATTTG |
SalI; 3' end of gfpmut1 |
| TM2535 |
CCATATCGATGCATCATCATCATCATCACGAAACGAGATTTCTAATGGAAAAAG |
ATG start codon; ClaI; His6-tag; 5’ end of ydfG |
| TM2536 |
CCATAAGCTTTCAATCTGCTGCGGGCATTTTC |
HindIII; 3’ end of ydfG |
| TM2545 |
CCATTCTAGATGCATCATCATCATCATCACGAAACGAGATTTCTAATGGAAAAAG |
ATG start codon; XbaI; His6-tag; 5’ end of ydfG |
| Clean deletions |
|
|
| TM2130 |
GCGGGGATCCTCTTACATTTATTAGTCC |
BamHI; upstream of PliaI |
| TM2131 |
CATTTGCCGCTTTTGTCTGGGCAGATCCTCCTTTCGTTTTC |
3' end of PliaI; 3' end of liaH |
| TM1055 |
CCAGACAAAAGCGGCAAATG |
3' end of liaH |
| TM1058 |
CCATGAATTCGAATGCGGACGTCCGTCACGC |
EcoRI; inside the liaG gene |
| TM2132 |
GCGAATTGATACGTGCGGGCAGATCCTCCTTTCGTTTTC |
upstream of liaI gene; upstream of liaG |
| TM2133 |
CCGCACGTATCAATTCGC |
upstream of liaG |
| TM2134 | GCTAGAATTCTGCCGGCTGTTTTGGAG | EcoRI; center of liaG gene |
a Relevant restriction sites are shown in italics, complementary regions for joining PCR are underlined. The sequence for optimized SD sequences of the liaI promoter is indicated by bold type, the start codon is bold italic. The His-tags are in italics and underlined.
Construction of markerless deletion mutant strains
Several markerless deletions of the liaIH operon (including its promoter and terminator) were constructed using the vector pMAD [43]. Genomic regions of approximately 1 kb up- and downstream of the regions to be deleted were amplified using the primers listed in Table 3. The two fragments were fused in a second joining PCR reaction, and the resulting fragment was cloned into pMAD via BamHI and EcoRI, generating the plasmids pAT101 (ΔliaIH) and pAT102 (ΔliaIHTerminator). For generating the deletion mutants, the procedure described by Arnaud et al. was applied [43]. In brief, B. subtilis 168 was transformed with pAT101 or pAT102 (Table 4) and incubated for two days at 30°C on LB agar plates containing X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside; 100 μg mL-1) with MLS selection. Individual blue colonies were selected and incubated for 6 to 8 h at 42°C in LB medium with MLS selection, resulting in the integration of the plasmids into the chromosome. Blue colonies were again picked from LB (X-Gal) plates and incubated at 30°C for 6 h in LB medium without selection. Subsequently, the liquid culture was shifted to 42°C for 3 h, and the cells were then plated on LB (X-Gal) plates, this time without selective pressure. White colonies that had lost the plasmids were picked and checked for MLS sensitivity. Finally, strains TMB1151 (ΔliaIH) and TMB1152 (ΔliaIHTerminator) were analyzed by PCR and sequencing to confirm the integrity of the desired genetic modifications.
Table 4.
Vectors and plasmids used in this study
| Plasmid | Genotype/propertiesa | Primer pair(s) used for cloning | Reference |
|---|---|---|---|
| pDG1662 |
cat, spc, bla, amyE' … 'amyE integrative vector |
|
[28] |
| pGP380 |
erm, bla, Strep-Tag, PdegQ36, replicative vector |
|
[29] |
| pMAD |
erm, ori(pE194-Ts), MCS-PclpB-bgaB, ori(pBR322), bla |
|
[43] |
| pSG1151 |
bla, cat, gfpmut1 |
|
[44] |
| pAT6200 |
pDG1662 derivative; spc gene deleted |
|
This work |
| pLIKE-int |
pAT6200 derivative; PliaI(opt); integrative protein expression vector |
TM2064/TM1980 |
This work |
| pLIKE-rep |
pGP380 derivative; PliaI(opt); replicative protein expression vector |
TM1991/TM1992 |
This work |
| pAT6203 |
pLIKE-int, PliaI(opt) translationally fused to gfp |
TM1981/TM1982 |
This work |
| pAT3803 |
pLIKE-rep, PliaI(opt) translationally fused to gfp |
TM1993/TM1994 |
This work |
| pAT101 |
pMAD ΔliaIH up/down overlap |
TM2130/ TM2131, TM1055/ TM1058 |
This work |
| pAT102 |
pMAD ΔliaIHTerminator up/down overlap |
TM2130/ TM2132, TM2133/ TM2134 |
This work |
| pKSLIKEr01 |
pLIKE-rep, PliaI(opt) translationally fused to His6-ydfG |
TM2545/TM2536 |
This work |
| pKSLIKEi01 | pLIKE-int, PliaI(opt) translationally fused to His6-ydfG | TM2535/TM2536 | This work |
a Resistance cassettes: erm, erythromycin; bla, ampicillin; cat, chloramphenicol; spc, spectinomycin.
Plasmid and strain construction
Bacterial strains used in this study are derivates of the laboratory wild type strain B. subtilis W168 and are listed in Table 2. Plasmids used in this study are listed in Table 4. The promoter of the liaIH operon for integrative and replicative vectors was obtained from strain B. subtilis W168 by PCR, using primers TM2064/TM1980 and TM1991/TM1992 (Table 3), respectively. During the amplification, bases in the ribosome-binding site (RBS) were mutated to a strong B. subtilis Shine-Dalgarno (SD) sequence (TAAGGAGG) [27] to yield the optimized liaI promoter PliaI(opt).
The integrative expression vector pLIKE-int, containing PliaI with an optimized SD site (PliaI(opt)) was generated in two steps. First, the B. subtilis integrative vector pDG1662 was treated with BstBI to remove the spectinomycin resistance gene. The truncated (6141 bp) fragment was self-ligated, yielding vector pAT6200. During this step, the multiple cloning site (MCS), containing unique BamHI, HindIII, and EcoRI sites was expanded by an additional unique ClaI restriction site, which is required for introducing genes at the ATG start codon (see Figure 2 for details): a PCR product encompassing PliaI(opt) was digested with BsaI and BamHI and cloned into pAT6200 digested with BamHI, resulting in pLIKE-int. The ClaI restriction site is recommended to use for reconstruction of the ATG start codon, but it is not strictly necessary. The use of BamHI, HindIII, or EcoRI has the disadvantage of fusing additional amino acids to the N-terminus of the target protein which can cause undesired disabilities.
To construct the replicative expression vector pLIKE-rep, again harboring PliaI(opt), the promoter fragment was amplified by PCR using primers TM1991/TM1992 (Table 3). After digest of the PCR product with EcoRI and XbaI, the promoter region was ligated into the corresponding sites of pGP380, resulting in vector pLIKE-rep. For cloning of a gene into pLIKE-rep, XbaI must be used as restriction enzyme to generate the ATG start codon (see Figure 2).
For the determination of the properties of the two expression vectors, the genes gfpmut1 and ydfG were used. The gfpmut1 gene was amplified using primers TM1981/TM1982 and TM1993/TM1994, respectively (Table 3), using plasmid pSG1151 as the template. The 720-bp amplicon obtained was cloned into ClaI/HindIII-digested pLIKE-int or XbaI/SalI-digested pLIKE-rep, resulting in translational fusions with PliaI(opt) in pAT6203 and pAT3803, respectively (Table 4). Next, the B. subtilis strains W168, TMB604, TMB1151, and TMB1152 (Table 2) were transformed with the pAT6203 integrative plasmid. The resulting strains were designated TMB1172, TMB1174, TMB1153, TMB1318, respectively (Table 2). Strains bearing the replicative pAT3803 GFP-expression plasmid were constructed by transformation of the above strains with plasmid DNA and selection for MLS resistance, resulting in strains TMB1176, TMB1178, TMB1342, and TMB1343, respectively. The ydfG gene was amplified from Bacillus licheniformis genomic DNA using primers TM2545/TM2536 and TM2535/TM2536, respectively (Table 3). The PCR product was cloned into ClaI/HindIII digested pLIKE-int or XbaI/HindIII digested pLIKE-rep, resulting in plasmids pKSLIKEi01 and pKSLIKEr01, respectively (Table 4). Next, the B. subtilis strain TMB1151 was transformed with pKSLIKEr01 replicative plasmid and TMB1152 was transformed with the linearized pKSLIKEi01 integrative plasmid, resulting in strains TMB1566 and TMB1570 (Table 2).
Activation of PliaI by bacitracin and analysis of gfp gene expression
For bacitracin-mediated induction of gene expression, the appropriate B. subtilis strains were inoculated from overnight LB cultures into a final volume of 150 μL LB medium in a 96-well plate with optical bottom (Sarstedt) and were incubated in a Synergy™ 2 multimode microplate reader (Biotek) at 37°C with constant medium shaking. When the culture reached an OD600 of 0.45, bacitracin (30 μg mL-1 final concentration) was added to one half of the wells (induced sample), and the other half was left untreated (uninduced control). Plates were covered with lids to prevent evaporation and incubated for 4 h. Growth was monitored by measuring absorbance at 600 nm. Fluorescence readings were taken from the bottom by using a GFP-specific filter pair (excitation 485/20 nm, emission 528/20 nm). Measurements were taken in 10 min intervals. To calculate expression levels, the natural fluorescence of three cultures of wild type B. subtilis strain 168 (containing no reporter gene) were averaged and subtracted from the raw fluorescence value of each reporter strain at the same OD600 value [45]. Determination of PliaI activity was calculated as described in [45] as the derivative of the fluorescence divided by the OD600 (dGFP/dt/OD600) for each time point. Expression values were averaged from three independent samples of the same time points ((P1+P2+P3)/3). Polynomial and exponential functions were used to fit the experimental GFP dataset; promoter activities (dGFP/dt/OD600) were calculated using these functions [45].
Western blotting
Total cytoplasmic proteins were prepared from 15 mL culture per time point by sonication. Proteins (20 μg per lane) were separated by SDS-PAGE, according to standard procedure [42]. After electrophoresis and equilibration of the gels in transfer buffer [15.2 g Tris; 72.1 g glycine; 750 mL methanol (100%) in a final volume of 5 L with deionized water] the proteins were blotted to a PVDF membrane using a mini-trans blot apparatus (Bio-Rad) according to standard procedure [42]. The LiaH antibody (polyclonal rabbit antisera raised against purified His10–LiaH [46]), GFP antibody (rabbit monoclonal antibody against the green fluorescent protein, Epitomics), and the secondary antibody (anti-rabbit IgG HRP conjugate, Promega) were diluted 1:20,000, 1:3,000, and 1:100,000, respectively. For LiaH/GFP detection, AceGlowTM (PeqLab) was used according to the manufacturer’s instructions. Blots were documented on a QUANTUM-ST4-3026 chemiluminescence documentation system (PeqLab).
Overproduction of YdfG
For the overexpression of ydfG, strains TMB1566 and TMB1570 were grown in LB medium at 37°C until they reached an OD600 of ~0.4-0.5. Cultures were split and one half was induced with 30 μg ml-1 bacitracin for 30 min. The other half was left untreated. 20 ml of each culture was harvested by centrifugation and cell pellets were kept at −80°C until further use. For total protein preparation, the cell pellets were resuspended in 1 ml of cold disruption buffer (50 mM Tris–HCl, 100 mM NaCl, pH 7.5) and cells were disrupted by sonication on ice. Proteins (10 μg per lane) were separated by 14% tricine SDS-PAGE, according to standard procedure [47] and gels were subsequently stained by colloidale Coomassie staining solution [48].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AAT carried out all experiments with the exception of the overexpression experiment and those acknowledged below. KS performed the overexpression experiment with YdfG. AAT, KS, and TM conceived the study and wrote the manuscript. MRS participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Anna A Toymentseva, Email: tojmencevaaa@mail.ru.
Karen Schrecke, Email: karen.schrecke@campus.lmu.de.
Margarita R Sharipova, Email: Margarita.Sharipova@ksu.ru.
Thorsten Mascher, Email: mascher@bio.lmu.de.
Acknowledgments
The authors are indebted to S. Hübner for invaluable assistance in establishing the BioTek microplate reader set-up in our laboratory, as well as with the corresponding data analysis. Moreover, we would like to thank D. Wolf, and S. Jordan for Bacillus strains used in this study and F. Kalamorz for the SDS-PAGE picture shown in Figure 1B. This work was supported by grant MA2837/1-3 from the Deutsche Forschungsgemeinschaft (to TM) and a Federal grant "Science and teaching program of innovative Russia" for 2009–2013 (to MRS). AAT was supported by a DAAD research fellowship “Forschungsstipendien für Doktoranden und Nachwuchswissenschaftler” (325-A/09/84065).
References
- Schallmey M, Singh A, Ward OP. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50:1–17. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- Barbe V, Cruveiller S, Kunst F, Lenoble P, Meurice G, Sekowska A, Vallenet D, Wang T, Moszer I, Medigue C, Danchin A. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology. 2009;155:1758–1775. doi: 10.1099/mic.0.027839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JM, Liebermeister W, Jules M, Uhr M, Muntel J, Botella E, Hessling B, Kleijn RJ, Le Chat L, Lecointe F. et al. Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science. 2012;335:1099–1103. doi: 10.1126/science.1206871. [DOI] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S. et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Ling Lin F, Zi Rong X, Wei Fen L, Jiang Bing S, Ping L, Chun Xia H. Protein secretion pathways in Bacillus subtilis: implication for optimization of heterologous protein secretion. Biotechnol Adv. 2007;25:1–12. doi: 10.1016/j.biotechadv.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Westers L, Westers H, Quax WJ. Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta. 2004;1694:299–310. doi: 10.1016/j.bbamcr.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189. doi: 10.1016/S0065-2164(07)62006-1. [DOI] [PubMed] [Google Scholar]
- Bhavsar AP, Zhao X, Brown ED. Development and characterization of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl Environ Microbiol. 2001;67:403–410. doi: 10.1128/AEM.67.1.403-410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers RS, Veening J-W, Van Wieringen M, Kuipers OP, Kleerebezem M. Development and characterization of a subtilin-regulated expression system in Bacillus subtilis: strict control of gene expression by addition of subtilin. Appl Environ Microbiol. 2005;71:8818–8824. doi: 10.1128/AEM.71.12.8818-8824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad B, Savchenko RS, Breves R, Hofemeister J. A T7 promoter-specific, inducible protein expression system for Bacillus subtilis. Mol Gen Genet. 1996;250:230–236. doi: 10.1007/BF02174183. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Pan JG, Park SH, Choi SK. Development of a stationary phase-specific autoinducible expression system in Bacillus subtilis. J Biotechnol. 2010;149:16–20. doi: 10.1016/j.jbiotec.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Liu HB, Chui KS, Chan CL, Tsang CW, Leung YC. An efficient heat-inducible Bacillus subtilis bacteriophage 105 expression and secretion system for the production of the Streptomyces clavuligerus beta-lactamase inhibitory protein (BLIP) J Biotechnol. 2004;108:207–217. doi: 10.1016/j.jbiotec.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Le Thuy AT, Schumann W. A novel cold-inducible expression system for Bacillus subtilis. Protein Expr Purif. 2007;53:264–269. doi: 10.1016/j.pep.2006.12.023. [DOI] [PubMed] [Google Scholar]
- Wenzel M, Müller A, Siemann-Herzberg M, Altenbuchner J. Self-inducible Bacillus subtilis expression system for reliable and inexpensive protein production by high-cell-density fermentation. Appl Environ Microbiol. 2011;77:6419–6425. doi: 10.1128/AEM.05219-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierau I, Kleerebezem M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol. 2005;68:705–717. doi: 10.1007/s00253-005-0107-6. [DOI] [PubMed] [Google Scholar]
- Wu CM, Lin CF, Chang YC, Chung TC. Construction and characterization of nisin-controlled expression vectors for use in Lactobacillus reuteri. Biosci Biotechnol Biochem. 2006;70:757–767. doi: 10.1271/bbb.70.757. [DOI] [PubMed] [Google Scholar]
- Vavrova L, Muchova K, Barak I. Comparison of different Bacillus subtilis expression systems. Res Microbiol. 2010;161:791–797. doi: 10.1016/j.resmic.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Mascher T, Margulis NG, Wang T, Ye RW, Helmann JD. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol Microbiol. 2003;50:1591–1604. doi: 10.1046/j.1365-2958.2003.03786.x. [DOI] [PubMed] [Google Scholar]
- Jordan S, Hutchings MI, Mascher T. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol Rev. 2008;32:107–146. doi: 10.1111/j.1574-6976.2007.00091.x. [DOI] [PubMed] [Google Scholar]
- Schrecke K, Staroń A, Mascher T. In: Two component systems in bacteria. Gross R, Beier D, Hethersett N, editor. UK: Horizon Scientific Press; 2012. Two-component signaling in the Gram-positive envelope stress response: intramembrane-sensing histidine kinases and accessory membrane proteins. [Google Scholar]
- Jordan S, Junker A, Helmann JD, Mascher T. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: Identification of inhibitor proteins, regulator binding sites and target genes of a conserved cell envelope stress-sensing two-component system. J Bacteriol. 2006;188:5153–5166. doi: 10.1128/JB.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T, Zimmer SL, Smith TA, Helmann JD. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob Agents Chemother. 2004;48:2888–2896. doi: 10.1128/AAC.48.8.2888-2896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Kalamorz F, Wecke T, Juszczak A, Mader U, Homuth G, Jordan S, Kirstein J, Hoppert M, Voigt B. et al. In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol. 2010;192:4680–4693. doi: 10.1128/JB.00543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard M, Stein T. Microtiter plate bioassay to monitor the interference of antibiotics with the lipid II cycle essential for peptidoglycan biosynthesis. J Microbiol Methods. 2008;75:70–74. doi: 10.1016/j.mimet.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Staroń A, Finkeisen DE, Mascher T. Peptide antibiotic sensing and detoxification modules of Bacillus subtilis. Antimicrob Agents Chemother. 2011;55:515–525. doi: 10.1128/AAC.00352-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellanoweth RL, Rabinowitz JC. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol. 1992;6:1105–1114. doi: 10.1111/j.1365-2958.1992.tb01548.x. [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis. Gene. 1996;180:57–61. doi: 10.1016/S0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- Herzberg C, Weidinger LA, Dörrbecker B, Hübner S, Stülke J, Commichau FM. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics. 2007;7:4032–4035. doi: 10.1002/pmic.200700491. [DOI] [PubMed] [Google Scholar]
- Mitrophanov AY, Groisman EA. Positive feedback in cellular control systems. BioEssays. 2008;30:542–555. doi: 10.1002/bies.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Rietkötter E, Hoyer D, Mascher T. Bacitracin sensing in Bacillus subtilis. Mol Microbiol. 2008;68:768–785. doi: 10.1111/j.1365-2958.2008.06194.x. [DOI] [PubMed] [Google Scholar]
- Ming LJ. Structure and function of "metalloantibiotics". Med Res Rev. 2003;23:697–762. doi: 10.1002/med.10052. [DOI] [PubMed] [Google Scholar]
- Wiegert T, Homuth G, Versteeg S, Schumann W. Alkaline shock induces the Bacillus subtilis σW regulon. Mol Microbiol. 2001;41:59–71. doi: 10.1046/j.1365-2958.2001.02489.x. [DOI] [PubMed] [Google Scholar]
- Wecke T, Halang P, Staroń A, Dufour YS, Donohue TJ, Mascher T. Extracytoplasmic function sigma factors of the widely distributed group ECF41 contain a fused regulatory domain. Microbiology Open. 2012;1:194–213. doi: 10.1002/mbo3.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersohn A, Brigulla M, Haas S, Hoheisel JD, Völker U, Hecker M. Global analysis of the general stress response of Bacillus subtilis. J Bacteriol. 2001;183:5617–5631. doi: 10.1128/JB.183.19.5617-5631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietiäinen M, Gardemeister M, Mecklin M, Leskela S, Sarvas M, Kontinen VP. Cationic antimicrobial peptides elicit a complex stress response in Bacillus subtilis that involves ECF-type sigma factors and two-component signal transduction systems. Microbiology. 2005;151:1577–1592. doi: 10.1099/mic.0.27761-0. [DOI] [PubMed] [Google Scholar]
- le Tam T, Eymann C, Albrecht D, Sietmann R, Schauer F, Hecker M, Antelmann H. Differential gene expression in response to phenol and catechol reveals different metabolic activities for the degradation of aromatic compounds in Bacillus subtilis. Environ Microbiol. 2006;8:1408–1427. doi: 10.1111/j.1462-2920.2006.01034.x. [DOI] [PubMed] [Google Scholar]
- Marciniak BC, Trip H, Kuipers OP, Van-der Veek PJ. Comparative transcriptional analysis of Bacillus subtilis cells overproducing either secreted proteins, lipoproteins or membrane proteins. Microb Cell Fact. 2012;11:66. doi: 10.1186/1475-2859-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood CR, Cutting SM. Molecular Biological Methods for Bacillus. Chichester: John Wiley & Sons; 1990. [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning - a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Arnaud M, Chastanet A, Debarbouille M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol. 2004;70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene. 1999;227:101–110. doi: 10.1016/S0378-1119(98)00580-0. [DOI] [PubMed] [Google Scholar]
- Botella E, Fogg M, Jules M, Piersma S, Doherty G, Hansen A, Denham EL, Le Chat L, Veiga P, Bailey K. et al. pBaSysBioII: an integrative plasmid generating gfp transcriptional fusions for high-throughput analysis of gene expression in Bacillus subtilis. Microbiology. 2010;156:1600–1608. doi: 10.1099/mic.0.035758-0. [DOI] [PubMed] [Google Scholar]
- Jordan S, Rietkötter E, Strauch MA, Kalamorz F, Butcher BG, Helmann JD, Mascher T. LiaRS-dependent gene expression is embedded in transition state regulation in Bacillus subtilis. Microbiology. 2007;153:2530–2540. doi: 10.1099/mic.0.2007/006817-0. [DOI] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9:255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]