

Abstract
The surface-exposed NadA adhesin produced by a subset of capsular serogroup B strains of Neisseria meningitidis is currently being considered as a vaccine candidate to prevent invasive disease caused by a hypervirulent lineage of meningococci. Levels of NadA are known to be controlled by both transcriptional regulatory factors and a component of human saliva, 4-hydroxyphenylacetic acid. Herein, we confirmed the capacity of a DNA-binding protein termed FarR to negatively control nadA expression. We also found that a known transcriptional regulator of farR in N. gonorrhoeae termed MtrR can have a negative regulatory impact on farR and nadA expression, especially when over-expressed. MtrR-mediated repression of nadA was found to be direct, and its binding to a target DNA sequence containing the nadA promoter influenced formation and/or stability of FarR::nadA complexes. The complexity of the multi-layered regulation of nadA uncovered during this investigation suggests that N. meningitidis modulates NadA adhesin protein levels for the purpose of interacting with host cells yet avoiding antibody directed against surface exposed epitopes.
Introduction
Neisseria meningitidis is a Gram-negative obligate human pathogen that colonizes the nasopharynx in 10–35% of adults [1]. For reasons not currently understood, commensal meningococcal (MC) colonization develops into an invasive disease causing septicemia and meningitis in 0.5 per 100,000 persons in the United States and up to 1,000 per 100,000 persons in sub-Saharan African epidemics [2]. The speed of disease progression results in up to 10–15% mortality even with antibiotic therapy [3], while often leaving survivors with permanent neurological complications [4]. Vaccines against the capsular polysaccharide of the most common disease-associated serotypes (A, C, W135, and Y) are available, leaving the hypervirulent and immune-evasive serotype B as a current focus for vaccine research [5].
Adhesion to the mucosal surface of the nasopharynx is the first step in successful colonization, mediated by a variety of factors, with type IV pili [6], [7], [8] and Opa and Opc proteins [9], [10] produced in the greatest abundance. Recently, a non-fimbrial “Oca” family (Oligomeric coiled-coil adhesin) neisserial adhesin termed NadA was identified in 50% of hypervirulent MC capsular serogroup B lineages [11], but not in other capsular serogroup strains. Comprised of a leader peptide, globular “head” domain, α-helix intermediate region, and a C-terminal membrane anchor, NadA forms highly stable multimeric coiled-coil structures along the helical stalk, positioning the globular “head” for host cell interaction [12]. Importantly for consideration as a vaccine candidate, recombinant NadA lacking the C-terminal anchor elicits a bactericidal antibody response with epitopes accessible in encapsulated MC. Although nadA allele sequences differ between strains, varied antigen expression, not diversity, influences immune sera titer levels and protection [11]. Accordingly, the identification of factors influencing NadA levels at the gene expression level is critical for optimizing the efficacy of a NadA-targeted vaccine. Furthermore, understanding nadA expression may offer clues into the signals involved in converting a passive co-inhabitant of the human mucosal lining into an invasive and fatal septic infection.
MC uses a multi-tiered approach to control nadA expression. Maximum levels of the NadA protein are observed in stationary-phase in a growth-dependent manner [11], with expression of nadA varying widely among MC strains [13], [14]. Upstream from the promoter are multiple tetranucleotide (TAAA) repeats whose number corresponds with varied nadA expression [13], [15]. These repeats are phase variable, likely caused by slipped-strand mispairings during replication [16]. Several regulatory proteins bind to the nadA promoter (Figure 1), including integration host factor (IHF) and ferric uptake regulatory protein (Fur), though nadA expression is unchanged in a Fur null mutant [14]. Recently, a MarR-family transcriptional regulator, termed FarR and NadR in separate publications [14], [17], was identified as a repressor of nadA, further expanding the list of nadA regulatory factors. This DNA-binding protein was first identified in the gonococcus (GC) and was shown to repress expression of the farAB-encoded efflux pump that is responsible for high levels of fatty acid resistance [18]. In contrast, MC FarR does not affect fatty acid resistance through FarAB, perhaps due to naturally high fatty acid resistance expressed by this pathogen [19]. Interestingly, however, MC FarR does bind to its farAB promoter region with relatively high affinity and represses farAB expression as shown by RT-PCR [20]. Because FarR regulates expression of farAB in both MC and GC, while nadA is present only in a subset of MC populations, we will continue to use the nomenclature of FarR for the repressor of nadA based on its more universal activity on farAB in both GC and MC.
Figure 1. Schematic of nadA, farR, and mtrR promoter regions used for lacZ fusions and DNA probes.

Representations of the nadA (A), farR (B), and mtrR (C) promoter regions showing regulatory protein binding sites, intergenic sequences of interest, and primer annealing locations with oligonucleotide sizes. White arrows represent each open reading frame with the translation start codon noted by “ATG” and a vertical line to indicate if a primer overlaps the start codon. Grey arrows represent the respective primers including oligonucleotide sizes with the following nomenclature: DNA probe for EMSAs - “orf”_prom_F & R; promoter region with start codon fused with lacZ - “orf”_F_Bam and “orf”_R_lacZ. IHF and Fur are binding sites for integration host factor and ferric uptake regulatory protein, respectively.
The small molecule 4-hydroxyphenylacetic acid (4HPA) was identified as an inducer or de-repressor of nadA by relieving the DNA-binding activity of FarR [14]. Being a colonizer of the oropharynx, MC is washed in saliva, in which 4HPA is a common metabolite [21], possibly leading to increased expression of nadA and subsequent invasive disease. Curiously, FarR-controlled targets in GC are directly and indirectly regulated by the TetR family regulator MtrR. Repression of farR by MtrR indirectly up-regulates farAB [18], while the gene encoding glutamine synthetase (glnA) is directly regulated by both FarR and MtrR [22]. Therefore, we questioned whether MtrR similarly affects nadA expression in MC, adding to the growing list of regulatory factors targeting nadA. Here we confirm that FarR is the primary repressor of nadA, yet MtrR, when expressed at elevated levels, directly represses nadA as well. Furthermore, DNA-binding and DNase I protection assays suggest that MtrR influences FarR binding at the nadA promoter similar to the phenomenon seen in glnA expression in GC [22], suggesting a higher complexity to Neisserial regulatory schemes that is conserved across species.
Results and Discussion
Control of nadA, farR, and mtrR expression in MC strain M7
Co-regulation and competitive regulation between FarR and MtrR has been shown previously for multiple targets in GC [18], [22] but not MC. Given previous observations that in MC FarR can regulate nadA, we asked if MC MtrR can control farR expression. We also tested if MC MtrR could control nadA directly. To investigate possible influences of MtrR on nadA and/or farR expression in MC, the expression profiles for these genes were determined using a promoterless lacZ-fusion expression system (Figure 1) employed previously to monitor gene expression in GC [23]. Using translational lacZ fusions to each gene's promoter (ranging from 427 to 524 bp; Figure 1), mtrR expression was compared to farR and nadA across multiple growth phases of broth–grown cultures or from overnight, agar-grown cultures (Figure 1) of MC strain M7, which is a capsule-deficient mutant of strain NMB that is used for biosafety purposes [24]. This was done because earlier work [25] reported that farR was maximally expressed between late-log and stationary phase, while nadA expression peaks at stationary phase [11]. Importantly, the lacZ fusions did not significantly impact growth rates in broth for the M7-derived strains (Figure 2A), suggesting that any expression profile differences are not growth rate-dependent. Aliquots from different growth phases (Figure 2A; boxed A, B, and C) were assessed for β-galactosidase activity and compared against the activity of overnight cultures grown on GCB agar plates (Figure 2B). The results showed that agar-grown MC had higher levels of expression for all three genes compared to broth-grown strains, and this was especially true for nadA. With respect to agar-grown cultures, we noted that nadA expression was considerably greater than farR or mtrR with the latter being the most poorly expressed gene (Figure 2B). Based on these results, all subsequent gene expression studies were performed on cultures grown overnight on GCB agar plates.
Figure 2. Growth phase-dependent expression of nadA, farR, and mtrR in N. meningitidis M7.

(A) Growth curve of strain M7 expressing lacZ fused to nadA (solid line; circle timepoints), farR (dotted line; triangle timepoints), and mtrR (dashed line; square timepoints) promoter regions measured by OD600 optical density. Boxed A, B, and C; timepoints for sample harvest. (B) Specific activity of β-galatosidase activity of lacZ fusions as indicated. Samples harvested from liquid culture at various growth phases (A, B, C) were compared with O/N growth on agar plates. Inset; magnified view of mtrR-lacZ expression. NS, not significant; **, P<0.01.
Having established that the lacZ fusion technology could be employed to monitor gene expression in strain M7, we next asked if MtrR can regulate farR as it does in GC and if, in turn, it can modulate nadA expression. We first asked if loss of MtrR impacts MC farR expression levels and for this purpose constructed an M7 farR-lacZ fusion strain (427 bp promoter region; Figure 1) in an mtrR null mutant that carries the non-polar aphA-3 cassette within the mtrR coding sequence. With this fusion strain, we noted a small but significant (p<0.01; Figure 3) increase in farR-lacZ expression, which was reversed by complementation when the M7 mtrR allele was expressed ectopically under its own promoter or an IPTG-inducible promoter. Having observed a significant, albeit modest, influence of MtrR on farR-lacZ expression, we next asked if nadA expression, known to be negatively controlled by FarR [17], would be impacted due to loss of MtrR using a nadA-lacZ fusion (514 bp promoter region; Figure 1). We first confirmed that loss of FarR significantly increased nadA-lacZ expression (strain JC5AZ; Figure 4A), and this was reversed by complementation with the wild-type farR gene from strain M7 (strain JC6AZ). Interestingly, loss of MtrR alone (strain JC2AZ) resulted in a consistent, albeit small, increase in nadA-lacZ expression, though not to significant levels, with restoration of nadA to wild-type levels when mtrR was expressed ectopically under its own promoter (strain JC3AZ; Figure 4B). Furthermore, complementation with the wild-type mtrR sequence under the control of an IPTG-inducible promoter significantly (p<0.01) reduced nadA-lacZ expression by 26% (strain JC4AZ; Figure 4B). Based on these results, we concluded that FarR-mediated repression of nadA-lacZ was greater than that mediated by MtrR, but that MtrR could impact nadA expression by a FarR-independent mechanism. In order to further test this, we compared nadA-lacZ expression in a ΔfarR/ΔmtrR double mutant (strain JC7AZ) against the single ΔfarR mutant (strain JC5AZ). Here, no significant impact on nadA-lacZ expression was observed, further supporting a dominant repression of nadA by FarR. However, IPTG-induction of mtrR in the absence of FarR (strain JC9AZ) did result in a significant reduction in nadA (p<0.01) compared to both the ΔfarR/ΔmtrR double mutant (strain JC7AZ) and ΔfarR mutant (strain JC5AZ; Figure 4C). Taken together, the data suggested that MtrR, when overexpressed, has an influence on nadA expression that is independent from FarR-modulated expression in MC strain M7. What effects MtrR may have on nadA expression in other MC strains that differ in either mtrR or nadA expression is not yet clear and the subject of future study.
Figure 3. MtrR-regulation of farR.
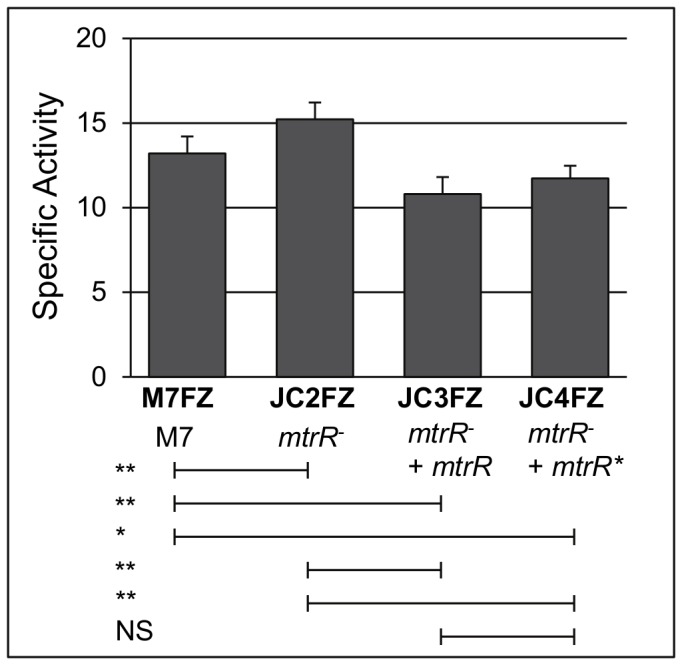
Specific activity of β-galactosidase activity of farR-lacZ in the strains M7 wild-type, ΔmtrR, and ΔmtrR complemented with the native and inducible-promoter alleles (superscript asterisk), respectively. NS, not significant; *, P<0.05; **, P<0.01.
Figure 4. Expression of nadA by FarR and MtrR.

(A–C) Specific activity of β-galactosidase activity of nadA-lacZ in various M7 backgrounds as indicated. Strains over-expressing MtrR marked with a superscript asterisk. NS, not significant; *, P<0.05; **, P<0.01. (D) Western immunoblot analysis of NadA, FarR, and MtrR levels. Protein samples grown overnight on GC agar plates, collected, and analyzed by electropheoresis through 6% (NadA) or 12% (FarR and MtrR) SDS-PAGE gels followed by immunoblot with the respective antisera. Molecular weight standards are listed to the left. Coomassie-stained gel provided for protein level comparison. Arrows represent minor immunoreactive bands used to determine NadA steady-state differences across strains.
Western blot analysis was performed to confirm the results observed with translational nadA promoter-lacZ fusions (Figure 4D). As expected, FarR was absent in the farR null mutant strains (JC5AZ, JC7AZ, and JC9AZ), but was present in the complemented mutants that expressed farR ectopically (strains JC6AZ and JC8AZ); although FarR levels were reduced in the complemented strains, repression of nadA was still evident (Figure 4A, C, D; arrow). NadA migrates in its multimeric form even under denaturing/reducing conditions [11], complicating discrimination of differences in NadA protein levels when analyzing the dominant band alone; therefore, minor NadA-dependent bands, observed elsewhere [20], were used for analysis of steady state levels of NadA (Figure 4D; arrows). Despite repeated attempts, MtrR could not be detected by immunoblot in any strain except when over-expressed (strains JC4AZ and JC9AZ), suggesting that MtrR is maintained at low levels in N. meningitidis. This low level of MtrR may explain why enhanced expression of farR in the mtrR null mutant was modest (Figure 4) and why its ability to repress nadA could only be observed when mtrR was over-expressed ectopically (strain JC4AZ; Figure 4B). In agreement with the data obtained with the nadA promoter translational fusions, steady-state levels of NadA exceeded wild-type in the absence of FarR (Figure 4D; arrows point to minor bands used for comparison). Unfortunately, differences in NadA levels were not discernible when comparing +mtrR to ΔmtrR strains in the +farR (strains M7AZ, JC2AZ, JC3AZ, and JC4AZ) or ΔfarR (strains JC5AZ, JC7AZ and JC9AZ) backgrounds (Figure 4D). Assessing differences in steady-state levels of NadA in other clinical MC strains following the loss of mtrR requires further study.
FarR and MtrR binding to a target DNA sequence upstream of nadA
The data obtained using translational fusion strains bearing an over-expressed wild-type mtrR allele in an mtrR null mutant background indicated that elevated levels of MtrR can exert negative regulatory influences on nadA expression in MC strain M7. Based on this hypothesis, we asked if MtrR can bind target DNA sequences upstream of nadA by employing electrophoretic mobility shift assays (EMSA) using FarR or MtrR fused to maltose-binding protein (MBP) at their C-terminus;maps describing the various DNA probes used are shown in Figure 1. As described previously [14], FarR was found to bind the target nadA sequence (457 bp) in a specific manner with at least three DNA-protein complexes observed (Figure 5A, arrows). Furthermore, the results from binding specificity assays suggested that FarR has greater affinity for the nadA-promoter than to its own (443 bp) or farAB promoter-bearing sequences (497 bp and shown in Figure 5B); importantly, a probe lacking a FarR-binding site (rnpB; 609 bp) was unable to compete with the labeled nadA probe for complexing with FarR.
Figure 5. DNA binding properties of FarR-MBP.

(A) Successive increases of FarR-MBP incubated with 10 ng nadA promoter region to assess binding by gel-shift analysis. Arrows; various complexes of DNA and FarR-MBP. (B) Competition assays. 32P-labeled nadA promoter (384 bp) was incubated with 0.5 µg FarR-MBP and competed with unlabeled nadA, farR (333 bp), farAB (435 bp), and rnpB (354 bp) at 25, 50, and 100 times molar excess of labeled probe (lanes 3 through 14). The competing probe used is listed below each panel. Arrow; 32P-labeled probe competed away from FarR-MBP by unlabeled probe. Lane 1, labeled probe alone; lane 2, labeled probe and 0.5 µg FarR-MBP without competitor.
Having verified that the nadA probe could be recognized in a specific manner with an MC DNA-binding protein (FarR), we next asked if nadA could bind MtrR. First, we confirmed by EMSA the DNA-binding capacity of the MtrR-MBP fusion protein by evaluating its ability to bind a known target DNA sequence, namely the promoter-bearing region upstream of MC mtrCDE (811 bp). In GC, MtrR is a repressor of the mtrCDE-encoded antimicrobial efflux pump by virtue of its binding between the −10 and −35 promoter elements [26], [27]. We found that as little as 1.0 µg of MC MtrR-MBP incubated with MC mtrCDE promoter-bearing probe resulted in virtually a complete shift (Figure 6A; arrow) of the target sequence. Importantly, a similar shifting of the nadA probe by MtrR was observed (Figure 6B; arrow). Specificity of MtrR-binding to the nadA probe was confirmed by use of competitive EMSA. Although the heterologous unlabeled rnpB promoter-bearing probe did to some extent compete for binding, especially at a high concentration (100×), the mtrCDE and nadA unlabeled probes proved to be more effective competitors at a lower concentration (25×) with the mtrCDE probe appearing to be superior (Figure 6C).
Figure 6. DNA binding properties of MtrR-MBP.

Successive increases of MtrR-MBP incubated with 10 ng mtrCDE (A) or nadA (B) promoter regions to assess binding by gel-shift analysis. Arrow; primary complex of DNA and MtrR-MBP. (C) Competition assays. 32P-labeled nadA promoter (384 bp) was incubated with 1.0 µg MtrR-MBP and competed with unlabeled nadA, mtrCDE (552 bp), and rnpB (354 bp) at 25, 50, and 100 times molar excess of labeled probe (lanes 3 through 14). The competing probe used is listed below each panel. Arrow; 32P-labeled probe competed away from MtrR-MBP by unlabeled probe. Lane 1, labeled probe alone; lane 2, labeled probe and 0.5 µg MtrR-MBP without competitor.
The ability of both FarR and MtrR to bind the nadA promoter-bearing region in a specific manner was reminiscent of their ability to bind the DNA sequence upstream of glnA of GC (22). As the binding of either protein to the glnA target can impact binding of the other we asked if a similar situation might exist for the MC-derived nadA target. To test this possibility, we pre-incubated the nadA probe with a fixed concentration of one protein and then introduced increasing amounts of the second protein. In the absence of a competing protein, both MtrR and FarR exhibited a distinct shift of the probe (Figure 7; arrowhead and arrows, respectively). As FarR-MBP concentrations increased following pre-incubation of nadA target DNA with MtrR-MBP, the MtrR shift remained relatively unchanged, suggesting that FarR-MBP does not compete with MtrR-MBP. In contrast, increasing amounts of MtrR-MBP changed the electrophoretic mobility of FarR-MBP:DNA complexes, suggesting that MtrR-MBP can significantly influence the formation or stability of FarR::nadA complexes.
Figure 7. MtrR influences FarR::DNA complexes.

Shown is an EMSA evaluating the binding of MtrR–MBP and FarR-MBP to 32P-labeled nadA promoter based on order of protein introduction. Lane assignments from left to right: probe alone; probe plus 1.0 mg MtrR-MBP and increasing amounts of FarR-MBP (0, 0.13, 0.25, 0.50, 1.0, 2.5, and 5.0 mg); probe alone; probe plus 0.5 mg MtrR-MBP and increasing amounts of FarR-MBP (0, 0.13, 0.25, 0.50, 1.0, 2.5, and 5.0 mg). The arrowhead shows the position of MtrR:DNA complex lacking FarR while the arrows show positions of FarR:DNA complexes lacking MtrR.
In order to learn the mechanism by which MtrR could influence the formation of FarR::nadA complexes, we used DNase I protection assays to determine if their respective binding sites might be in close proximity. We confirmed FarR-MBP-binding to the three sites (data not shown) described by Metruccio et al. [14], which include the −10 promoter and TAAA phase-variable regions (Figure 8B). In repeated DNase I protection assays, clear evidence for a sequence(s) capable of recognizing MtrR could not be obtained. However, in these experiments a DNase I hypersensitive site was identified positioned at the end of the phase-variable TAAA repeats (Figure 8B; asterisk at nucleotide position 209). The presence of this suggests an interaction of MtrR with a sequence upstream of nadA that could influence binding of FarR.
Figure 8. Identification of a DNase I hypersensitive site at the nadA promoter in the presence of MtrR.

(A) Increasing amounts of MtrR-MBP (0, 1, 5, 10 mg) were incubated with the nadA promoter prior to DNase I incubation. Site of DNase I hypersensitivity is denoted with an asterisk. The nucleotide sequence (G, A, T, C) is listed adjacent to the lanes. (B) Nucleotide sequence of the nadA promoter. Colored boxes, FarR-MBP binding sites 1, 2 & 3; Asterisk, DNase I hypersensitive site. The transcription start site (+1) and translational start site (ATG, bold and underlined) are indicated.
While N. meningitidis colonizes up to 35% of humans [1], fewer than 1% of the population develops an invasive infection [2], suggesting that the bacterium focuses on a more commensal lifecycle. The ability to effectively transition between passive residence and active infection relies on tight transcriptional regulation involving an array of external and internal control systems. In GC, the transcriptional regulators FarR and MtrR have been well-characterized for their role in antimicrobial resistance, allowing for host persistence [28]. FarR represses expression of the fatty-acid efflux pump FarAB [29]; MtrR represses expression of the mtrCDE, which encodes an antimicrobial efflux pump [26], [30]. Recently, MC FarR was shown to repress expression of nadA [17], whose gene product is a highly immunogenic adhesin and invasin associated with hypervirulent strains of serotype B MC [11], [12]. Interestingly, GC MtrR has also been shown to repress GC FarR, thereby influencing transcription of farAB [18]. As this regulation of a regulator is not unique to GC [22], we explored whether MtrR likewise modulates farR in MC, thus affecting nadA expression.
Our results suggest that MC employs a dual-repressor approach to control nadA expression. Using lacZ translational fusions, EMSA, and DNase I protection assays, we confirmed earlier work [14], [17], [25] that FarR is a negative regulator of nadA due to its ability to bind target DNA upstream of the coding region (Figure 7B) and affect subsequent expression (Figure 4A). Complicating this regulatory scheme, our results indicate that MtrR can exert an influence on nadA directly by interacting with the upstream DNA sequence and indirectly through its ability to reduce farR expression (Figure 3). With respect to the first mode of MtrR regulation over nadA, our DNA-binding studies indicate that MtrR can bind upstream of this gene in a specific manner (Figure 6C) and can impact the formation and/or stability of FarR::DNA complexes (Figure 7) when its level exceeded that of FarR. The stronger influence of FarR on nadA expression is likely due to its ability to recognize three target sites (Figure 7B) while under the conditions employed in the DNase I protection assay. A possible site for MtrR binding could only be surmised by the presence of a DNase I hypersensitive site (Figure 7A). Interestingly, this site is positioned within a tract of tetranucleotide repeats and a FarR-binding site (Figure 7B).
Under what conditions might MtrR-mediated regulation of nadA have biologic relevance given the strong regulatory action of FarR? We propose several potential mechanisms: The development of mutations impacting FarR regulation of nadA would require alternative mechanisms of transcriptional regulation that could in part be fulfilled by MtrR. Thus, mutations in FarR that reduce its DNA-binding activity or mutations in FarR-binding sites could enhance nadA expression unless other regulatory processes are available. Alternatively, mutations that enhance MtrR levels or interactions with nadA-binding sites might repress nadA expression above that seen by FarR alone. Precedent for clinical isolates of Neisseria bearing regulatory mutations impacting gene expression exists in that gonococcal strains isolated from patients frequently contain mutations in mtrR and these can cause dysregulation of the mtrCDE-encoded efflux pump operon. Furthermore, cis-acting regulatory mutations can influence transcription of mtrR and/or mtrCDE or directly enhance expression of the mtrCDE-encoded efflux pump [28], [31], [32], [33]. In M7, mtrR expression is typically low, yet overexpression of mtrR results in almost 30% repression of nadA (Figure 4B and D). Accordingly, it will be important to evaluate MC clinical isolates to determine if they may develop mutations impacting nadA expression directly or indirectly; the latter being due to mutations in farR or mtrR. With the expression of nadA varying significantly between MC strains [13], [14], mutations affecting farR and/or mtrR expression may have more profound effects on nadA expression than those observed here. Deletion of the Correia element or IHF binding site upstream from mtrR affects expression of mtrCDE, which is an MtrR target [32]; Enriquez et al. observed several MC isolates with Correia element deletions upstream from mtrR, including one serotype B, suggesting that these mutations are not an exception [34]. We propose that this multi-layered regulation of nadA, which now includes direct regulation by MtrR, reflects an effort by MC to balance levels of the NadA adhesion important for interacting with host cells yet avoiding potentially protective antibody responses.
Materials and Methods
Bacterial strains and growth conditions
All N. meningitidis strains listed in Table 1 are derivations of strain M7 constructed for this study; M7 is a stable capsule-negative variant of strain NMB and was used for biosafety purposes. MC were cultured on GCB agar (Difco Laboratories, Detroit, MI) with defined supplements I and II [35] at 37°C under 3.8% (vol/vol) CO2. For growth-phase analysis, MC were grown in a shaking incubator at 37°C in GCB broth with sodium bicarbonate and defined supplements I and II as previously described [35].
Table 1. Bacterial strains and plasmids used in this study.
| Strain | Relevant genotype or remarks | Source |
| Neisseria meningitidis | ||
| M7 | Unencapsulated NMB derivative | [24] |
| JC2 | M7 mtrR::kana | This study |
| JC3 | As JC2 but mtrR+ delivered using vector pGCC3 | This study |
| JC4 | As JC3 but mtrR+ delivered using vector pGCC4 | This study |
| JC5 | M7 farR::spcb | This study |
| JC6 | As JC5 but farR+ delivered using vector pGCC3 | This study |
| JC7 | As JC5 but mtrR::kan | This study |
| JC8 | As JC7 but farR+ delivered using vector pGCC3 | This study |
| JC9 | As JC7 but mtrR+ delivered using vector pGCC4 | This study |
| M7AZ | As M7 but with a nadA promoter translationally-fused to lacZ and delivered by pLES94 termed nadA-lacZ | This study |
| JC2AZ | As JC2 but with a nadA-lacZ | This study |
| JC3AZ | As JC3 but with a nadA-lacZ | This study |
| JC4AZ | As JC4 but with a nadA-lacZ | This study |
| JC5AZ | As JC5 but with a nadA-lacZ | This study |
| JC6AZ | As JC6 but with a nadA-lacZ | This study |
| JC7AZ | As JC7 but with a nadA-lacZ | This study |
| JC8AZ | As JC8 but with a nadA-lacZ | This study |
| JC9AZ | As JC9 but with a nadA-lacZ | This study |
| M7RZ | As M7 but with an mtrR promoter translationally-fused to lacZ and delivered by pLES94 termed mtrR-lacZ | This study |
| M7FZ | As M7 but with an farR promoter translationally-fused to lacZ and delivered by pLES94 termed farR-lacZ | This study |
| JC2FZ | As JC2 but with a farR-lacZ | This study |
| JC3FZ | As JC3 but with a farR-lacZ | This study |
| JC4FZ | As JC4 but with a farR-lacZ | This study |
| Escherichia coli | ||
| DH5α | [F− Φ80dlacZΔM15 Δ(lacZYA-argF)U169 endA1 recA1 hsdR17(rΚ − mΚ +) deoR thi-1 supE44 λ− gyrA96 relA1] | [41] |
| Top10 | [F− mcrA Δ(mrr-hsdRMS-mcrBC)Φ80 lacZΔM15 ΔlacX74 recA1 deoR araD139 Δ(ara-leu)7697 galU galK rpsL (Strr endAI nupG)] | Invitrogen |
| Plasmids | ||
| pCR®2.1-TOPO® | pUC-derived cloning vector; AmpR | Invitrogen |
| pUC18K | Source of aphA-3 cassette; KanR | [36] |
| pHP45Ω | Source of spc cassette; SpcR | [42] |
| pLES94 | pUC18K-derivative allowing a translational fusion to a promoterless lacZ fusion and insertion between neisserial proA and proB genes. | [23] |
| pGCC3 | NCIS vector for insertion of genes directed under their native promoter between neisserial lctp and aspC genes. | [37] |
| pGCC4 | As pGCC3 except inserted genes are directed under an IPTG-inducible promoter | [37] |
| pMal-c2 | IPTG-inducible expression vector for fusion of proteins to maltose binding protein (MBP) and cytoplasmic expression | NE Biolabs |
Strain construction and verification
For construction of strain JC2, overlapping PCR products were generated to replace mtrR with aphA-3 [36] by allelic exchange at the native N. meningitidis locus, conferring kanamycin resistance. Specifically, primers mtrC_R_out_5′ and mtrR_R_Kan_5′ovhg generated product A; kan_F_mtrR_5′ovhg and kan_R_mtrR_3′ovhg generated product B; and mtrR_F_Kan_3′ovhg and NMB1718_FWD generated product C (Table 2). Products A, B, and C were used as template with flanking primers mtrC_R_out_5′ and NMB1718_FWD (Table 2) to generate the final PCR product used for transformation of wild-type M7 with selection on kanamycin at 50 µg ml−1. The substitution of mtrR with aphA-3 was verified by PCR (data not shown) and Western blot (Figure 4). For construction of JC5, PCR products generated by FarR_prom_F and FarR _Sma_R (Table 2) were subcloned into vector pCR®2.1 (Invitrogen) generating pJC5α with selection on ampicillin at 100 µg ml−1. PCR products from FarR_ Sma_F & FarR_pmalC_Xba_R (Table 2) were purified, digested with SmaI, and ligated into pJC5α at the SmaI site, generating pJC5β, and sequenced for accuracy. Finally, pHP45Ω was digested with SmaI to liberate the spc cassette then ligated into the SmaI site of pJC5β generating pJC5 used to transform M7 with selection on spectinomycin at 60 µg ml−1. The interruption of farR with spc was verified by PCR (data not shown) and Western blot (Figure 4). Complementation of JC2 and JC5 was accomplished by delivering the parent gene ectopically using vector pGCC3 or pGCC4 [37]. Briefly, primers mtrR_F_Pac_GC3 and mtrR_R_Pme or mtrR_F_Pac_GC4 and mtrR_R_Pme (Table 2) were used to amplify the mtrR allele then digested with appropriate restriction enzymes and ligated into pGCC3 and pGCC4, respectively. Similarly, farR_F_Pac_GC3 and farR_R_Pme (Table 2) were used to amplify the farR allele for subsequent digestion and ligation into pGCC3. All constructs were verified by sequencing prior to transformation. Transformants were selected on erythromycin at 1 µg ml−1.
Table 2. Oligonucleotides used.
| Primer name | Sequence 5′ → 3′ |
| kan_F_mtrR_5′ovhg | AAA CGC CAT TAT GGC TAA AAT GAG AAT ATC ACC |
| kan_R_mtrR_3′ovhg | CAA GGC TTG ACT AAA ACA ATT CAT CCA GTA AAA TA |
| mtrR_R_Kan_5′ovhg | CAT TTT AGC CAT AAT GGC GTT TTC GTT TCG G |
| mtrR_F_Kan_3′ovhg | ATT GTT TTA GTC AAG CCT TGG TAG CAA TGC |
| mtrC_R_out_5′ | GAA CAG GCG TTT TTG GAT GAT GC |
| NMB1718_FWD | GCC CAC ATC GTT ATT CTC ATA AAG GC |
| mtrR_R_Pme | GGG TTT AAA CTT ATT TCC GGC GCA GGT CGG |
| mtrR_F_Pac_GC3 | GCC ATT AAT TAA CCT ATC TGT CTG GTT TGA TGT AAA GGG |
| mtrR_F_Pac_GC4 | GGT TAA TTA ACC GCC CTC ATC AAA CCG ACC |
| farR_ Sma_F | CTG ATA CAG GCC CGG GAA GCC CTG ATG |
| farR _Sma_R | CAT CAG GGC TTC CCG GGC CTG TAT CAG |
| farR_F_Pac_GC3 | GGT TAA TTA AGA TGC GGC GGC TTTT GTT TTT TCT GG |
| farR_R_Pme | GGG TTT AAA CTT ACG AGT TCA ACG CAT CC |
| nadA_F_Bam | ATA TGG ATC CGT CGA CGT CCT CGA TTA CG |
| nadA_lacZ_R | ATA TGG ATC CTG TTT CAT GCT CAT TAC C |
| mtrR_F_Bam | CGG GAT CCC GAG CCA TTA TTT ATC CTA TCT GTC |
| mtrR_lacZ_R | GGT TGG ATC CAT AAT GGC GTT TTC GTT TCG GG |
| farR_F_Bam | ATA TGG ATC CGG CGG CTT TTG TTT TTT CTG G |
| farR_lacZ_R | CGC AGG ATC CGA TTG GGT AGG CAT TGT TCA AG |
| farR_pmalC_F | ATG CCT ACC CAA TCA AAA CAT GCG |
| farR_pmalC_Xba_R | TTA CTC TAG ATT ACG AGT TCA ACG CAT CCT CG |
| mtrR_pmalC_F | ATG AGA AAA ACC AAA ACC GAA GCC |
| mtrR_pmalC_Xba_R | CAA GTC TAG ATT ATT TCC GGC GCA GGT CG |
| rnpB1F | CGG GAC GGG CAG ACA GTC GC |
| rnpB1R | GGA CAG GCG GTA AGC CGG GTT C |
| farAB_prom_F | ATG TGG GAG GTT TTC GAA CCA CG |
| farAB_prom_R | CGT GTG CGT ATC CAT AAG ATT GGG |
| farR_prom_F | CCG CTA TGT AGA GAA TCA AGC GG |
| farR_prom_R | TTG GGT AGG CAT TGT TTA AGT CTC C |
| nadA_prom_F | GTC GAC GTC CTC GAT TAC GAA GG |
| nadA_prom_R | ATG CAT GCT CAT TAC CTT TGT GAG TGG |
| KH9_3 | AGA CGA CAG TGC CAA TGC AAC G |
| mtrR_R_out_5′ | TTG CGG TAA AAG GTT TCC AAG GC |
Construction of lacZ reporter fusions, β-galactosidase assay and immunoblot analysis
All lacZ fusions used in this study were prepared in pLES94 and performed as described previously [18], [23] using appropriate primers. For nadA-lacZ, primers nadA_F_Bam and nadA_lacZ_R were used; for farR-lacZ, primers farR_F_Bam and farR_lacZ_R were used (Table 2); and for mtrR-lacZ, primers mtrR_F_Bam and mtrR_lacZ_R were used (Table 2) Constructs encoding a lacZ fusion were grown overnight on GCB agar with supplements, 1 mM IPTG, and 5 mM 4-hydroxyphenylacetic acid, when appropriate. Cells were harvested directly from plates or used to inoculate GCB broth with appropriate supplements and 1 mM IPTG then grown through stationary phase for growth-phase analysis of protein expression. Cells collected from overnight plates were resuspended in phosphate-buffered saline pH 7.2 (PBS), centrifuged for 2 min at 13,000 rpm, and stored overnight at −20°C after the supernatant was removed. From liquid cultures, 5 mL aliquots were removed at the indicated growth phases (Figure 1) and centrifuged for 15 min at 5,000 rpm. After removing the supernatant, cell pellets were resuspended in PBS, centrifuged for 2 min at 13,000 rpm, and stored overnight at −20°C. To determine β-galactosidase specific activities, cell pellets were resuspended in 50/50 PBS and Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgCl2, 50 mM β-Mercaptoethanol; pH 7.0). After addition of 20 µL 0.1% SDS and 40 µL chloroform, samples were vortexed and incubated at room temperature for 5 min. Protein concentrations were quantified by Nanodrop1000 (Nanodrop Technologies,Wilmington, DE). To 200 µL of Z-buffer, 30 µL of each cell lysate and 70 µL of ONPG (2-Nitrophenyl-β-D-galactopyranoside; Sigma, St. Louis, MO) at a concentration of 4 mg ml−1 in Z-buffer was added. Following a color change, the reaction was stopped with 500 µL of 1 M Na2CO3 solution. The reactions were centrifuged at room temperature for 5 min at 13,000 rpm to remove cell debris and 200 µL of supernatant was transferred to a 96-well microtiter plate and analyzed at 420 nM by a PerkinElmer Victor X3 microplate reader. For data analysis, specific activity was calculated using the formula: {[(OD420 * v)/(4500 nL nmoles−1 cm−1×1 cm)]/t/mg protein} with v being the volume used and t being the reaction time. All reactions were performed in triplicate and repeated at least 3 times. Statistical analysis was performed using multivariate ANOVA followed by Tukey HSD post-hoc pairwise comparison using SAS 9.2 software (The SAS Institute, Cary, NC).
Verification of protein absence, overexpression, and comparison between strains were assessed by immunoblot. Total protein was quantified by Nanodrop1000 (Nanodrop Technologies) prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) separation [38] and Western immunoblotting [39]. Rabbit polyclonal antibodies were used at the following dilutions: anti-NadA, 1∶2,000; anti-FarR, 1∶5,000; and anti-MtrR, 1∶1,000. Anti-rabbit alkaline phosphatase-conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA) was used at 1∶7,500.
Purification of MtrR and FarR
Fusion and purification of N. meningitidis M7 MtrR and FarR to maltose-binding protein (MBP) were performed per manufacturer's guidelines (New England Biolabs, Beverly, MA) and as described previously [27] with some exceptions. Primers mtrR_pmalC_F & mtrR_pmalC_Xba_R and primers farR_pmalC_F & farR_pmalC_Xba_R were used to PCR amplify the mtrR and farR alleles, respectively, before XbaI digestion and blunt-end ligation into pMal-c2 (New England Biolabs). Both constructs were sequenced for accuracy. Factor Xa digestion was not performed due to protein stability issues; therefore, all DNA-binding investigations utilizing these proteins maintained an intact MBP fusion, which has been successful in prior investigations [27]. Analysis of eluted fractions by SDS-PAGE revealed a 65-kDa band consistent with the expected size an MtrR-MBP fusion (data not shown). For consistency, N. meningitidis M7 FarR was likewise fused to MBP and purified, in which the purified protein band migrated to about 60 kDa (data not shown).
DNA-binding studies
Electrophoretic mobility shift assays (EMSA) and competitive EMSAs using FarR-MBP and MtrR-MBP were performed essentially as previously described [22], [27], [40] with some modifications. Briefly, unlabeled and 32P-labeled probes were generated with the following primers: nadA, nadA_prom_F and nadA_prom_R; mtrCDE, KH9_3 and mtrR_R_out_5′; farR, farR_prom_F and farR_prom_R; farAB, farAB_prom_F and farAB_prom_R; and rnpB, rnpB1F and rnpB1R (Table 2). Appropriate PCR products were end labeled with [γ-32P] dATP using T4 polynucelotide kinase (New England Biolabs, Beverly, MA). The labeled products (10 ng) were each incubated with purified FarR-MBP or MtrR-MBP in a 30 µL reaction volume [10 mM Tris-HCl (pH 7.5), 0.5 mM dithiothreitol, 0.5 mM EDTA, 1 mM MgCl2, 50 mM NaCl, 0.05 µg/mL poly(dI-dC)] at room temperature for 30 min. Loading buffer (Epicentre, Madison, WI) was added to each sample then separated by 6% polyacrylamide gel at 4°C, followed by autoradiography. Competitive EMSAs were performed similarly, but unlabeled probes generated from the same primer sets as labeled probes were included.
DNase I protection assays were performed as previously described [22], [27], [40] with slight modifications. Target DNA probes were generated with PCR pimers nadA_prom_F and nadA_prom_R (Table 2). The 5′ end was labeled with T4 polynucleotide kinase as described above for EMSA probes. Purified MtrR-MBP was incubated with target DNA for 15 min at 37°C. Dnase I in loading buffer [10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 4 mM CaCl2, 1 mM dithiothreitol, 40% glycerol] was added to each reaction for 1 min at 37°C. The reactions were stopped with DNase I stop buffer (95% ethanol and 7.5 mM ammonium acetate), plunged in an ethanol dry-ice bath for 15 min, and precipitated overnight at −80°C. Pellets were washed in 70% (vol/vol) ethanol, dried, and resuspended in loading buffer (Epicentre). Resuspended reaction mixtures were loaded on 6% denaturing polyacrylamide gel and resolved by autoradiography.
Acknowledgments
We thank Virginia Stringer for excellent technical assistance, Lane Pucko for help with manuscript preparation, and Oliver Kurzai for the gift of antiserum to FarR and NadA.
Funding Statement
This work was supported by NIH grant AI021150-28 and a VA Merit award to W.M.S. W.M.S. is the recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cartwright KA, Stuart JM, Jones DM, Noah ND (1987) The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica . Epidemiol Infect 99: 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stephens DS, Greenwood B, Brandtzaeg P (2007) Epidemic meningitis, meningococcaemia, and Neisseria meningitidis . Lancet 369: 2196–2210. [DOI] [PubMed] [Google Scholar]
- 3. Sharip A, Sorvillo F, Redelings MD, Mascola L, Wise M, et al. (2006) Population-based analysis of meningococcal disease mortality in the United States: 1990–2002. Pediatr Infect Dis J 25: 191–194. [DOI] [PubMed] [Google Scholar]
- 4.Cartwright KA (1995) Meningococcal Carriage and Disease. In: Cartwright KA, editor. Meningococcal Disease. Chichester: John Wiley & Sons. pp. 115–146.
- 5. Virji M (2009) Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7: 274–286. [DOI] [PubMed] [Google Scholar]
- 6. Nassif X, Beretti JL, Lowy J, Stenberg P, O'Gaora P, et al. (1994) Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc Natl Acad Sci U S A 91: 3769–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheuerpflug I, Rudel T, Ryll R, Pandit J, Meyer TF (1999) Roles of PilC and PilE proteins in pilus-mediated adherence of Neisseria gonorrhoeae and Neisseria meningitidis to human erythrocytes and endothelial and epithelial cells. Infect Immun 67: 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Virji M, Kayhty H, Ferguson DJ, Alexandrescu C, Heckels JE, et al. (1991) The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol Microbiol 5: 1831–1841. [DOI] [PubMed] [Google Scholar]
- 9. Simon D, Rest RF (1992) Escherichia coli expressing a Neisseria gonorrhoeae opacity-associated outer membrane protein invade human cervical and endometrial epithelial cell lines. Proc Natl Acad Sci U S A 89: 5512–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Virji M, Saunders JR, Sims G, Makepeace K, Maskell D, et al. (1993) Pilus-facilitated adherence of Neisseria meningitidis to human epithelial and endothelial cells: modulation of adherence phenotype occurs concurrently with changes in primary amino acid sequence and the glycosylation status of pilin. Mol Microbiol 10: 1013–1028. [DOI] [PubMed] [Google Scholar]
- 11. Comanducci M, Bambini S, Brunelli B, Adu-Bobie J, Arico B, et al. (2002) NadA, a novel vaccine candidate of Neisseria meningitidis . J Exp Med 195: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Capecchi B, Adu-Bobie J, Di Marcello F, Ciucchi L, Masignani V, et al. (2005) Neisseria meningitidis NadA is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol Microbiol 55: 687–698. [DOI] [PubMed] [Google Scholar]
- 13. Martin P, Makepeace K, Hill SA, Hood DW, Moxon ER (2005) Microsatellite instability regulates transcription factor binding and gene expression. Proc Natl Acad Sci U S A 102: 3800–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Metruccio MM, Pigozzi E, Roncarati D, Berlanda Scorza F, Norais N, et al. (2009) A novel phase variation mechanism in the meningococcus driven by a ligand-responsive repressor and differential spacing of distal promoter elements. PLoS Pathog 5: e1000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin P, van de Ven T, Mouchel N, Jeffries AC, Hood DW, et al. (2003) Experimentally revised repertoire of putative contingency loci in Neisseria meningitidis strain MC58: evidence for a novel mechanism of phase variation. Mol Microbiol 50: 245–257. [DOI] [PubMed] [Google Scholar]
- 16. Murphy GL, Connell TD, Barritt DS, Koomey M, Cannon JG (1989) Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell 56: 539–547. [DOI] [PubMed] [Google Scholar]
- 17. Schielke S, Huebner C, Spatz C, Nagele V, Ackermann N, et al. (2009) Expression of the meningococcal adhesin NadA is controlled by a transcriptional regulator of the MarR family. Mol Microbiol 72: 1054–1067. [DOI] [PubMed] [Google Scholar]
- 18. Lee EH, Rouquette-Loughlin C, Folster JP, Shafer WM (2003) FarR regulates the farAB-encoded efflux pump of Neisseria gonorrhoeae via an MtrR regulatory mechanism. J Bacteriol 185: 7145–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schielke S, Schmitt C, Spatz C, Frosch M, Schubert-Unkmeir A, et al. (2010) The transcriptional repressor FarR is not involved in meningococcal fatty acid resistance mediated by the FarAB efflux pump and dependent on lipopolysaccharide structure. Appl Environ Microbiol 76: 3160–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fagnocchi L, Pigozzi E, Scarlato V, Delany I (2012) In the NadR regulon, adhesins and diverse meningococcal functions are regulated in response to signals in human saliva. J Bacteriol 194: 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahama U, Oniki T, Murata H (2002) The presence of 4-hydroxyphenylacetic acid in human saliva and the possibility of its nitration by salivary nitrite in the stomach. FEBS Lett 518: 116–118. [DOI] [PubMed] [Google Scholar]
- 22. Johnson PJ, Stringer VA, Shafer WM (2011) Off-target gene regulation mediated by transcriptional repressors of antimicrobial efflux pump genes in Neisseria gonorrhoeae . Antimicrob Agents Chemother 55: 2559–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silver LE, Clark VL (1995) Construction of a translational lacZ fusion system to study gene regulation in Neisseria gonorrhoeae . Gene 166: 101–104. [DOI] [PubMed] [Google Scholar]
- 24. Stephens DS, Swartley JS, Kathariou S, Morse SA (1991) Insertion of Tn916 in Neisseria meningitidis resulting in loss of group B capsular polysaccharide. Infect Immun 59: 4097–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schielke S, Spatz C, Schwarz RF, Joseph B, Schoen C, et al. (2011) Characterization of FarR as a highly specialized, growth phase-dependent transcriptional regulator in Neisseria meningitidis . Int J Med Microbiol 301: 325–333. [DOI] [PubMed] [Google Scholar]
- 26. Hagman KE, Pan W, Spratt BG, Balthazar JT, Judd RC, et al. (1995) Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology 141 (Pt 3) 611–622. [DOI] [PubMed] [Google Scholar]
- 27. Lucas CE, Balthazar JT, Hagman KE, Shafer WM (1997) The MtrR repressor binds the DNA sequence between the mtrR and mtrC genes of Neisseria gonorrhoeae . J Bacteriol 179: 4123–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zalucki YM, Mercante AD, Cloward JM, Ohneck EA, Kandler J, et al. (2011) Function and regulation of Neisseria gonorrhoeae efflux pumps. Microbial Efflux Pumps: Current Research: Horizon Scientific and Caister Academic Press.
- 29. Lee EH, Shafer WM (1999) The farAB-encoded efflux pump mediates resistance of gonococci to long-chained antibacterial fatty acids. Mol Microbiol 33: 839–845. [DOI] [PubMed] [Google Scholar]
- 30. Hagman KE, Shafer WM (1995) Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae . J Bacteriol 177: 4162–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohneck EA, Zalucki YM, Johnson PJ, Dhulipala V, Golparian D, et al. (2011) A novel mechanism of high-level, broad-spectrum antibiotic resistance caused by a single base pair change in Neisseria gonorrhoeae. MBio 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rouquette-Loughlin CE, Balthazar JT, Hill SA, Shafer WM (2004) Modulation of the mtrCDE-encoded efflux pump gene complex of Neisseria meningitidis due to a Correia element insertion sequence. Mol Microbiol 54: 731–741. [DOI] [PubMed] [Google Scholar]
- 33. Shafer WM, Balthazar JT, Hagman KE, Morse SA (1995) Missense mutations that alter the DNA-binding domain of the MtrR protein occur frequently in rectal isolates of Neisseria gonorrhoeae that are resistant to faecal lipids. Microbiology 141 (Pt 4) 907–911. [DOI] [PubMed] [Google Scholar]
- 34. Enriquez R, Abad R, Chanto G, Corso A, Cruces R, et al. (2010) Deletion of the Correia element in the mtr gene complex of Neisseria meningitidis. J Med Microbiol 59: 1055–1060. [DOI] [PubMed] [Google Scholar]
- 35. Shafer WM, Guymon LF, Lind I, Sparling PF (1984) Identification of an envelope mutation (env-10) resulting in increased antibiotic susceptibility and pyocin resistance in a clinical isolate of Neisseria gonorrhoeae . Antimicrob Agents Chemother 25: 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Menard R, Sansonetti PJ, Parsot C (1993) Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol 175: 5899–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skaar EP, Lazio MP, Seifert HS (2002) Roles of the recJ and recN genes in homologous recombination and DNA repair pathways of Neisseria gonorrhoeae . J Bacteriol 184: 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 39. Folster JP, Dhulipala V, Nicholas RA, Shafer WM (2007) Differential regulation of ponA and pilMNOPQ expression by the MtrR transcriptional regulatory protein in Neisseria gonorrhoeae . J Bacteriol 189: 4569–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Folster JP, Johnson PJ, Jackson L, Dhulipali V, Dyer DW, et al. (2009) MtrR modulates rpoH expression and levels of antimicrobial resistance in Neisseria gonorrhoeae . J Bacteriol 191: 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Laboratory Press.
- 42. Prentki P, Krisch HM (1984) In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29: 303–313. [DOI] [PubMed] [Google Scholar]