

Abstract
For many biomedical applications, there is need for porous implant materials. The current article focuses on a method for preparation of drug-eluting porous structures for various biomedical applications, based on freeze drying of inverted emulsions. This fabrication process enables the incorporation of any drug, to obtain an “active implant” that releases drugs to the surrounding tissue in a controlled desired manner. Examples for porous implants based on this technique are antibiotic-eluting mesh/matrix structures used for wound healing applications, antiproliferative drug-eluting composite fibers for stent applications and local cancer treatment, and protein-eluting films for tissue regeneration applications. In the current review we focus on these systems. We show that the release profiles of both types of drugs, water-soluble and water-insoluble, are affected by the emulsion's formulation parameters. The former's release profile is affected mainly through the emulsion stability and the resulting porous microstructure, whereas the latter's release mechanism occurs via water uptake and degradation of the host polymer. Hence, appropriate selection of the formulation parameters enables to obtain desired controllable release profile of any bioactive agent, water-soluble or water-insoluble, and also fit its physical properties to the application.
Keywords: controlled release, poly (dl-lactic-co-glycolic acid), tissue engineering, porosity, biomaterials
Introduction: Techniques for Preparation of Porous Structures for Biomedical Applications
For many biomedical applications, there is need for porous implant materials. Some of the many applications in which porous biomaterials are used include artificial blood vessels,1,2 skin,3,4 bone5,6 and cartilage7,8 reconstruction, periodontal repair9 and drug delivery systems.10
In the most basic sense, porosity is sought to promote new tissue formation by providing an appropriate surface to encourage cellular attachment and an adequate space to host cells as they develop into tissue. However, recent studies have demonstrated how cells are highly sensitive to geometrical constraints from their microenvironment, which regulate tissue formation by affecting cell migration, proliferation, and also differentiation.11-13
The manner in which a bulk material of an implant is distributed from the macro down to the micro and nano-scales often corresponds to the tissue, cellular and molecular scales, respectively. Such hierarchical porous architecture defines the mechanical properties of the scaffold as well as the initial void space that is available for regenerating cells to form new tissues, new blood vessels and the passageways for mass transport via diffusion or convection.14,15
Porous materials have to fulfill specific requirements which are application-dependant. For example, for skin growth and wound healing the optimum pore size is in the range of 20–120 µm,16 whereas for bone ingrowth, the optimum pore size is in the range of 75–250 µm.17 For ingrowth of fibrocartilagenous tissue, the recommended pore size is somewhat larger and ranges 200–300 µm.17 Larger voids are required to allow for vascularization of a developing tissue, but at the same time, it is important to identify the upper limits in pore size since large pores may compromise the mechanical properties of the scaffolds by increasing void volume.18
In contrast to tissue engineering constructs described above, in biomaterials loaded with therapeutic agents, pores with size less than 10 µm in diameter are needed to administer release of the agent by a slow, local, continuous and controlled flux.10 In complex systems such multifunctional devices which act as scaffolds with controlled release, there may come a need to combine different pore sizes within the same structure. Besides pore size, other parameters which are linked to porosity, such as pore interconnectivity (% of non-isolated pores), pore interconnection throat size, and changes in porosity due to degradability also play an important role.19,20
Some of the main techniques used to prepare porous biomaterials are outlined below.
Particulate-leaching techniques
Particulate leaching has been widely used to fabricate scaffolds for tissue engineering applications. In this method, small particles of salt,20,21 sugar22-24 or another substance (porogen) of the desired size are transferred into a mold. A polymer solution, or ceramic slurry, is then cast into the porogen-filled mold. After the evaporation of the solvent and/or solidification of the matrix, the porogen is leached away using water,25 or burnt out,26 to form the pores of the scaffold. Alternatively to solvent casting, a polymer can also be melt molded in the presence of a porogen which is then leached in a similar way. The pore size and shape attained in this method can be controlled by the size and geometry of the porogen and the porosity is controlled by the porogen/polymer ratio. Pore sizes between 50–200 µm and porosities up to 90% have been reported.21,23,24, Although salt/sugar fusion in humid environment can be employed to get scaffolds with enhanced interconnectivity, pore shape and inter-pore openings are usually difficult to control using this method.24 Another disadvantage of these fabrication methods is the exposure of the matrix material to organic solvents or elevated temperatures, which may be harmful to cells or bioactive agents if they are to be incorporated in the material during fabrication.
Gas has also been used as a porogen. The process begins with the formation of solid discs of polymer which are placed in a chamber and exposed to high pressure CO2 for three days, at which time the pressure is rapidly decreased to atmospheric pressure. Porosities of up to 93% and pore sizes of up to 100 µm can be obtained using this technique, but the pores are largely unconnected, especially on the surface of the foam.27 While this fabrication method requires no leaching step and uses no harsh chemical solvents, the high temperatures involved in the disc formation prohibit the incorporation of cells or bioactive molecules and the unconnected pore structure make cell seeding and migration within the foam difficult. Nam et al.28 reported a technique which includes both gas foaming and particulate leaching aspects which does not result in the creation of a nonporous outer skin. Ammonium bicarbonate is added to a solution of polymer in methylene chloride or chloroform. Vacuum drying causes the ammonium bicarbonate to sublime while immersion in water results in concurrent gas evolution and particle leaching. Porosities as high as 90% with pore sizes from 200–500 µm are attained using this technique.
Phase separation techniques
Under certain conditions a homogeneous multi-component system may become thermodynamically unstable and separate into more than one phase in order to lower the system free energy. A polymer solution may separate in such way into two phases, a polymer-rich phase and a polymer-lean phase.29 Alternatively, phase separation may be induced by mechanical shearing, or emulsification of two or more phases.30 After the solvents are removed, often by vacuum or freeze-drying, the polymer-rich phase solidifies to become the scaffold, while the polymer lean phase becomes a void. Manipulation of the thermodynamics and kinetics of phase separations leads to a wide variety of morphologies of the phase-separated domains, which greatly impacts the architecture of the scaffold. The pores formed using such techniques usually have small diameters on the order of a few to tens of microns, which can be unsuitable for certain tissue engineering applications but extremely advantageous in designing controlled drug release systems.
Another advantage of the phase separation technique is the ability to incorporate sensitive bioactive agents such as growth factors growth directly into the scaffold without loss in bioactivity due to exposure to harsh solvents or elevated temperatures.31
Textile technologies
Fibers are a fundamental unit of most tissues, and collagen fibers are the most abundant protein in the body. It is not surprising that natural and synthetic fiber-based structures have been widely used for biomedical applications.
Fibers can be formed in three-dimensional structures such as knitted, braided, woven, and nonwoven. The orientation of fibers in these structures may range from highly regular to completely random. The final structure of the fibers affects the behaviors of the fibers when they are applied. Most often, the porosity of a textile is determined by the void space between fibers, but porosity could also occur in the fibers themselves.32,33
Woven structures are porous and more stable structure compared with other textile structures. Some applications of wovens include arterial grafts,34 cartilage reconstruction35 and rotator cuff repair.36 As a disadvantage, wovens can be unraveled at the edges when they are cut squarely or obliquely for implantation. Knit structures are flexible and highly porous and have an inherent ability to resist unraveling when cut. Due to the high level of conformability and porosity, knitted fabrics are ideal candidates for vascular implants.37 Other applications include aortic valves,38 tracheal cartilage reconstruction39 and ligament reconstruction.40 Braided structures are mostly used as sutures and ligaments41 because the spaces between the yarns, which cross each other, make them porous but still enable them to withstand high loads during the healing process. A braided structure has also been used in nerve guide constructs.42 Non-woven structures may have a wide range of porosities and their isotropic structure provides good mechanical and thermal stability.43 They can easily compress and expand. These advantages make them a suitable material for many tissue-engineering applications ranging from heart tissue44 to a corneal graft.45 Emerging nano-fabrication methods such as electro-spinning now enable to produce non-wovens from synthetic nano-scale fibers which are dimensionally similar to collagen fibers and thus allow stronger interfacing with the host tissue.11
Sintering
Porous metals have been used as coatings for fixation of dental and orthopedic implants since they encourage bone growth and enhance fixation. The most common approach in fabrication of porous metal and metal alloys are sintering of loose powder,46,47 or slurry sintering.48,49 The process of sintering involves heating alloy beads and a substrate to about a half of the alloys melting temperature to enable diffusive mechanisms to form necks that join the beads to one another and to the surface. Loose powder sintering yields relatively small pores (< 20 μm), and low porosities (< 40%).47,49,50 In order to increase porosity and pore size, the metal powder can be mixed with a porogen such as ammonium hydrogen carbonate as which is later burnt out leaving behind voids. This process enables to increase the porosity to 74%.51 The pores attained in this method are a mixed population of 5–20 μm pores as resulting from conventional sintering and much larger pores 300–800 μm, as resulting from the presence of the porogen.
Rapid prototyping techniques
Rapid prototyping techniques have attracted much interest in recent years as powerful tools to fabricate scaffolds. These scaffolds are built layer by layer, through material deposition on a stage, either in a molten phase52,53 (known as fused deposition modeling) or in droplets together with a binding agent54 (referred to as 3D Printing). These methods can be applied to an extended range of basic materials including polymers,52 metals53 and ceramics.55 The 3D outcomes of this process can be guaranteed to have 100% interconnected pores if during fabrication the layers are deposited as interpenetrating networks. Another advantage of these methods is the ability to incorporate cells within the structure during fabrication.56
Part of the mentioned above methods can serve for preparation of implants and scaffolds loaded with drugs, that in addition to their regular role (of support for example) they also release drug molecules in a controlled desired manner to the surrounding tissue, and therefore induce healing effects. In such cases it is necessary to incorporate the drug molecules in the porous structure during the process of scaffold formation and to be able to control their release profile. It is also important to preserve the drug's activity during the process of encapsulation in the porous structure. This is not simple because many drugs and all protein lose their activity when they are exposed to organic solvents or elevated temperature. Protein incorporation during the process of preparation is still a challenge in all mentioned above methods. Also, most of the suggested methods do not describe how the drug release profile from the porous structure can be controlled and fit the application.
The current article focuses on a method for preparation of drug-eluting porous structures for various biomedical applications, based on freeze drying of inverted emulsions. Any bioactive agent (drug or protein) can be incorporated during the process of preparation, without losing the activity. Examples are given for controlled release of hydrophilic drugs, hydrophobic drugs and proteins.
Drug-Eluting Porous Structures Based on Freeze-Dried Inverted Emulsions
Emulsions
An emulsion is a metastable mixture of two immiscible liquids such as oil and water in the form of droplets of one substance (discontinues phase) in the other (continues phase). Emulsions are generally categorized into two groups: oil in water (O/W), where water is the continuous phase, and water in oil (W/O) where water is the discontinuous phase, i.e., inverted emulsion. Emulsions are obtained by activating shear forces between the phases, leading to the fragmentation of one phase into the other. The outward pressure (Laplace pressure) of the formed droplets is inversely proportional to the droplet diameter and the droplet diameter therefore decreases as shear forces are increased.57
During destabilization, an emulsion goes through several consecutive and parallel steps, which eventually lead to separation. At first, the droplets move due to diffusion or stirring to the fusion of two Brownian driven adjacent droplets, irreversibly, and if the repulsion potential is too weak, they become aggregated to each other. This process is called flocculation. The single droplets are now replaced by twins or multiplets, which are separated by a thin film. The thickness of the thin film is reduced due to the van der Waals attraction, and when a critical value of its dimension is reached, the film bursts and the two droplets unite to a single droplet in a process called coalescence. The decrease in free energy caused during the process of thinning of the interdroplet film determines the contact angle.57,58 In parallel to the processes described above, the droplet also rises through the continuous phase (creaming) or sinks to the bottom of the continuous phase (sedimentation) due to differences in density of the dispersed and continuous mediums.57,59
The presence of surface active agents (surfactants) stabilizes an emulsion since they reduce the interfacial tension between the two immiscible phases. Proteins are widely used as emulsion stabilizers in the food industry.60,61 It has been reported that metastable “water in oil” emulsions can be stabilized by bovine serum albumin.60,62,63 Hydrophilic polymers, such as poly(vinyl alcohol) and poly(ethylene glycol), act as surfactants due to their amphiphilic molecular structure, thus increasing the affinity between the aqueous and organic phases.64-66
The concept of freeze-dried inverted emulsions
In the current study we developed a special technique termed freeze drying of inverted emulsions, and studied the effects of process and formulation parameters on the obtained microstructure and on the resulting drug release profile and other properties that are relevant for the application. The inverted emulsions used in our study are prepared by homogenization of two immiscible phases: an organic solution containing a known amount of poly (dl-lactic-co-glycolic acid) (PDLGA) in chloroform, and an aqueous phase containing, double-distilled water. Homogenization of the two phases is usually performed for the duration of 90 sec at an average rate of 16,000 RPM using a homogenizer. Both, process parameters and formulation parameters, are controllable and affect the microstructure and properties. The “process parameters” are the homogenization rate and duration and are termed as kinetic parameters, and the “formulation parameters” are the polymer content of the organic phase, the polymer's molecular weight, the copolymer composition (glycolic acid: lactic acid), the organic: aqueous (O:A) phase ratio, the drug content and incorporation of surfactants. These are termed “themodynamic parameters,” due to their strong effect on the microstructure through the emulsion's stability, as will be explained in details and examples below. The formulation parameters were found to be more important than the process parameters in determining the microstructure.67-72
After preparing the inverted emulsions they can be poured into a dish, followed by immediate freezing in a liquid nitrogen bath so as to form a porous drug-loaded film. It can also coat any structure (dense fiber, stent or any bulky 3D structure). The following freeze drying process enables to preserve the micro/nano-structure of the inverted emulsion and get a solid implant encapsulated with drug molecules. The whole process of preparation in described in Figure 1. Examples for implant structures are presented in Figure 2. These include a porous film (Fig. 2A), a composite mesh/matrix structure composed of a mesh made of dense fibers and porous matrix (Fig. 2B), and a core/shell composite fiber (Fig. 2C). All porous elements in these structures are prepared using the freeze drying of inverted emulsion technique. Their microstructure is shown in high SEM magnification in a separate circled part of Figure 2.

Figure 1. A schematic representation of the freeze drying of inverted emulsion process.

Figure 2. SEM micrographs of biodegradable drug-loaded porous structures derived from freeze-dried inverted emulsions: (A) cross section of a film, (B) composite mesh/matrix structure and (C) cross section of core/shell fiber. High magnification of the porous structure is shown in the circle.
The freeze-drying of inverted emulsions technique is unique in being able to preserve the liquid structure in solids and was employed in our studies in order to produce highly porous micro and nano-structures, as those presented in Figure 2, that can be used as basic elements or parts of various implants and scaffolds for tissue regeneration. This fabrication process enables the incorporation of both water-soluble and water-insoluble drugs into the film in order to obtain an “active implant” that releases drugs to the surrounding in a controlled manner and therefore induces healing effects in addition to its regular role (of support, for example). Water-soluble bioactive agents are incorporated in the aqueous phase of the inverted emulsion, whereas water-insoluble drugs are incorporated in the organic (polymer) phase. Sensitive bioactive agents, such as proteins, can also be incorporated in the aqueous phase. This prevents their exposure to harsh organic solvents and enables the preservation of their activity.
There are numerous medical applications for our freeze-dried drug-eluting structures. For example: porous films, fibers, or composite structures loaded with water-soluble drugs, such as antibiotics, can be used for wound dressing applications, treatment of periodontal diseases, meshes for Hernia repair, as well as coatings for fracture fixation devices. Fibers loaded with water insoluble drugs such as antiproliferative agents can be used as basic elements of drug-eluting stents and also for local cancer treatment. Films and fibers loaded with growth factors can be used as basic elements of highly porous scaffolds for tissue regeneration. These structures for the suggested applications were investigated by us and selected examples are presented in the three following chapters. We will also show how appropriate selection of the formulation (thermodynamic) parameters enables to obtain desired controllable release profile of any bioactive agent, water-soluble or water-insoluble, that fits the application.
Porous Structures with Controlled Release of Water-Soluble Drugs
Water-soluble agents, such as many antibiotic drugs, are incorporated in the aqueous phase of the inverted emulsion and therefore, after the freeze drying process are located on the pore walls of the highly porous solid structures. In such structures relatively high burst release can be obtained when immersed in aqueous surrounding, due to the high water solubility of these drugs. Their location in the pores (rather than in the polymeric domains), as a result of the process of preparation, also tend to increase the burst release. Therefore it is extremely important to be able to control the release profile of such drugs through structuring of the porous matrix. Such structuring effects are obtained by choosing the appropriate formulation parameters. The effects of the formulation parameters on the microstructure and on the resulting drug release profile were investigated by us. In this study we chose to focus on antibiotic release from wound dressing structures, prepared using the freeze drying of inverted emulsion technique. We present here the effect of structuring on the antibiotic release profile and on the mechanical and physical properties of the wound dressings. The biological performance and in-vivo results are presented as well.
In addition to the wound healing applications, antibiotic release from porous structures can be used for other medical applications, such as treatment of periodontal diseases, meshes for hernia repair and coatings for fracture fixation devices. Water-soluble drugs can even be used for broader range of applications. Hence, this study of porous structures with controlled release of water-soluble drugs is beneficial for many biomedical applications.
Antibiotic-eluting composite wound dressings
The skin is regarded as the largest organ of the body and has many different functions. Wounds with tissue loss include burn wounds, wounds caused as a result of trauma, diabetic ulcers and pressure sores. The regeneration of damaged skin includes complex tissue interactions between cells, extracellular matrix molecules and soluble mediators in a manner that results in skin reconstruction. The moist, warm, and nutritious environment provided by wounds, together with diminished immune functioning secondary to inadequate wound perfusion, may allow build-up of physical factors such as devitalized, ischemic, hypoxic, or necrotic tissue and foreign material, all of which provide an ideal environment for bacterial growth.73
The main goal in wound management is to achieve rapid healing with functional and esthetic results. An ideal wound dressing can restore the milieu required for the healing process, while protecting the wound bed against penetration of bacteria and environmental threats. The dressing should also be easy to apply and remove. Most modern dressings are designed to maintain a moist healing environment, and to accelerate healing by preventing cellular dehydration and promoting collagen synthesis and angiogenesis.74 Nonetheless, over-restriction of water evaporation from the wound should be avoided, since accumulation of fluid under the dressing may cause maceration and facilitate infection. The water vapor transmission rate (WVTR) from the skin has been found to vary considerably depending on the wound type and healing stage, increasing from 204 gm−2 d−1 for normal skin to 278 and as much as 5,138 gm−2 d−1 for first degree burns and granulating wounds, respectively.75 The physical and chemical properties of the dressing should therefore be adapted to the type of wound as well as to the degree of wound exudation.
A range of dressing formats based on films, hydrophilic gels and foams are available or have been investigated. Thin semi-permeable polyurethane films coated with a layer of acrylic adhesive, such as Optsite® (Smith and Nephew) and Bioclussive® (J and J), are typically used for minor burns, post-operative wounds, and a variety of minor injuries including abrasions and lacerations. Gels such as carboxymethylcellulose-based Intrasite Gel® (Smith and Nephew) and alginate-based Tegagel® (3M) are used for many different types of wounds, including leg ulcers and pressure sores. These gels promote rapid debridement by facilitating rehydration and autolysis of dead tissue. Foam dressings, such as Lyofoam (Mölnlycke Healthcare) and Allevyn (Smith and Nephew) are used to dress a variety of exudating wounds, including leg and decubitus ulcers, burns, and donor sites.
Films and gels have a limited absorbance capacity and are recommended for light to moderately exudating wounds, whereas foams are highly absorbent and have a high WVTR and are therefore considered more suitable for wounds with moderate to heavy exudation.76 The characteristics of the latter are controlled by the foam texture, pore size, and dressing thickness.
Infection is defined as a homeostatic imbalance between the host tissue and the presence of microorganisms at concentrations that exceeds 105 organisms per gram of tissue or the presence of β-hemolytic streptococci.77,78 The main goal of treating the various types of wound infections should be to reduce the bacterial load in the wound to a level at which wound healing processes can take place. Otherwise, the formation of an infection can seriously limit the wound healing process, can interfere with wound closure and may even lead to bacteremia, sepsis and multi-system failure. Evidence of bacterial resistance is on the rise, and complications associated with infections are therefore expected to increase in the general population.
Bacterial contamination of a wound seriously threatens its healing. In burns, infection is the major complication after the initial period of shock, and it is estimated that about 75% of the mortality following burn injuries is related to infections rather than to osmotic shock and hypovolemia.79 Bacteria in wounds are able to produce a biofilm within approximately 10 h. This biofilm protects them against antibiotics and immune cells already in the early stages of the infection process.80 The rapidity of biofilm growth suggests that efforts to prevent or slow the proliferation of bacteria and biofilms should begin immediately after creation of the wound. This has encouraged the development of improved wound dressings that provide an antimicrobial effect by eluting germicidal compounds such as iodine (Iodosorb®, Smith and Nephew), chlorohexidime (Biopatch®, J and J) or most frequently silver ions (e.g., Acticoat® by Smith and Nephew, Actisorb® by J and J and Aquacell® by ConvaTec). Such dressings are designed to provide controlled release of the active agent through a slow but sustained release mechanism which helps avoid toxicity yet ensures delivery of a therapeutic dose to the wound. Some concerns regarding safety issues related to the silver ions included in most products have been raised. Furthermore, such dressings still require frequent change, which may be painful to the patient and may damage the vulnerable underlying skin, thus increasing the risk of secondary contamination.
Bioresorbable dressings successfully address this shortcoming, since they do not need to be removed from the wound surface once they have fulfilled their role. Biodegradable film dressings made of lactide-caprolactone copolymers such as Topkin® (Biomet) and Oprafol® (Lohmann and Rauscher) are currently available. Bioresorbable dressings based on biological materials such as collagen and chitosan have been reported to perform better than conventional and synthetic dressings in accelerating granulation tissue formation and epithelialization.81,82 However, controlling the release of antibiotics from these materials is challenging due to their hydrophilic nature. In most cases, the drug reservoir is depleted in less than two days, resulting in a very short antibacterial effect.83,84
The effectiveness of a drug-eluting wound dressing is strongly dependent on the rate and manner in which the drug is released.85 These are determined by the host matrix into which the antibiotic is loaded, the type of drug/disinfectant and its clearance rate. If the agent is released quickly, the entire drug could be released before the infection is arrested. If release is delayed, infection may set in further, thus making it difficult to manage the wound. The release of antibiotics at levels below the minimum inhibitory concentration (MIC) may lead to bacterial resistance at the release site and intensify infectious complications.86,87 A local antibiotic release profile should therefore generally exhibit a considerable initial release rate in order to respond to the elevated risk of infection from bacteria introduced during the initial shock, followed by a sustained release of antibiotics at an effective level, long enough to inhibit latent infection.83
There is currently no available synthetic dressing that combines the advantages of occlusive dressings with biodegradability and intrinsic topical antibiotic treatment. In order to obtain this combination of properties we have recently developed and studied a composite wound dressing based on the concept of core/shell (matrix) composite structures. Its characteristics are described here.
Composites are made up of individual materials, matrix and reinforcement. The matrix component supports the reinforcement material by maintaining its relative positions and the reinforcement material imparts its special mechanical properties to enhance the matrix properties. Taken together, both materials synergistically produce properties unavailable in the individual constituent materials, allowing the designer to choose an optimum combination. In our application, a reinforcing polyglyconate mesh affords the necessary mechanical strength to the dressing, while the porous Poly(dl-lactic-co-glycolic acid) (PDLGA) binding matrix is aimed to provide adequate moisture control and release of antibiotics in order to protect the wound bed from infection and promote healing. Both structural constituents are biodegradable, thus enabling easy removal of the wound dressing from the wound surface once it has fulfilled its role. This new structural concept in the field of wound healing is presented in Figure 2B.
The freeze-drying of inverted emulsions technique which was used to create the porous binding matrix is unique in its ability to preserve the liquid structure in the solid state.88 The viscous emulsion, consisting of a continuous PDLGA/chloroform solution phase and a dispersed aqueous drug solution, formed good contact with the mesh during the dip-coating process. Consequently, an unbroken solid porous matrix was deposited by the emulsion following freeze-drying (Fig. 2B). The freeze-drying of inverted emulsions technique has several advantages. First, it enables attaining a thin uninterrupted barrier, which unlike mesh or gauze alone can better protect the wound bed against environmental threats and dehydration. Second, it entails very mild processing conditions which enable the incorporation of sensitive bioactive agents such as antibiotics.10,89 and even growth factors88 to help reduce the bio-burden in the wound bed and accelerate wound healing. Third, the microstructure of the freeze-dried matrix can be customized through modifications of the emulsion’s formulation to exhibit different attributes, namely different porosities or drug release profiles. Such structuring effects are described in this chapter. The mechanical and physical properties of these new wound dressings and their biological performance are also presented. Finally, a guinea pig model was used to evaluate the effectiveness of these antibiotic-eluting dressings and the main conclusions are brought here.
Structure-controlled release effects
The controlled release of antibiotics from wound dressings is challenging, since various related design considerations need to be addressed. Specifically, porosity which is desired to provide adequate gaseous exchange and absorption of wound exudates90 may act as a two-edged sword; allowing rapid water penetration which typically leads to a rapid release of the water soluble active agent within several hours to several days.91,92 Structural effects on the controlled release of gentamicin and ceftazidime from our composite structures were extensively studied.10,70 and the most important results are presented here.
As mentioned above, the emulsion's formulation parameters which determine the porous matrix structure and also the resulting properties are the organic:aqueous (O:A) phase ratio, the drug content in the aqueous phase, the polymer content in the organic phase, the polymer's initial molecular weight (MW) and also surfactants incorporated in the emulsion so as to increase its stability. The characteristic features of our studied samples are presented in Table 1. The basic formulations were used for the microstructure-release profile study. A highly interconnected porous structure poses almost no restriction to outward drug diffusion once water penetrates the matrix, and drug release in this case is most probably governed by the rate of water penetration into the matrix. Hence, the antibiotic release from our reference formulation (formulation 1, Fig. 3A, ○) clearly demonstrates the prominent effect of pore connectivity on the burst release of the antibiotics, i.e., release of drug within the first 6 h. Samples with relatively low emulsion's O:A phase ratio (up to 8:1) typically demonstrate much pore connectivity (Fig. 3B) and their in vitro release patterns display a burst release of approximately 95% (Fig. 3A, ○). In contradistinction, porous shell structures derived from higher O:A phase ratios (for example 12:1), display reduced pore connectivity and a lower pore fraction (Fig. 3C and Table 1), resulting in a significant half-fold decrease in the burst release of antibiotics to approximately 45% (Fig. 3A, △).
Table 1. Structural characteristics of the ceftazidime-loaded porous matrix70.
| Formulation | O:A | Drug loading* (w/w) | Polymer content in the organic phase**(w/v) | Polymer MW (KDa) | Surfactant** | Freeze-dried emulsion | ||
|---|---|---|---|---|---|---|---|---|
| |
|
|
|
|
|
|
Porosity (%) |
Pore diameter (μm) |
| Basic Formulations |
(1) Reference |
6:1 |
|
15% |
100 |
None |
68 |
1.5 ± 0.6 |
| (2) High O:A |
12:1 |
5% |
15% |
100 |
None |
45 |
1.6 ± 0.4 |
|
| (3) High polymer content |
6:1 |
5% |
20% |
100 |
None |
22 |
1.2 ± 0.9 |
|
| (4) High polymer MW |
6:1 |
5% |
15% |
240 |
None |
16 |
0.5 ± 0.4 |
|
| Formulations with Surfactants | (5) BSA1: ref., stabilized with BSA |
6:1 |
5% |
15% |
83 |
BSA (1% w/v in the aqueous phase) |
63 |
1.4 ± 0.3 |
| (6) BSA2: high O:A, stabilized with BSA |
12:1 |
5% |
15% |
83 |
BSA (1% w/v in the aqueous phase) |
35 |
1.4 ± 0.3 |
|
| (7) SPAN: high O:A, stabilized with Span | 12:1 | 5% | 15% | 83 | Span80 (1% w/v in the organic phase) | 45 | 1.1 ± 0.3 | |
Relative to the polymer weight, **relative to the liquid phase volume (organic or aqueous)

Figure 3. (A) Controlled release of the antibiotic drug ceftazidime from composite structures based on various formulations. ○, the reference formulation (formulation 1): 5% w/w ceftazidime and 15% w/v polymer (75/25 PDLGA, MW = 100 KDa), O:A = 6:1; △, formulation 2: increased O:A phase ratio (12:1); formulation 3: increased polymer MW (240KDa); ◇, formulation 4: increased polymer content in the organic phase (20%). (B–E) SEM fractographs showing the effect of a change in the emulsion’s formulation parameters on the microstructure of the binding matrix for formulations 1–4, respectively.10
An increase in the polymer's molecular weight (MW) from 100 KDa to 240 KDa resulted in a tremendous effect on the shell microstructure. The porosity of the shell in this case was reduced to only 16% (Fig. 3D and Table 1). Since high viscosity increases the shear forces during the process of emulsification and also reduces the tendency of droplets to move, it is expressed in a significantly smaller pores and relatively thick polymeric domain between them. These changes in microstructure reduced the burst release of the encapsulated antibiotics to approximately 30% and enabled a continuous moderate release over a period of one month (Fig. 3A,☐).
Finally, an increase in the emulsion's polymer content to 20% w/v also resulted in a dramatic decrease in the burst release (Fig. 3A, ◇). A higher polymer content in the organic phase results in denser polymer walls between pores after freeze-drying (Fig. 3E) and therefore poses better constraint on the release of drugs out of pores. Interestingly, samples containing a 20% polymer content exhibited a three-phase release pattern: an initial burst release, a continuous release at a declining rate during the first two weeks until release of 50% of the encapsulated drug, followed by a third phase of release of a similar nature reaching 99% release after 42 d. The second phase of release is governed by diffusion, whereas the third phase is probably governed by degradation of the host polymer which enables trapped drug molecules to diffuse out through newly formed elution paths. In other cases described thus far, drug release was governed primarily by diffusion, since almost the entire amount of drug was released before polymer degradation would in fact be able to affect the release profile. Thus, when drug diffusion out of the shell is restricted as in the case of high polymer content, and a considerable amount of drug still remains within the porous matrix, polymer degradation will contribute to further release the antibiotics, which leads to an additional release phase.
Other modifications to the emulsion formulation included the addition of surfactants. Surfactants promote stabilization of the emulsion by reduction of interfacial tension between the organic and aqueous phases, resulting in refinement of the microstructure. We examined three matrix formulations loaded with surfactants (listed in Table 1), which display distinctly different micro-structural features (Fig. 4A–C and Table 1). The effect of the O:A phase ratio was examined on formulations containing bovine serum albumin (BSA) as surfactant. As expected, a higher O:A phase ratio, i.e., lower aqueous phase quantity, resulted in a smaller porosity of the solid structure. However, both microstructures were homogenous and characterized by a similar average pore size. The stabilization effect of Span 80 was even higher than that obtained using BSA, and therefore resulted in a smaller pore size (Table 1). The release profile of antibiotics from wound dressings varied considerably with the changes in formulation (Fig. 4D). Ceftazidime release from the dressings based on the BSA1 formulation was relatively short, reaching almost complete release of the encapsulated drug within 24 h. An increase in the emulsion's O:A phase ratio from 6:1 to 12:1 reduced the burst release. Specifically, burst release values of 97% and 57% were recorded after 6 h for formulations BSA1 and BSA2, respectively, after which the release of the antibiotics from BSA2 dressings continued for 5 d at a decreasing rate. The ceftazidime release profile from the SPAN formulation was totally different. It exhibited a low burst release of 6% during the first 6 h of incubation and then a release pattern of a nearly constant rate for 10 d. Surfactant incorporation can contribute to the achievement of more than merely a stabilizing effect, by binding to antibiotics and thus counteracting drug depletion. We have found, for instance, that dressings containing mafenide in combination with albumin as surfactant display a lower burst release and a moderate release rate.10

Figure 4. (A–C) SEM fractographs demonstrating the microstructure of wound dressings based on formulations BSA1, BSA2 and SPAN, respectively. (D) The controlled release of the antibiotic drug ceftazidime from the three studied wound dressings and (E) water vapor transmission rates, corresponding to each sample, together with these obtained from a dense (non-porous) PDLGA (50/50, MW 100 KDa) film () and from an uncovered surface.70
In summary, we demonstrated the release of antibiotic contents at high (> 90%), intermediate (40–60%) and low (~5%) burst release rates and release spans ranging from several days to three weeks. The versatility of the drug release profiles was obtained through the effects of the inverted emulsion’s formulation parameters on the porous structure. In particular, lower burst release rates and longer elution durations can be achieved through structuring toward a reduced pore size, pore connectivity and total porosity.
Physical and mechanical properties
Moisture management
Successful wound healing requires a moist environment. Two parameters must therefore be determined: the water uptake ability of the dressing and the water vapor transmission rate (WVTR) through the dressing. An excessive WVTR may lead to wound dehydration and adherence of the dressing to the wound bed, whereas a low WVTR might lead to maceration of healthy surrounding tissue and buildup of a back pressure and pain to the patient. A low WVTR may also lead to leakage from the edges of the dressing which may result in dehydration and bacterial penetration.93,94 It has been claimed that a burn dressing should ideally possess a WVTR in the range of 2,000–2,500 g/m2/d, half of that of a granulating wound.93 In practice, however, commercial dressings do not necessarily conform to this range, and have been shown to cover a larger spectrum of WVTR, ranging from 90 (Dermiflex®, J&J) to 3,350 g/m2/d (Beschitin®, Unitika).90 Clearly, the WVTR is related to the structural properties (thickness, porosity) of the dressing as well as to the chemical properties of the material from which it is made.
In this part of the study, we examined the specific emulsion formulations that included surfactants (BSA1, BSA2, SPAN, see Table 1). These were chosen based on emulsion stability and resultant microstructure (Fig. 4A–C), and also on drug release profiles (Fig. 4D). Evaporative water loss through the various dressings was linearly dependant on time (R2 > 0.99 in all cases), resulting in a constant WVTR, between 480–3452 g/m2/d, depending on the formulation (Fig. 4E). These results demonstrate how the WVTR can be customized based on modifications of the porous matrix's microstructure. The lowest value is similar to that reported for film type dressings (e.g., Tegaderm, 491 ± 44 g/m2/d),95 while the highest value is similar to that of foam type dressings (e.g., Lyofoam, 3052 ± 684 g/m2/d).95 Further investigation of O:A phase ratios between 6:1 and 12:1 with albumin may generate a WVTR specifically in the 2000–2500 g/m2/d range. A WVTR of 2641 ± 42 g/m2/d which was achieved for 12:1 O:A with the surfactant Span80 (formulation 7) is close to this range and seems the most appropriate.
Water uptake by the wound dressing may occur either as the result of water entry into accessible voids in the porous matrix structure (hydration effect), or as the polymer matrix material gradually uptakes water and swells (swelling effect). Our water uptake patterns for wound dressings based on formulations loaded with BSA demonstrated both these effects.70 Both types of wound dressing (formulations 5 and 6) demonstrated a 3-stage water uptake pattern.
Mechanical properties
The mechanical properties of a wound dressing are an important factor in its performance, whether it is to be used topically to protect cutaneous wounds or as an internal wound support, e.g., for surgical tissue defects or hernia repair. Furthermore, in the clinical setting, appropriate mechanical properties of dressing materials are needed to ensure that the dressing will not be damaged by handling. Porous structures typically possess inferior mechanical properties compared with dense structures, yet in wound healing applications porosity is an essential requirement for diffusion of gasses, nutrients, cell migration and tissue growth. Most wound dressings are therefore designed according to the bi-layer composite structure concept and consist of an upper dense “skin” layer to protect the wound mechanically and prevent bacterial penetration and a lower spongy layer designed to adsorb wound exudates and accommodate newly formed tissue. Our new dressing design integrates both structural/mechanical and functional components (e.g., drug release and moisture management) in a single composite layer.70 It combines relatively high tensile strength and modulus together with good flexibility (elongation at break). It actually demonstrated better mechanical properties than most other dressings currently used or studied, as demonstrated in Table 2.
Table 2. Mechanical properties of various wound dressings70.
| Material/format | Elastic modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| BSA1 (composite polyglyconate mesh, coated with PDLGA porous matrix) |
126 ± 27 |
24.2 ± 4.5 |
55 ± 5 |
| Electrospun poly-(L-lactide-co-ɛ-caprolactone) (50:50) mat28 |
8.4 ± 0.9 |
4.7 ± 2.1 |
960 ± 220 |
| Electrospun gelatin mat |
490 ± 52 |
1.6 ± 0.6 |
17.0 ± 4.4 |
| Electrospun collagen mat |
|
11.4 ± 1.2 |
|
| Resolut® LT regenerative membrane (Gore). Glycolide fiber mesh coated with an occlusive PDLGA membrane |
|
11.7 |
20 |
| Kaltostat® (ConvaTec) Calcium/Sodium Alginate fleece | 1.3 ± 0.2 | 0.9 ± 0.1 | 10.8 ± 0.4 |
The initial mechanical properties of natural polymers such as collagen or gelatin can be satisfactory. However, considerable degradation of these properties is expected to occur rapidly due to hydration96 and enzymatic activity.97 The results of the three weeks degradation study of our wound dressings show a significant decrease only in Young's modulus (Fig. 5). The maximal stress and strain of our composite wound dressing (24 MPa and 55%, respectively) are dictated mainly by the mechanical properties of the reinforcing fibers which fail first during breakage. At these time periods they are not subjected to considerable degradation, which explains the constancy in these properties. In contradistinction, the Young's modulus of the dressings is considerably affected by the properties of the binding matrix that makes up the largest part of the cross-sectional area. The degradation of the matrix material which is clearly in progress after two weeks of exposure to PBS thus leads to a decrease in Young's modulus. The mechanical properties of our wound dressings are superior to those reported before, and remain good even after three weeks of degradation (Young's modulus of 69 MPA, maximal stress 24 MPa and maximal strain 61%), as demonstrated in Figure 5.

Figure 5. (A) Tensile stress-strain curves for wound dressings immersed in water for 0 (–0–), 1 (–1–), 2 (–2–), and 3 (–3–) weeks. (B) Young’s modulus, (C) tensile strength and (D) maximal tensile strain as a function of immersion time. Comparison was made using ANOVA and significant differences are indicated (*).70
In summary, the mechanical properties of our wound-dressing structures were found to be superior, combining relatively high tensile strength and ductility, which changed only slightly during three weeks of incubation in an aqueous medium. The parameters of the inverted emulsion as well as the type of surfactant used for stabilizing the emulsion were found to affect the microstructure of the binding matrix and the resulting physical properties, i.e., water absorbance and water vapor transmission rate.
Biological performance
Bacterial inhibition
The strategy of drug release to a wound depends on the condition of the wound. After the onset of an infection, it is crucial to immediately respond to the presence of large numbers of bacteria (> 105 CFU/mL) which may already be present in the biofilm,80 and which may require antibiotic doses of up to 1,000 times those needed in suspension.98,99 Following the initial release, sustained release at an effective level over a period of time can prevent the occurrence of latent infection. We have shown that the proposed system can comply with these requirements (see “Structure-controlled release effects”).
The time-dependent antimicrobial efficacy of these antibiotic-eluting wound dressing formulations was tested in vitro by two complementary methods. The first method is based on the corrected zone of inhibition test (CZOI),69 which is also termed the disc diffusion test. According to this method presence of bacterial inhibition in an area that exceeds the dressing material (CZOI > 0) can be considered beneficial. This method gives a good representation of the clinical situation, where the dressing material is applied to the wound surface, allowing the drug to diffuse to the wound bed. The results from this method are dependent on the rate of diffusion of the active agent from the dressing, set against the growth rate of the bacterial species growing on the lawn, and are highly dependent on the physicochemical environment. The second method is actually a release study from selected wound dressings in the presence of bacteria, which was performed in order to study the effect of drug release on the kinetics of residual bacteria.69 This method, which is termed viable counts, provides valuable information on the kill rate, which is a key comparator for different formulations and physicochemical conditions.
The bacterial strains Staphylococcus aureus (S. aureus), Staphylococcus albus (S. Albus) and Pseudomonas aeruginosa (P. aeruginosa) were used in this study. The minimal inhibitory concentration of the antibiotics gentamicin and ceftazidime against these strains are presented in Table 3. The results for wound dressings stabilized with BSA using the CZOI method are presented in Figure 6. Wound dressings containing gentamicin demonstrated excellent antimicrobial properties over two weeks, with bacterial inhibition zones extending well beyond the dressing margin at most times (Fig. 6A–C). Interestingly, inhibition zones around dressing materials containing gentamicin remained close to constant over time and for the different drug loads. The largest CZOI were measured for the Gram-positive bacteria (S. aureus. and S. albus) and especially for S.albus. Despite having the lowest minimal inhibitory concentration (MIC) (Table 3), The Gram-negative P. aeruginosa was least inhibited, and exhibited the smallest CZOI (Fig. 6). This was not the case for ceftazidime-loaded materials, for which CZOI were found to decrease over time, and with lower drug loads. In contradistinction to gentamicin-loaded materials, ceftazidime was found to be most effective against P. aeruginosa and less effective against S. albus and S. aureus, and in good correlation with their MIC's (Table 3).
Table 3. Minimum inhibitory concentrations of antibiotics69.
| Microorganism |
MIC (μg/mL) |
|
|---|---|---|
| Gentamicin | Ceftazidime | |
|
Pseudomonas aeruginosa |
2.5 |
6.3 |
|
Staphylococcus albus |
3 |
12.5 |
| Staphylococcus aureus | 6.3 | 12.5 |

Figure 6. Histograms showing the effect of drug release on corrected zone of inhibition (CZOI) around (1% w/v) BSA loaded wound dressings (n = 3) containing 5% (w/w), 10% (w/w) and 15% (w/w) drug, as a function of pre-incubation time in PBS. (A–C) gentamicin-loaded wound dressings, (D–F) ceftazidime-loaded dressings. The bacterial strain (P. aeruginosa, S. albus and S. aureus) is indicated.69
Cell cytotoxicity
In order to complete the results of bacterial inhibition, it is also necessary to ensure that the dressing material we developed is not toxic to the cells that participate in the healing process. Previous studies have shown that dressing materials may impose a toxic effect on cells, caused by the dressing material itself, its processing, or due to the incorporation of antimicrobials.100,101 We assessed cell viability by observations of cell morphology, and by use of the Alamar-Blue assay, which is comparable to the MTT assay in measuring changes in cellular metabolic activity.102 This method involves the addition of a non-toxic fluorogenic redox indicator to the culture medium. The oxidized form of AB has a dark blue color and little intrinsic fluorescence. When taken up by cells, the dye becomes reduced and turns red. This reduced form of AB is highly fluorescent. The extent of the AB conversion, which is a reflection of cell viability, can be quantified spectrophotometrically at wavelengths of 570 and 600 nm. The AB assay is advantageous in that it does not necessitate killing the cells (as in the MTT assay), thus enabling day by day monitoring of the cell cultures. The AB assay was performed on human fibriblast cell cultures before introducing the dressing materials and then every 24 h for 3 d.
We saw no difference in the appearance of the cell cultures over the three days during which they were exposed to the dressing material devoid of antibiotics. The AB assay also shows a stable preservation of cellular viability. Thus, we are assured that the dressing material itself and its processing by freeze-drying of inverted emulsions do not inflict a toxic effect. Similar results were obtained for all the dressing materials containing antibiotics. No more than a 10% reduction in the metabolic activity of cell cultures was measured and in most cases metabolic activity even increased as the cells became more confluent (Fig. 7). These results are promising, when compared with studies reporting the similar testing of commonly used silver-based dressing materials. Burd et al. and Paddle-Leinek et al. have reported that such dressings induce a mild to severe cytotoxic effect on keratinicytes and fibroblasts grown in culture, which correlated with the silver released to the culture medium.101,103 Specifically, it was shown that commercial dressings such as Acticoat™, Aquacel® Ag and Contreet® Ag reduce fibroblast viability in culture by 70% or more. All silver dressings were shown to delay wound reepithelialization in an explant culture model, and Aquacel® Ag and Contreet® Ag were found to significantly delay reepithelialization in a mouse excisional wound model.103 These findings emphasize the superiority of the proposed new antibiotic-eluting wound dressings over dressings loaded with silver ions.

Figure 7. Histograms demonstrating changes in the viability of dermal fibroblast cultures (Alamar Blue assay) in the presence of wound dressing discs (D = 10 mm): (A) BSA-stabilized wound dressings (n = 3) containing 5% or 15% (w/w) gentamicin. (B) BSA and Span stabilized wound dressings containing 5% or 15% ceftazidime. Dressing materials devoid of antibiotics and pristine cell cultures served as control.69
In summary the microbiological studies showed that the investigated antibiotic-eluting wound dressings are highly effective against the three relevant bacterial strains. Despite severe toxicity to bacteria, the dressing material was not found to have a toxic effect on cultured fibroblasts, indicating that the new antibiotic-eluting wound dressings represent an effective and selective treatment option against bacterial infection.
In vivo study
The guinea pig is often used as a dermatological and infection model.104-107 Research on guinea pigs has included topical antibiotic treatment,108 delivery of delayed-release antibiotics109 and investigation of wound dressing materials.110,111 A deep partial skin thickness burn is an excellent wound model for the evaluation of wound healing, not only for contraction and epithelialization of the peripheral area such as in third degree burns, but also for evaluation of the recovery of skin appendages, to serve as the main source for the re-epithelization, which completes the healing process. The metabolic response to severe burn injury in guinea pigs is very similar to that of the human post-burn metabolic response.112 Furthermore, bacterial colonization and changes within the complement component of the immune system in human burn victims is analogous to guinea pigs affected by severe burns.105 Such a model was therefore used in the current study to evaluate the effectiveness of our novel composite antibiotic-eluting wound dressing. Four groups of guinea pigs were used in this study.113 After infliction of second degree burns each animal was seeded with Pseudomonas aeruginosa and then treated with the relevant treatment option, as follows:
Group 1 was treated with a neutral non-adherent dressing material (Melolin®, Smith and Nephew). Melolin® consists of three layers: a low adherent perforated film, a highly absorbent cotton/acrylic pad and a hydrophobic backing layer. According to the manufacturer, it allows for rapid drainage of wound exudate, thus reducing trauma to the healing tissue. This group is termed “melolin.”
Group 2 was treated with our composite dressing, derived from emulsion formulation containing 15% w/v PDLGA with 6:1 O:A phase ratio and 1% w/v BSA, which did not contain antibiotics. This group is termed “control.”
Group 3 was treated with a composite dressing derived from emulsion formulation containing 15% w/v PDLGA with 6:1 O:A phase ratio and 1% w/v BSA, which contained also 10% w/w gentamicin. The gentamicin release profile from this dressing demonstrated a relatively high burst release of antibiotics (68%), followed by a gradual release in a decreasing rate over time (Fig. 8A). This group is termed “fast release,” due to the provided fast gentamicn release rate

Figure 8. (A) cumulative release of gentamicin from wound dressings derived from emulsions with 10% drug contents, that were used in the animal study: (blue square) formulation based on 6:1 O:A phase ratio, stabilized with 1% (w/v) BSA (“fast release”). (Green circle) Formulation based on 12:1 O:A phase ratio, stabilized with 1% (w/v) Span 80 (“slow release”). (B) Representative photographs of wounds, ten and 14 d after treatment with the four types of wound dressings: Melolin® (group 1), “control” (group 2), “fast release” (group 3) and “slow release” (group 4).111
Group 4 was treated with a composite dressing derived from emulsion formulation containing 15% w/v PDLGA with 12:1 O:A phase ratio and 1% w/v sorbitan monooleate (Span 80), which contained also 10% w/w gentamicin. The gentamicin release from this dressing demonstrated a considerably lower burst release (4%) and a longer overall release of gentamicin, with an almost constant release rate for 4 weeks. This group is termed “slow release,” due to the provided slow gentamicn release rate (Fig. 8A).
In all studied groups the wound dressing materials remained in position over the course of treatment and were not disrupted. The dressing material created good contact with the skin, turning transparent in the exudating regions of the wound. All dressing materials used in the study were easily removed from the wound. Notable degradation of the binding matrix occurred in the regions subject to exudation, creating visible voids between the supporting fibers. This finding was supported by SEM photographs of different regions of the retrieved dressing material. The dressing's margin demonstrated negligible degradation while its center demonstrated advanced degradation. The fibrous mesh remained intact despite degradation of the binding matrix.113
Second degree burn wounds were evaluated macroscopically by two quantitative parameters ten and 14 d after infliction of the burns: (1) percentage of the original area subjected to burn injury which was still an open wound and (2) wound contraction as depicted by the total wound area (epithelialized and non-epithelialized) as a percentage of the original area subjected to burn injury. Representative photographs of wounds treated with the various dressing materials and the two endpoints are presented in Figure 8B. As demonstrated, controlled release of gentamicin had a beneficial effect on wound closure. Ten days after the infliction of burns, an 88% of re-epithelization was observed with the fast release formulation and a 95% of re-epithelization with the slow release formulation (Fig. 9A). Despite a half-fold decrease in the open wound area compared with Melolin®, the superiority of the fast-release formulation was not proven statistically. However, the non-epithelialized area under the slow-release formulation was significantly smaller than with all other formulations (p ≤ 0.05), and 88% smaller than with Meolin®. All wounds were almost fully epithelialized two weeks after the infliction of burns.
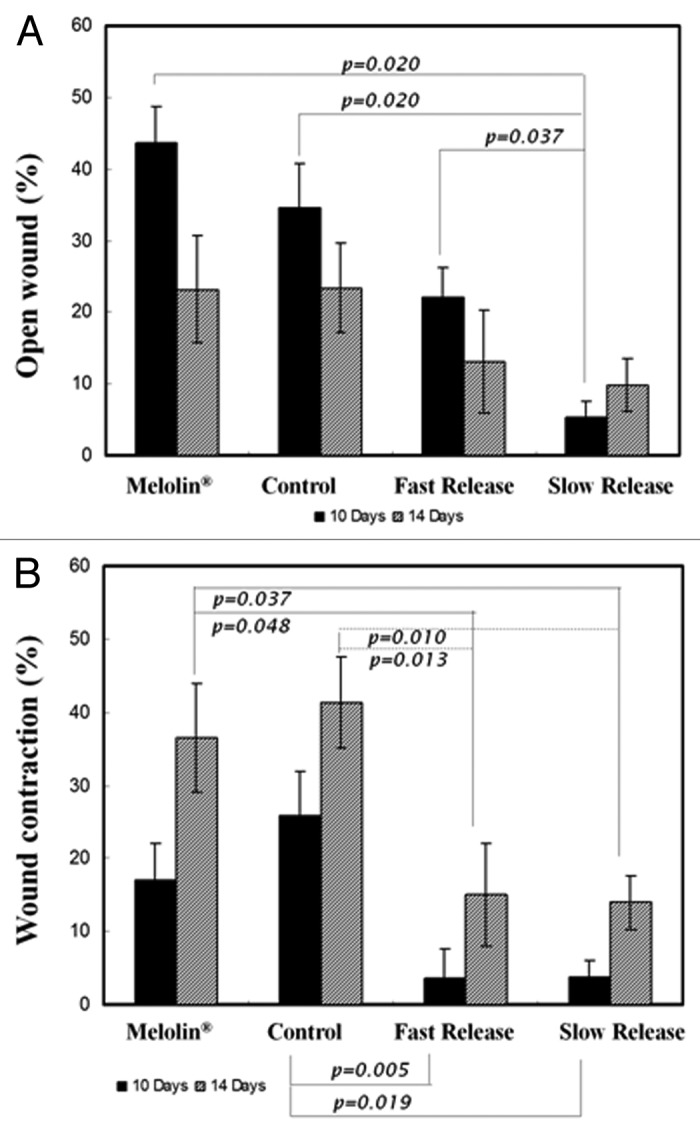
Figure 9. (A) Percentage of open wound measured at 10 and 14 d, with respect to the inflicted wound area (mean ± SEM), (B) Wound contraction as percentage of total wound area measured at 10 and 14 d, with respect to the inflicted wound area (mean ± SEM).111
Wound contraction is an ancient survival mechanism that allows animals to overcome injury and reduce the size of a wound without further treatment. However, it is an unfavorable process in humans, since it can lead to disfigurement of the skin and poor aesthetic results. It may also lead to loss of the normal flexibility of the skin- a fixed deformity that entails a functional disability, especially of the skin over the joints. Visible wound contraction is not usually evident until 5–9 d after injury, since significant fibroblast invasion into the wound area must occur before the onset of contraction. Contraction is generally enhanced when the healing process is delayed. It is therefore advisable to cause wound closure as soon as possible.
After ten days, fast and slow gentamicin-eluting dressing materials demonstrated less than 4% contraction compared with 17% and 26% contraction measured for the wounds treated with the dressing material devoid of antibiotics and Melolin®, respectively (Fig. 9B). After 14 d, wound contraction increased in wounds treated with the non-antibiotic-eluting materials (37% and 41%, respectively), while contraction in wounds treated with controlled release of gentamicin increased mildly to 15% and 14% for the fast and slow releasing formulations, respectively, which was significantly lower than with the non-antibiotic-eluting materials (p ≤ 0.05).
To summarize, in vivo evaluation of the antibiotic-eluting wound dressings in a contaminated wound demonstrated its ability to accelerate wound healing compared with an unloaded format of the wound dressing and a non-adherent dressing material (Melolin®). Wound contraction was reduced significantly, and better quality scar tissue was formed. The gold standard local treatment with topical antibacterial agents, e.g., silverol®, requires daily or twice-daily replacements of the dressing material, which are time consuming and painful to the patient. As written above, several of the dressing materials used today that provide controlled release of silver ions as an antibacterial agent have been shown to induce a toxic effect on cells, which can delay wound healing.100,101,103,114 The current dressing material shows promising results. It does not require bandage changes and offers a potentially valuable and economic approach for treating the life-threatening complication of burn-related infections.
Porous Structures with Controlled Release of Water-Insoluble Drugs
Water-insoluble drugs do not tend to diffuse out from their host polymeric structure and therefore they are released slowly in an aqueous environment and it is hard to control their release profile. Thus, when encapsulated in highly porous structures, their release profile can be more controllable, due to the relatively high surface area for diffusion. In the current study we chose to focus on controlled release of antiproliferative drugs, which are extremely hydrophobic, from our freeze-dried inverted emulsions and on the resulting biological effects. The potential applications of antiproliferative drug release are mainly coatings for drug-eluting vascular stents and local cancer treatment. However, the concept of release of water-insoluble drugs from our highly porous structures can be used also for many other biomedical applications.
Antiproliferative Drug-Eluting Core/Shell Fiber Structures
Drug-eluting fibers
Drug-eluting fibers may efficiently deliver antiproliferative drugs locally at the tumor resection site or a few cm from the tumor to help target tumor metastases. The advantages of fibers include ease of fabrication, high surface area, wide range of possible physical structures, and localized delivery of the bioactive agent to the target. Two basic types of drug-eluting fibers have been reported: monolithic fibers and reservoir fibers.115-122
• Monolithic fibers: in these systems the drug is dissolved or dispersed throughout the polymer fiber. For example: curcumin, paclitaxel and dexamethasone were melt spun with PLLA to generate drug-loaded fibers115 and aqueous drugs were solution spun with PLLA.116 Various steroid-loaded fiber systems have demonstrated the expected first order release kinetics.118,119
• Reservoir fibers: these are hollow fibers, where drugs such as dexamethasone and methotrexane were added to the internal section of the fiber post melt extrusion.120-122
The main disadvantage of monolithic fibers is poor mechanical properties, due to drug incorporation in the fiber. Furthermore, many drugs and all proteins cannot tolerate the high temperatures involved in the fabrication process of monolithic fibers. Reservoir fibers also do not exhibit good mechanical properties.
The general goal of our study was therefore to develop and investigate a novel drug-eluting bioresorbable core/shell fiber platform that will successfully serve as a basic element for medical implants. The concept of core/shell fibers is based on location of the drug molecules in a separate compartment (“shell”) around a melt spun “core” fiber (Fig. 2C). Such fiber platform is designed to combine good mechanical properties with the desired drug release profile. Preparation of the porous coating was based on the freeze-drying of water in oil (inverted) emulsions technique, described in “Introduction: Techniques for Preparation of Porous Structures for Biomedical Applications.” The shell is highly porous, designed to provide a large surface area for diffusion and thus control the antiproliferative drug release. As written above, most antiproliferative drugs are hydrophobic and are therefore released slowly in an aqueous environment. Furthermore, most antiproliferative drugs are highly cytotoxic. Therefore, maintaining the drug concentration between the effective and the toxic levels, in a single dosage, is a complex task when incorporating hydrophobic/cytotoxic drugs.
When loaded with antiproliferative agents, our new fibers are designed for two purposes. The first is use as basic elements of endovascular stents in order to mechanically support blood vessels while delivering drugs directly to the blood vessel wall for prevention of restenosis. The second application offers local treatment of cancer post tumor resection in conjunction with standard treatment.
Restenosis and stents
Restenosis (re-narrowing of the blood vessel wall) and cancer are two different pathologies that have drawn extensive research attention over the years. Antiproliferative drugs such as paclitaxel inhibit cell proliferation and are therefore effective in the treatment of cancer as well as neointimal hyperplasia, which is known to be the main cause of restenosis.
Drug-eluting stents significantly reduce the incidence of in-stent restenosis, which was once considered a major adverse outcome of percutaneous coronary stent implantations. Localized release of antiproliferative drugs interferes with the pathological proliferation of vascular smooth muscle cells (VSMC), which is the main cause of in-stent restenosis.123
Current drug-eluting biodegradable or biostable stent coatings exhibit side effects due to delayed or incomplete healing and are far from optimal in terms of controlled release of drugs within the therapeutic range. Biodegradable stents may overcome current DES endothelial related limitations and suggest a larger drug reservoir if they could provide mechanical stability along the healing period. Nevertheless, these stents cannot carry enough drug because of the trade-off between the mechanical properties and drug loading. Although both types of drug-eluting stents have long been studied, there is still no such drug release device, biodegradable or stable, that can provide controlled release of a drug within the therapeutic dosage with safe healing of the tissue. We present a new approach for the basic elements of biodegradable endovascular stents that mechanically support the blood vessels while delivering drugs for prevention of restenosis directly to the blood vessel wall. Our novel fiber systems, derived from drug-loaded emulsions, may provide targeted and controlled drug release without interfering with the mechanical properties of the device. The highly porous coating can also be applied successfully on metal stents.
Local cancer treatment
Conventional approaches to treating cancer are mainly surgical excision, irradiation and chemotherapy. In cancer therapy, surgical treatment is usually performed on patients with a resectable carcinoma. An integrated therapeutic approach, such as the addition of a delivery system loaded with an antiproliferative drug at the tumor resection site, is desirable.124,125
The concept of drug-eluting devices for cancer treatment has been studied extensively, and systems explored so far for localized antiproliferative drug delivery in cancer treatment include wafers, microspheres and fibers. However, current solutions include non-selectivity of the drug, sub-optimal control over drug release, and problems in drug incorporation. Our delicate fibers are designed to combine good strength with flexibility and can therefore be handled easily and implanted in the desired location during and post-surgery. Since these fibers are very delicate, they may also be used stereotactically, obviating the need for surgery. The main advantages of our composite drug-loaded fibers include ease of fabrication and high surface area for controlled release. Furthermore, an integrated therapeutic approach for cancer treatment may be highly advantageous and may provide high local concentrations of antiproliferative drugs at the tumor resection site in a controlled manner. This method could prevent re-growth and metastasis of tumors and may enable passage of drugs directly through the BBB, which is crucial in cases of glioblastoma, a pathology for which there is still no effective treatment.
The drugs used in the current study
Several antiproliferative drugs were examined in the current study, the mostly used were paclitaxel and Farnesylthiosalicylate. Paclitaxel is the most popular antiproliferative agent. It was originally isolated from a trace compound found in the bark of the Pacific Yew (Taxus brevifolia).126 Its anti-tumor activity was detected in 1967 by the US National Cancer Institute (NCI) and it was later found to be a promising novel antineoplastic drug. It was approved by the FDA for ovarian cancer in 1992, for advanced breast cancer in 1994 and for early stage breast cancer in 1999. Paclitaxel eventually became a standard medication in oncology.126,127 It acts to inhibit mitosis in dividing cells by binding to microtubules and causes the formation of extremely stable and non-functional microtubules. Slow release of perivascularly applied paclitaxel totally inhibits intimal hyperplasia and prevents luminal narrowing following balloon angioplasty. However, paclitaxel's narrow toxic-therapeutic window may cause side effects during therapy.127
Farnesylthiosalicylate (FTS, Salirasib) is a new, rather specific, nontoxic drug which was developed at the Tel-Aviv University.128 It acts as a Ras antagonist,129,130 which in its active form (GTP-bound) promotes enhanced cell proliferation, tumor cell resistance to drug-induced cell death, enhanced migration and invasion. Ras is therefore considered an important target for cancer therapy as well as for therapy of other proliferation diseases, including restenosis. The apparent selectivity of FTS for active (GTP-bound) Ras and the absence of toxic or adverse side effects were proven in animal models129 and in humans (Concordia Pharmaceuticals, Inc.). FTS was found to be a potent inhibitor of intimal thickening in the rat carotid artery injury model which serves as a model for restenosis, while it does not interfere with endothelial proliferation.129 The incorporation of the new drug FTS into a stent coating may overcome the incomplete healing and lack of endothelial coverage associated with current drug-eluting stents.
In the current study we investigated the effects of the inverted emulsion's parameters, i.e., polymer content, drug content, organic to aqueous (O:A) phase ratio and copolymer composition on the shell microstructure and on the release profile of both drugs, paclitaxel and FTS, from the fibers. Our results showed that the effect of the copolymer composition, i.e., the relative quantities of lactic acid and glycolic acid in the copolymer, on the drug release profile and on the shell microstructure was the most pronounced of all parameters tested. In addition, we found the optimal formulation which enabled us to obtain a relatively stable emulsion for each drug (FTS or paclitaxel), as may be inferred from the shell's bulk porous microstructure. 50/50 PDLGA and 75/25 PDLGA were chosen as host polymers due to their relatively fast degradation rate in order to be able to release the hydrophobic antiproliferative agents at an appropriate rate.71,72
Release profiles of antiproliferative drugs from core/shell structures
As written above, the dense core of our composite fibers enables obtaining the desired mechanical properties and the drug is located in a porous shell so as not to affect the mechanical properties. The shell is highly porous so as to enable release of the relatively hydrophobic antiproliferative drugs in a desired manner. In order to characterize our drug-eluting core/shell fiber platform, we studied paclitaxel and FTS release from the fibers in light of the shells' morphology and degradation and weight loss profiles. We also studied the activity of the drugs post-fabrication and then the overall effect of the system as a tumor-targeted antiproliferative release device using cancer cell lines. The trial setting measured the antiproliferative property of the fibers and is believed to predict in vivo models for local treatment of cancer and restenosis.
The diameter of the treated core fibers (i.e., without the coating) was in the range of 200–250 μm and a shell thickness of 30–70 μm was obtained. The shell’s porous structure contained round-shaped pores in all the specimens that were based on relatively stable emulsions, usually within the 2–7 μm range, with a porosity of 67–85%. The encapsulation efficiency of the studied samples was in the range of 17 and 68% for the FTS-incorporated coatings and in the range of 30% and 75% for the paclitaxel-incorporated coatings (Table 4). The structural characteristics of the shell and encapsulation efficiency values of the examined specimens are also summarized in Table 4. The 50/50 PDLGA is less hydrophobic than the 75/25 PDLGA due to its higher glycolic acid content. The interfacial tension (difference between the surface tensions of the organic and the aqueous phases) of the 50/50 PDLGA emulsion is therefore lower and the inverted emulsion is more stable. This results in a lower shell pore size of the 50/50 PDLGA for both paclitaxel and FTS-loaded fibers (Table 4).
Table 4. The structural characteristics of the shell structures loaded with antiproliferative agents and their encapsulation efficiency values133.
| Sample | Pore diameter (μm) | Porosity(%) | Encapsulation efficiency (%) |
Drug amount (μg/cm fiber) |
|---|---|---|---|---|
|
FTS-loaded samples 50/50 PDLGA 75/25 PDLGA |
2.9 ± 1.1 4.5 ± 1.3 |
84.2 ± 4.5 76.2 ± 2.3 |
56.7 ± 7.9 51.8 ± 4.3 |
0.336 ± 0.82 0.374 ± 0.57 |
|
Paclitaxel-loaded samples 50/50 PDLGA 75/25 PDLGA |
4.1 ± 1.3 6.4 ± 2.3 |
67.0 ± 6.0 69.0 ± 6.0 |
53 ± 1.2 48 ± 0.7 |
0.122 ± 0.45 0.113 ± 0.12 |
The drug release profiles from a shell based on 50/50 PDLGA and from a shell based on 75/25 PDLGA are presented in Figure 10 for fibers loaded with paclitaxel (Fig. 10A) and FTS (Fig. 10B). The degradation profiles of the two copolymers are presented in Figure 11A and their weight loss profiles are presented in Figure 11B. Paclitaxel's cumulative release exhibited the following three phases:

Figure 10. The effect of the copolymer composition on the cumulative drug release profile from core/shell fiber structures (Green triangle, 50/50 PDLGA; blue circle, 75/25 PDLGA): (A) Paclitaxel release, (B) FTS release. Plots of dMt/dt vs. sqrt (1/t) for the first 5 weeks of release (in the small frames) indicate diffusion controlled region.71,72

Figure 11. Degradation profile: (A) weight loss profile, (B) and water uptake, (C) green triangle, 50/50 PDLGA; blue circle, 75/25 PDLGA porous structures.133
(1) The first phase of release (phase a) occurred during weeks 1–8, in which the drug was released in an exponential manner, i.e., the rate of release decreased with time. Such a release profile is typical of diffusion-controlled systems. A minor initial burst release was obtained during the first day of release. Paclitaxel release from the porous shell was relatively slow for both types of host polymer, 50/50 PDLGA and 75/25 PDLGA, mainly due to paclitaxel's extremely hydrophobic nature. Moreover, the release rate decreased with time, since the drug had a progressively longer distance to pass and a lower driving force for diffusion.
(2) The second phase of release (phase b) occurred during weeks 5–20, in which the drug was released at a constant rate. The rate of paclitaxel release from the 50/50 PDLGA host polymer was significantly higher than that obtained from the 75/25 PDLGA. This difference is attributed to a difference in the degradation rate of these two copolymers, which assists the drug's diffusion. The 50/50 copolymer degrades faster than the 75/25 copolymer and therefore releases the drug at a faster rate. The degradation rate of the 50/50 PDLGA is indeed significantly higher than that of the 75/25 PDLGA, as inferred by the slope of their molecular weight profile (Fig. 11A).
(3) The third phase of release (phase c) occurred during weeks 21–38, when the porous shell structure was already destroyed due to intensive degradation. In fact, at this stage most of the shell remains are no longer attached to the core fiber and the core fiber also undergoes erosion.
Most of the encapsulated paclitaxel was released from the 50/50 PDLGA shell during phases a and b. However, in our system the highly hydrophobic paclitaxel is probably attached to the surface of the hydrophobic 75/25 PDLGA even after intensive degradation. It is clear that the paclitaxel release profile during phases a and b corresponds to the degradation profile of the porous host PDLGA shell. Intensive degradation of the host polymer is necessary in order to obtain release of the highly hydrophobic bulky paclitaxel. Overall, about 10 μg paclitaxel, corresponding to 90% of the loaded drug, was released from the 50/50 PDLGA shell, whereas about 4 μg paclitaxel, corresponding to 30% of the loaded drug, was released from the 75/25 PDLGA shell. Other investigators working on paclitaxel-eluting systems also reported its relatively slow release rate from various polymeric systems.131,132
The FTS release profiles from our fiber platform differ from the paclitaxel release profiles. They exhibited a burst effect accompanied by a release rate which decreased with time (Fig. 10B). The 50/50 PDLGA fiber released 62% of the encapsulated drug during the first day of release, whereas the 75/25 PDLGA fiber released only 30%. This difference is attributed mainly to differences in the hydrophilic/hydrophobic balance of these two copolymers. The 50/50 PDLGA copolymer contains more glycolic acid groups and fewer lactic acid groups along the polymer chain and is therefore less hydrophobic than the 75/25 PDLGA and probably exhibits higher water uptake during the initial phase of release. This enables more rapid water inflow which results in a higher burst release. Furthermore, the rate of release from the 50/50 PDLGA formulation is slightly higher than the rate obtained with the 75/25 PDLGA formulation and after two weeks of degradation the 50/50 formulation released 100% of the drug, whereas the 75/25 formulation released only 79%. Both polymers exhibited a small weight loss of less than 10% during the first 3 weeks of degradation, whereas after 3 weeks of degradation the 50/50 PDLGA exhibited a fast weight loss while the 75/25 PDLGA did not erode during the measured time period (Fig. 11B), as expected. These results indicate that most of the FTS is released from our porous coatings before they undergo massive weight loss.
The changes in the shell microstructures of both copolymers during exposure to the aqueous medium are presented in Figure 12A (50/50 PDLGA) and Figure 11B (75/25 PDLGA). The starting point (day 0) shows highly porous delicate structures with round pores for both samples. The pore size of the 50/50 PDLGA shell (Fig. 12A) is significantly smaller than that of the 75/25 PDLGA (Fig. 12B) and its porosity is higher (Table 4). Furthermore, the 50/50 PDLGA microstructure is highly interconnected compared with the 75/25 PDLGA, which enables more surface area for diffusion. The 50/50 PDLGA shell exhibited a rougher structure after seven days of degradation in the aqueous medium (Fig. 12A), whereas after 14 d of degradation it exhibited a completely dense (non-porous) structure (Fig. 12A). The 75/25 PDLGA also underwent a similar change in microstructure (Fig. 12B). However, in this case the entire process was slower and took approximately 126 d, due to the more hydrophobic nature of the copolymer, which is rich in lactic acid. The changes in microstructure are caused by early swelling and water uptake and subsequent degradation and erosion.

Figure 12. SEM fractographs of shell structures showing their microstructural changes with time: (A) 50/50 PDLGA at days 0, 7, 14, 28, (B) 75/25 PDLGA at days 0, 7, 14, 28, 56 and 126.133
The water uptake measurements indicate an increase of 40% in the 50/50 PDLGA's weight during the first 24 h, after which it stabilizes, whereas the 75/25 PDLGA exhibited a slower water uptake which lasts for 200 h (Fig. 11C). This further supports our hypothesis that the early structural changes are due to water uptake rather than degradation or erosion. Since the FTS diffusion through the fiber's shell occurs in an aqueous swollen phase, a relatively high water uptake, such as that of our 50/50 PDLGA porous shell, enables faster diffusion of the relatively small FTS molecules.133
It can therefore be concluded that higher glycolic acid content in the copolymer, i.e., a less hydrophobic copolymer, enables a greater initial surface area for diffusion and higher FTS release from the fibers mainly due to early swelling. Higher water uptake affects the microstructure and results in a higher burst release and a higher degradation rate of the host polymer, which assists diffusion. The contribution of the swelling and the resulting microstructural effects are more significant for the FTS-eluting systems than for the paclitaxel-eluting systems.
It is important to note that our drug delivery, water uptake and degradation results clearly show that although antiproliferative drugs are highly hydrophobic, their release profiles from biodegradable polymers can be totally different. Some drugs, such as paclitaxel, can be totally released only after intensive degradation of the host polymer, and other drugs, such as FTS, can be partially released even as a result of some water uptake, a short time after being immersed in an aqueous medium. In order to further investigate these systems, molecular simulations were performed so as to evaluate the physical properties, such as the solubility parameter, chemical structure, and molecular area of the investigated drugs.
The Hildebrand solubility parameter, δ,134 is defined as the square root of the cohesive energy density. Materials with similar δ values are likely to be miscible. The calculated solubility parameters of paclitaxel and FTS, using the discover simulation software, are 21.15 (J/cm3)1/2 and 21.06 (J/cm3)1/2, respectively. I.e., they are almost the same (Table 5). The molecular weight, calculated van der Waals volume and molecular areas of these drugs are also presented in Table 5. The 3D images of these drugs are presented in Figure 13. It should be noted that paclitaxel exhibit relatively high volumes (754 mμ3) and high molecular area (744 mμ2), while FTS exhibit much lower volume (365 mμ3) and lower molecular area (415 mμ2). Furthermore, paclitaxel has spherical and complex molecular shape while FTS exhibits a simpler straight linear shape (Fig. 13). The complex shape and large size of paclitaxel probably substantially reduce the diffusion coefficient of the drug molecules, since they lower the molecular mobility of the drug and thus delay its release. Additional water insoluble drugs in our study also show that small narrow drug molecules can diffuse out of the porous structure when some water uptake occurs, while bulky drug molecules need intensive degradation of the host polymer in order to diffuse out. This phenomenon can be attributed to the fact that the hydrophobic drug molecules are probably located in the polymeric domains of the porous structure rather than in the pores.135 Otherwise they would be released much faster from the partially connected porous structure.
Table 5. Physical properties of the hydrophobic drugs Paclitaxel and FTS135.
| Drug | Molecular weight (Da) | Solubility parameter (δ)(J/cm3) 0.5 | Van der Walls volume (μm3) | Molecular area (μm2) |
|---|---|---|---|---|
| Paclitaxel |
853.9 |
21.15 |
754 |
744 |
| FTS | 358.5 | 21.06 | 365 | 415 |
Figure 13. The 3D structures of sirolimus and paclitaxel. The various atoms are presented by colors as follows: H, white; C, gray; O, red; N, blue; S, yellow.135
In conclusion, highly porous structures for controlled release of hydrophobic drugs such as antiproliferative agents were developed and studied. The effects of both the polymer and the drug structure and physical properties on the drug release profile were studied through a combination of in vitro results and molecular simulations. We conclude that both drug and polymer chain structures strongly affect the release profile of hydrophobic drugs from relatively hydrophobic host polymers. The chemical structure of the polymer chain directly affects the drug release profile through water uptake in the early stages or degradation and erosion in later stages. It also affects the release profile indirectly through the polymer's 3D porous structure. However, this effect is minor. The hydrophobic drug molecules are probably located in the polymeric domains of the 3D structure, rather than in the pores. The drug volume and molecular area have a dominant effect on the drug's diffusion rate from the 3D polymeric porous structure.
Biological performance of the drug-eluting core/shell fibers
The fabrication process of a drug-release device may affect the activity of the incorporated drug. We examined the activity of post-fabrication paclitaxel and FTS on cell growth. Both drugs were extracted from the matrices and were then used in cell culture experiments, in which we compared the effect of the extracted drug with that of the control drug which was not exposed to any fabrication process. Drug activities were tested on three different cancer cell lines: H-Ras-transformed Rat-1 fibroblasts (EJ), U87 glioblastoma, and A549 lung cancer cells, all of which were previously shown to be sensitive to FTS.136-139 The cells were incubated with FTS (25, 50 or 100 nM) or with a single dose of paclitaxel (50 nM) or with the vehicle control. The FTS-treated and paclitaxel-treated cells were counted after four days and one day, respectively. Figure 14 shows that post-fabrication paclitaxel and control paclitaxel caused a marked decrease in the number of EJ, A549 and U87 cells relative to the control (60–90% decrease). The decrease in cell number was attributed to cell death, in line with the known cytotoxic action of paclitaxel. Importantly, there was no difference between the extent of activity of post-fabrication paclitaxel and control paclitaxel (p > 0.8, Fig. 14), indicating that the fabrication process did not affect paclitaxel's pharmacological activity.
Figure 14. Both control paclitaxel and post-fabrication paclitaxel induce a significant decrease in the cell count. Cells (EJ, A549 or U87) were plated at a density of 10 × 103 cells/well in a 24-well plate in tetraplicate (n = 2). One day after plating, 0.1% DMSO (control), 50 μM control paclitaxel (black columns), or 50 μM post-fabrication paclitaxel (white columns) were added for 24 h. The cells were then counted using a hemocytometer (*p < 0.01 compared with the control well). Paclitaxel's effect on the above-mentioned cell lines is presented in terms of mean cell viability ± standard deviation.133
We showed that post-fabrication FTS extracted from the core/shell fiber is as active as control FTS (Fig. 14). However, these studies did not examine the impact of FTS that elutes from the fibers directly onto the cells as would be the case in vivo. Moreover, we could not tell whether the amount of drug incorporated into the fiber is high enough to inhibit cell growth. In order to provide answers to these questions, we performed a set of experiments in which cells were directly exposed to the FTS-loaded core/shell fibers. Both slow and fast release fibers (Fig. 10) were used to allow a relatively slow and fast accumulation of FTS in the wells. A relatively slow release rate was obtained with shells based on 75/25 PDLGA, while a relatively fast FTS release rate was obtained with shells based on 50/50 PDLGA. We used U87, A549, and EJ cells in these experiments in order to document inhibition of cell growth and induction of cell death. Cells were imaged at various time points after being exposed to control (no drug) or FTS-eluting fibers. We chose not to test paclitaxel-eluting fibers because they are well-documented in the literature. Furthermore, paclitaxel’s cytotoxic mechanism of action was not damaged during fabrication, and the effect of the paclitaxel fibers is therefore predictable. Our results indicate that FTS-eluting composite fibers can effectively induce growth inhibition or cell death by a gradient effect and a dose-dependent manner (Fig. 15). We concluded that the combined effect of the targeted mechanism of FTS as a Ras inhibitor together with the localized and controlled release characteristics of the fiber is an advantageous antiproliferative quality.

Figure 15. FTS loaded core/shell fiber structures inhibit growth or induce cell death of glioblastoma cells by a gradient effect and dose-dependent manner. U87 cells were plated at a density of 8x103 cells/well in a 24-well plate in tetraplicate (n = 2). One day after plating, control fibers (not loaded with FTS), slow or fast FTS release fibers were added to each well (2 fibers, each with a length of 1 cm, as described in the materials and methods). (A) Images taken after 5 d of incubation with FTS fibers, in locations which are near and distant to the fiber (magnification × 100). (B) A single well edge to edge panoramic view shows a gradiential increase in the cell concentration with the increase in the distance from a slow release fiber (magnification × 100, cells were counted using an image analysis software). (C) Images were taken after 7 d of incubation with FTS fibers; the control fiber well presents high cell viability while the well containing the fast FTS fiber exhibited cell death (magnification × 100). Note that the dark shape is the actual fiber.133
Porous Structures with Protein Controlled Release
Tissue engineering is described as “an interdisciplinary field that applies the principles of engineering and life sciences towards the development of biological substitutes that restore, maintain, or improve tissue function or a whole organ.”140 One of the major approaches in tissue engineering is scaffolds that elute bioactive agents. Upon implantation of such scaffolds, cells from the body are recruited to the site, thus enabling tissue formation.141 Growth factors are essential for promoting cell proliferation and differentiation. However, direct administration of growth factors is problematic, due to their poor in vivo stability.142,143 It is therefore necessary to develop scaffolds with controlled delivery of bioactive agents that can achieve prolonged availability as well as protection of these bioactive agents, which may otherwise undergo rapid proteolysis.144,145 The main obstacle to successful incorporation and delivery of small molecules as well as proteins from scaffolds is their inactivation during the process of scaffold manufacture due to exposure to high temperatures or harsh chemical environments. Methods that minimize protein inactivation must therefore be developed. Three approaches to protein (growth factor) incorporation into bioresorbable scaffolds have recently been presented: (1) adsorption onto the surface of the scaffold,146 (2) composite scaffold/microsphere structures145,147 and (3) freeze-drying of inverted emulsions. The latter method, which was developed and studied by us is described in details in the current article. Sensitive bioactive agents, such as proteins, are incorporated in the aqueous phase of the inverted emuleion, and this prevents their exposure to harsh organic solvents and enables the preservation of their activity.
Effect of the emulsion parameters and host polymer
In the current study we investigated highly porous film scaffolds, such as the one presented in Figure 2A, was produced using the freeze-drying of inverted emulsions technique. Horseradish peroxidase (HRP) is a relatively inexpensive enzyme and was chosen as a model protein since it is very sensitive to solvents and elevated temperatures. Thus, if proteins such as HRP can be incorporated in the films without losing their activity, these films can be loaded with growth factors and can be used for building scaffolds for tissue regeneration applications. Proteins such as HRP, which contain defined hydrophobic/hydrophilic regions and an electrostatic charge,148 have a natural tendency to adsorb to the organic/aqueous interface. Proteins thus act similarly to block-co-polymer surfactants, which are widely used as emulsifiers.148,149 Our model protein HRP thus acts as a surfactant and our inverted emulsions are relatively stable in the range of formulation parameters used in this study.
In our study we first investigated the effects of the inverted emulsion's formulation parameters, i.e., HRP content, polymer content and organic:aqueous (O:A) phase ratio, and also the host polymer's parameters, i.e., copolymer composition and initial molecular weight. This part of the study actually enabled elucidation of the process-structure-release profile effects of our unique protein-eluting systems. The second phase of the study focused on the combined effect of at least two formulation/polymer parameters on the film's microstructure and on the resulting HRP release profile. An emulsion formulation containing 17.5% w/v 50/50 PDLGA (i.v. = 83 KDa) in the organic solution, 1% w/w HRP in the aqueous medium (relative to the polymer load), and an organic to aqueous (O:A) phase ratio of 4:1 v/v was used as the reference formulation. Most films exhibited a HRP release profile of an initial burst release accompanied by a decreased release rate with time.67 A dual pore size population is characteristic of most films, with large 12–18 μm pores and small 1.5–7 μm pores, and porosity in the range of 76–92%.
An increase in the polymer content and its initial molecular weight, O:A phase ratio and lactic acid content, or a decrease in the HRP content, all resulted in a decreased burst effect and a more moderate release profile (Figs. 16 and 17). The HRP content significantly affected the HRP release profile, through the driving force for diffusion. A decrease in the burst release and continuous release rate can also be achieved through the higher hydrophobic nature of the host polymer, i.e., by increasing the initial MW of the host polymer and its content in the organic phase of the emulsion. The former exhibited greater effectiveness than the latter. The O:A phase ratio and copolymer composition only slightly affected the release profile. An increase in the polymer content and initial MW resulted in a smaller diameter of the small pores, due to the emulsion's higher viscosity and shear forces.

Figure 16. In vitro release of HRP from the porous scaffolds demonstrating the effect of a change in the emulsion's formulation parameters compared with the reference formulation: (A) effect of HRP content: green diamond, 2% w/w; blue square, 1% w/w; red triangle, 0.5% w/w. (B) Effect of polymer content: yellow circle, 15% w/v; blue square, 17.5 w/v; empty green triangle, 25% w/v. (C) effect of O:A phase ratio: white square, 2:1, empty blue diamond, 6:1, x - 8:1.67

Figure 17. In vitro release of HRP from the porous scaffolds demonstrating the effect of a change in the host polymer compared with the reference sample: (A) effect of the copolymer composition: blue square, 50/50 PDLGA, red triangle, 75/25 PDLGA. (B) Effect of the initial molecular weight of the host polymer: green diamond, 50 KDa; blue square, 83 KDa; black circle, 185 KDa.67
Combined effect of parameters
As shown here, the HRP release profile from the studied series exhibited a medium-high burst release accompanied by relatively high release rates, and most of the encapsulated HRP was released within 3 weeks. Such profiles can be suitable for various biomedical applications. However, in certain cases relatively low burst effects and lower release rates are needed. Based on this first stage of research, we investigated the combined effects of changes in two or three parameters on the HRP release profile and on the film's microstructure. The formulation parameters and structural features of the studied samples are presented in Table 6 and their HRP release results are presented in Figure 18, compared with the reference sample.
Table 6. Structural features of HRP-eluting films with a change in 2 or 3 parameters (compared with the reference sample)67.
| Process parameters | Mean large pore diameter (μm) | Mean small pore diameter (μm) | Porosity |
|---|---|---|---|
| Reference sample 1% w/w HRP 50/50 PDLGA MW = 83 KDa 17.5% w/v polymer, O:A = 4:1 |
17.1 ± 2.7 |
4.3 ± 0.8 |
86.9 ± 2.6 |
| * Sample A 0.5% w/w HRP 25% w/v polymer |
12.5 ± 1.8 |
2.1 ± 0.6 |
81.0 ± 1.7 |
| * Sample B 0.5% w/w HRP MW = 185 KDa |
18.6 ± 3.4 |
2.8 ± 0.8 |
82.9 ± 1.1 |
| * Sample C 0.5% w/w HRP O:A = 8:1 |
16.4 ± 2.2 |
3.6 ± 1.5 |
85.1 ± 1.3 |
| * Sample D 0.5% w/w HRP 25% w/v polymer O:A = 8:1 |
12.4 ± 2.0 | 1.8 ± 0.7 | 76.9 ± 3.3 |
Only the parameters that are different from those of the reference sample are indicated.

Figure 18. In vitro release of HRP from the porous scaffolds demonstrating the effect of combined changes in the emulsion's formulation parameters compared with the reference formulation (blue square, 17.5% w/v polymer, i.v. = 83 KDa, 1% w/w HRP, O:A = 4:1). (A) Combined effect of a change in 2 parameters, 0.5% w/w HRP and: green diamond, 25% w/v polymer; black circle, 185 KDa; red triangle, O:A = 8:1. (B) White circle, combined effect of a change in three parameters (0.5% w/w HRP, 25% w/v polymer, O:A = 8:1). (C) X, composite film composed of a porous inner 50/50 PDLGA film (25% w/v polymer, MW = 83 KDa, O:A = 8:1 and 0.5% w/w HRP) and two external PDLLA layers (17.5% w/v polymer, MW = 80 KDa, O:A = 2:1, no HRP).67
All three studied films (samples A, B and C) with a simultaneous change in two parameters exhibited a relatively low burst release of 15–20% and a more moderate continuous release profile, with a release rate which decreased with time (Fig. 18A). All three release profiles are similar and approximately 90% of the encapsulated drug was released within 4 weeks. Also, it appears that the effect of a simultaneous change in two parameters resulted in a decrease in the burst release (from 57% to 15–20%), which is more effective than the “sum of the separate effects.” It is therefore suggested that these parameters have a synergistic effect on the release profile.
An additional sample (sample D), with a simultaneous change in three parameters: 0.5% w/w HRP, 25% w/v polymer and O:A = 8:1, was also studied. The HRP release profile from this film compared with that of the reference film is presented in Figure 18B. The HRP release profile from this film is similar to that obtained for the film after a change in two parameters (Fig. 18A), i.e., burst release of 14% and a release rate which decreases with time, but at a slower rate than that obtained for the film after a change in two parameters. After four weeks of incubation this film released 73% of the encapsulated HRP. We suggest that the changes in the HRP burst release and continuous profile are attributed mainly to differences in the driving force for diffusion (which depend on the HRP content), hydrophilicity/hydrophobicity of the host polymer and its water uptake during the first period of exposure to the aqueous medium. In our systems the latter are determined mainly by the initial MW of the host polymer and its content in the organic phase of the inverted emulsion.
Composite film with “sandwich” structure
At this advanced stage of research we also developed and studied a composite film. The rationale for developing this film was to combine desired release profile with cell growth into the scaffold. Since cell growth requires relatively large pores, which will probably not enable desired HRP release profile, it was clear that a film which contains at least two different layers should be developed. Our prototype composed three layers: a porous 50/50 PDLGA film with two PDLLA layers (on both sides). The parameters of the 50/50 PDLGA inner layer are: 25% w/v polymer, MW = 83 KDa, O:A = 8:1 and 0.5% w/w HRP. The parameters of the PDLLA outer layers are: 17.5% w/v polymer, MW = 80 KDa and O:A = 2:1. The outer film layers contained pores of approximately 100 μm and did not contain HRP. A unique HRP release profile was obtained for this composite film, which exhibited a very low burst release (approximately 8%) and a constant release rate of 65% of the encapsulated HRP during the four weeks of the in vitro study (Fig. 18C). It can be assumed that total release would be achieved after 6 weeks of incubation. The outer layers served as barriers for HRP release which decreased the release rate and enabled a constant rate of release. Furthermore, the relatively large pore size of these layers may be suitable for tissue growth. Our new platform for composite films thus has a high potential for use in tissue regeneration applications.
We have demonstrated that appropriate selection of the formulation's parameters can yield unique highly porous films with adjustable protein release behavior which can serve as scaffolds for bioactive agents in tissue regeneration applications.
Conclusion
In the current study we developed a special technique termed freeze drying of inverted emulsions, and studied the effects of process and formulation parameters on the obtained microstructure and on the resulting drug release profile and other properties that are relevant for the application. The inverted emulsions used in our study are prepared by homogenization of two immiscible phases: an organic solution containing a known amount of poly (dl-lactic-co-glycolic acid) (PDLGA) in chloroform, and an aqueous phase containing, double-distilled water. Water soluble drugs and proteins are incorporated in the aqueous phase while water insoluble drugs are incorporated in the organic phase. We investigated the three types of systems.
According to our study, a qualitative model describing the formulation→structure→drug release profile effects in our porous drug-eluting structures, prepared from freeze-dried inverted emulsions, can be summarized as follows (Fig. 19): there are two routes by which the emulsion's formulation affects the drug-release profile, direct and indirect.

Figure 19. Schematic representation of a qualitative model describing the drug release mechanisms from the porous drug eluting structures derived from freeze-dried emulsions.
Direct route
The emulsion formulation (especially the host polymer) affects the water uptake and swelling of the structure and therefore also the burst release of the drug molecules. This route is the main one for release of water insoluble drugs, such as the antiproliferative agents paclitaxel and FTS. When the water-insoluble drug is relatively small and narrow such as FTS, its diffusion through the polymeric structure is possible at early swelling stage. In such cases degradation of the host polymer may also affect the release rate, at a later stage. When a relatively big and extremely hydrophobic drug such as paclitaxel is incorporated into the porous structure, its diffusion through the host polymer is much slower and massive degradation and erosion of the host polymer must occur in order to enable it.
Indirect route
The effect of the emulsion formulation on the microstructure occurs also via an emulsion stability mechanism. The emulsion stability determines the surface area for diffusion through the microstructure, e.g., the surface area increases when porosity is high and pore size is low. These affect both the burst release and later release. This route is the main one for release of water-soluble drugs, such as the antibiotics ceftazidime and gentamicin, used in our study.
This model explains why the most important parameter which affects the release behavior of water-insoluble drugs is the copolymer composition (Lactic acid: glycolic acid). It affects the water uptake and swelling and therefore the FTS release profile (early mechanism). The copolymer composition affects the degradation rate of the polymer and therefore also the paclitaxel release profile (late mechanism). Hence, the copolymer composition plays a very important role in the drug release profile of water-insoluble drugs through the direct route. The other formulation parameters (O:A phase ratio, polymer and drug contents, and initial MW) exhibit a smaller effect on the water uptake and degradation rate of the host polymer, they only slightly affect the microstructure through emulsion stability. Therefore they almost do not affect the release profile of the water-insoluble drugs from our porous structures.
In contradistinction, our study shows that the release profile of water-soluble drugs is affected by most of the emulsion's formulation parameters, due to their effect on the emulsion's stability. The initial MW of the host polymer exhibited the most significant effect on the release profile of the antibiotic drugs and on the release profile of the protein (HRP), due to its effect on the porous microstructure, through the emulsion's viscosity and shear stresses. The polymer content also affected the release profile of the water-soluble drugs due to same phenomena. The O:A phase ratio, copolymer composition and drug content only slightly affected the release profile, due to their small effect on the emulsion's stability and microstructure of the resulting solid porous structure. The protein (HRP) release is a special case, where the drug acts as a surfactant which stabilizes the inverted emulsion and therefore its content affected the release profile through this indirect route. It is important to note that a stronger effect on the protein release profile can be achieved when two or three emulsion's parameters are changed. Using the “combined effect” phenomenon enabled us to reduce the protein burst release and release rate and thus, obtain release profiles that are beneficial for tissue engineering applications.
There are numerous medical applications for our freeze-dried drug-eluting structures. For example: porous films, fibers, or composite structures loaded with water-soluble drugs, such as antibiotics, can be used for wound dressing applications, treatment of periodontal diseases, meshes for Hernia repair, as well as coatings for fracture fixation devices. Fibers loaded with water insoluble drugs such as antiproliferative agents can be used as basic elements of drug-eluting stents and also for local cancer treatment. Films and fibers loaded with growth factors can be used as basic elements of highly porous scaffolds for tissue regeneration. We showed here that appropriate selection of the formulation parameters enables to obtain desired controllable release profile of any bioactive agent, water-soluble or water-insoluble, that fits the application. Desired physical properties can also be obtained through structuring effects.
Acknowledgments
The authors are grateful to the Israel Science Foundation (ISF, grant no. 1312/07) and to the Israel Ministry of Health (grant no. 3-3943) for supporting this research. We would like to thank Concordia Pharmaceuticals for kindly providing us with farnesylthiosalicylate (FTS, Salirasib).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/biomatter/article/22838
References
- 1.Yang J, Motlagh D, Webb AR, Ameer GA. Novel biphasic elastomeric scaffold for small-diameter blood vessel tissue engineering. Tissue Eng. 2005;11:1876–86. doi: 10.1089/ten.2005.11.1876. [DOI] [PubMed] [Google Scholar]
- 2.Buttafoco L, Engbers-Buijtenhuijs P, Poot AA, Dijkstra PJ, Daamen WF, van Kuppevelt TH, et al. First steps towards tissue engineering of small-diameter blood vessels: preparation of flat scaffolds of collagen and elastin by means of freeze drying. J Biomed Mater Res B Appl Biomater. 2006;77:357–68. doi: 10.1002/jbm.b.30444. [DOI] [PubMed] [Google Scholar]
- 3.Adekogbe I, Ghanem A. Fabrication and characterization of DTBP-crosslinked chitosan scaffolds for skin tissue engineering. Biomaterials. 2005;26:7241–50. doi: 10.1016/j.biomaterials.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 4.Powell HM, Boyce ST. Fiber density of electrospun gelatin scaffolds regulates morphogenesis of dermal-epidermal skin substitutes. J Biomed Mater Res A. 2008;84:1078–86. doi: 10.1002/jbm.a.31498. [DOI] [PubMed] [Google Scholar]
- 5.Ryan GE, Pandit AS, Apatsidis DP. Porous titanium scaffolds fabricated using a rapid prototyping and powder metallurgy technique. Biomaterials. 2008;29:3625–35. doi: 10.1016/j.biomaterials.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 6.Lin CY, Kikuchi N, Hollister SJ. A novel method for biomaterial scaffold internal architecture design to match bone elastic properties with desired porosity. J Biomech. 2004;37:623–36. doi: 10.1016/j.jbiomech.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 7.Woodfield TB, Van Blitterswijk CA, De Wijn J, Sims TJ, Hollander AP, Riesle J. Polymer scaffolds fabricated with pore-size gradients as a model for studying the zonal organization within tissue-engineered cartilage constructs. Tissue Eng. 2005;11:1297–311. doi: 10.1089/ten.2005.11.1297. [DOI] [PubMed] [Google Scholar]
- 8.Woodfield TB, Malda J, de Wijn J, Péters F, Riesle J, van Blitterswijk CA. Design of porous scaffolds for cartilage tissue engineering using a three-dimensional fiber-deposition technique. Biomaterials. 2004;25:4149–61. doi: 10.1016/j.biomaterials.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 9.Bottino MC, Thomas V, Schmidt G, Vohra YK, Chu TM, Kowolik MJ, et al. Recent advances in the development of GTR/GBR membranes for periodontal regeneration--a materials perspective. Dent Mater. 2012;28:703–21. doi: 10.1016/j.dental.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 10.Elsner JJ, Zilberman M. Antibiotic-eluting bioresorbable composite fibers for wound healing applications: microstructure, drug delivery and mechanical properties. Acta Biomater. 2009;5:2872–83. doi: 10.1016/j.actbio.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Stevens MM, George JH. Exploring and engineering the cell surface interface. Science. 2005;310:1135–8. doi: 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- 12.Théry M. Micropatterning as a tool to decipher cell morphogenesis and functions. J Cell Sci. 2010;123:4201–13. doi: 10.1242/jcs.075150. [DOI] [PubMed] [Google Scholar]
- 13.Reilly GC, Engler AJ. Intrinsic extracellular matrix properties regulate stem cell differentiation. J Biomech. 2010;43:55–62. doi: 10.1016/j.jbiomech.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Muschler GF, Nakamoto C, Griffith LG. Engineering principles of clinical cell-based tissue engineering. J Bone Joint Surg Am. 2004;86-A:1541–58. doi: 10.2106/00004623-200407000-00029. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh K, Ingber DE. Micromechanical control of cell and tissue development: implications for tissue engineering. Adv Drug Deliv Rev. 2007;59:1306–18. doi: 10.1016/j.addr.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Yannas IV. Tissue regeneration by use of collagen-glycosaminoglycan copolymers. Clin Mater. 1992;9:179–87. doi: 10.1016/0267-6605(92)90098-E. [DOI] [PubMed] [Google Scholar]
- 17.Jansen JA, von Recum AF. Textured and porous materials. In, Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, eds. UK: Biomaterials science: an introduction to materials and medicine. London UK: Elsevier Academic Press, 2004: 218-225. [Google Scholar]
- 18.Karageorgiou V, Kaplan D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials. 2005;26:5474–91. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Chevalier E, Chulia D, Pouget C, Viana M. Fabrication of porous substrates: a review of processes using pore forming agents in the biomaterial field. J Pharm Sci. 2008;97:1135–54. doi: 10.1002/jps.21059. [DOI] [PubMed] [Google Scholar]
- 20.de Groot JH, Nijenhuis AJ, Bruin P, Pennings AJ, Veth RPH, Klompmaker J. Use of porous biodegradable polymer implants in meniscus reconstruction. 1) Preparation of porous biodegradable polyurethanes for the reconstruction of meniscus lesions. Colloid Polym Sci. 1990;268:1073–81. doi: 10.1007/BF01410672. [DOI] [Google Scholar]
- 21.Reignier J, Huneault MA. Preparation of interconnected poly(ε-caprolactone) porous scaffolds by a combination of polymer and salt particulate leaching. Polymer (Guildf) 2006;47:4703–17. doi: 10.1016/j.polymer.2006.04.029. [DOI] [Google Scholar]
- 22.Wei G, Ma PX. Macroporous and nanofibrous polymer scaffolds and polymer/bone-like apatite composite scaffolds generated by sugar spheres. J Biomed Mater Res A. 2006;78:306–15. doi: 10.1002/jbm.a.30704. [DOI] [PubMed] [Google Scholar]
- 23.Lee M, Wu BM, Dunn JC. Effect of scaffold architecture and pore size on smooth muscle cell growth. J Biomed Mater Res A. 2008;87:1010–6. doi: 10.1002/jbm.a.31816. [DOI] [PubMed] [Google Scholar]
- 24.Vaquette C, Frochot C, Rahouadj R, Wang X. An innovative method to obtain porous PLLA scaffolds with highly spherical and interconnected pores. J Biomed Mater Res B Appl Biomater. 2008;86:9–17. doi: 10.1002/jbm.b.30982. [DOI] [PubMed] [Google Scholar]
- 25.Mikos AG, Thorsen AJ, Czerwonka LA, Bao Y, Langer R, Winslow DN, et al. Preparation and characterization of poly(L-lactic acid) foams. Polymer (Guildf) 1994;35:1068–77. doi: 10.1016/0032-3861(94)90953-9. [DOI] [Google Scholar]
- 26.Uchida A, Nade SM, McCartney ER, Ching W. The use of ceramics for bone replacement. A comparative study of three different porous ceramics. J Bone Joint Surg Br. 1984;66:269–75. doi: 10.1302/0301-620X.66B2.6323483. [DOI] [PubMed] [Google Scholar]
- 27.Mooney DJ, Baldwin DF, Suh NP, Vacanti JP, Langer R. Novel approach to fabricate porous sponges of poly(D,L-lactic-co-glycolic acid) without the use of organic solvents. Biomaterials. 1996;17:1417–22. doi: 10.1016/0142-9612(96)87284-X. l. [DOI] [PubMed] [Google Scholar]
- 28.Nam YS, Yoon JJ, Park TG. A novel fabrication method of macroporous biodegradable polymer scaffolds using gas foaming salt as a porogen additive. J Biomed Mater Res. 2000;53:1–7. doi: 10.1002/(SICI)1097-4636(2000)53:1<1::AID-JBM1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 29.Lo H, Ponticiello MS, Leong KW. Fabrication of controlled release biodegradable foams by phase separation. Tissue Eng. 1995;1:15–28. doi: 10.1089/ten.1995.1.15. [DOI] [PubMed] [Google Scholar]
- 30.Whang K, Thomas CH, Healy KE. A novel method to fabricate bioabsorbable scaffolds. Polymer (Guildf) 1995;36:837–42. doi: 10.1016/0032-3861(95)93115-3. [DOI] [Google Scholar]
- 31.Guan J, Stankus JJ, Wagner WR. Biodegradable elastomeric scaffolds with basic fibroblast growth factor release. J Control Release. 2007;120:70–8. doi: 10.1016/j.jconrel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pant HR, Neupane MP, Pant B, Panthi G, Oh HJ, Lee MH, et al. Fabrication of highly porous poly (ɛ-caprolactone) fibers for novel tissue scaffold via water-bath electrospinning. Colloids Surf B Biointerfaces. 2011;88:587–92. doi: 10.1016/j.colsurfb.2011.07.045. [DOI] [PubMed] [Google Scholar]
- 33.Elsner JJ, Zilberman M. Antibiotic-eluting bioresorbable composite fibers for wound healing applications: microstructure, drug delivery and mechanical properties. Acta Biomater. 2009;5:2872–83. doi: 10.1016/j.actbio.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Goldman M, McCollum CN, Hawker RJ, Drolc Z, Slaney G. Dacron arterial grafts: the influence of porosity, velour, and maturity on thrombogenicity. Surgery. 1982;92:947–52. [PubMed] [Google Scholar]
- 35.Valonen PK, Moutos FT, Kusanagi A, Moretti MG, Diekman BO, Welter JF, et al. In vitro generation of mechanically functional cartilage grafts based on adult human stem cells and 3D-woven poly(epsilon-caprolactone) scaffolds. Biomaterials. 2010;31:2193–200. doi: 10.1016/j.biomaterials.2009.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derwin KA, Codsi MJ, Milks RA, Baker AR, McCarron JA, Iannotti JP. Rotator cuff repair augmentation in a canine model with use of a woven poly-L-lactide device. J Bone Joint Surg Am. 2009;91:1159–71. doi: 10.2106/JBJS.H.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zilla P, Moodley L, Wolf MF, Bezuidenhout D, Sirry MS, Rafiee N, et al. Knitted nitinol represents a new generation of constrictive external vein graft meshes. J Vasc Surg. 2011;54:1439–50. doi: 10.1016/j.jvs.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 38.Van Lieshout M, Peters G, Rutten M, Baaijens FA. A knitted, fibrin-covered polycaprolactone scaffold for tissue engineering of the aortic valve. Tissue Eng. 2006;12:481–7. doi: 10.1089/ten.2006.12.481. [DOI] [PubMed] [Google Scholar]
- 39.Tatekawa Y, Kawazoe N, Chen G, Shirasaki Y, Komuro H, Kaneko M. Tracheal defect repair using a PLGA-collagen hybrid scaffold reinforced by a copolymer stent with bFGF-impregnated gelatin hydrogel. Pediatr Surg Int. 2010;26:575–80. doi: 10.1007/s00383-010-2609-2. [DOI] [PubMed] [Google Scholar]
- 40.Seo YK, Yoon HH, Song KY, Kwon SY, Lee HS, Park YS, et al. Increase in cell migration and angiogenesis in a composite silk scaffold for tissue-engineered ligaments. J Orthop Res. 2009;27:495–503. doi: 10.1002/jor.20752. [DOI] [PubMed] [Google Scholar]
- 41.Alan Barber F, Boothby MH, Richards DP. New sutures and suture anchors in sports medicine. Sports Med Arthrosc. 2006;14:177–84. doi: 10.1097/00132585-200609000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Bini TB, Gao S, Xu X, Wang S, Ramakrishna S, Leong KW. Peripheral nerve regeneration by microbraided poly(L-lactide-co-glycolide) biodegradable polymer fibers. J Biomed Mater Res A. 2004;68:286–95. doi: 10.1002/jbm.a.20050. [DOI] [PubMed] [Google Scholar]
- 43.Burger C, Hsiao BS, Chu B. Nanofibrous materials and their applications. Annu Rev Mater Res. 2006;36:333–68. doi: 10.1146/annurev.matsci.36.011205.123537. [DOI] [Google Scholar]
- 44.Zong X, Bien H, Chung CY, Yin L, Fang D, Hsiao BS, et al. Electrospun fine-textured scaffolds for heart tissue constructs. Biomaterials. 2005;26:5330–8. doi: 10.1016/j.biomaterials.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 45.Yan J, Qiang L, Gao Y, Cui X, Zhou H, Zhong S, et al. Effect of fiber alignment in electrospun scaffolds on keratocytes and corneal epithelial cells behavior. J Biomed Mater Res A. 2011 doi: 10.1002/jbm.a.33301. In press. [DOI] [PubMed] [Google Scholar]
- 46.Oh IH, Nomura N, Masahashi N, Hanada S. Mechanical properties of porous titanium compacts prepared by powder sintering. Scr Mater. 2003;49:1197–202. doi: 10.1016/j.scriptamat.2003.08.018. [DOI] [Google Scholar]
- 47.Nicula R, Lüthen F, Stir M, Nebe B, Burkel E. Spark plasma sintering synthesis of porous nanocrystalline titanium alloys for biomedical applications. Biomol Eng. 2007;24:564–7. doi: 10.1016/j.bioeng.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Li JP, Li SH, Van Blitterswijk CA, de Groot K. A novel porous Ti6Al4V: characterization and cell attachment. J Biomed Mater Res A. 2005;73:223–33. doi: 10.1002/jbm.a.30278. [DOI] [PubMed] [Google Scholar]
- 49.Yang D, Shao H, Guo Z, Lin T, Fan L. Preparation and properties of biomedical porous titanium alloys by gelcasting. Biomed Mater. 2011;6:045010. doi: 10.1088/1748-6041/6/4/045010. [DOI] [PubMed] [Google Scholar]
- 50.Banhart J. Manufacture, characterisation and application of cellular metals and metal foams. Prog Mater Sci. 2001;46:559–632. doi: 10.1016/S0079-6425(00)00002-5. [DOI] [Google Scholar]
- 51.Wang X, Li Y, Xiong J, Hodgson PD, Wen C. Porous TiNbZr alloy scaffolds for biomedical applications. Acta Biomater. 2009;5:3616–24. doi: 10.1016/j.actbio.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Moroni L, de Wijn JR, van Blitterswijk CA. 3D fiber-deposited scaffolds for tissue engineering: influence of pores geometry and architecture on dynamic mechanical properties. Biomaterials. 2006;27:974–85. doi: 10.1016/j.biomaterials.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 53.Hollander DA, von Walter M, Wirtz T, Sellei R, Schmidt-Rohlfing B, Paar O, et al. Structural, mechanical and in vitro characterization of individually structured Ti-6Al-4V produced by direct laser forming. Biomaterials. 2006;27:955–63. doi: 10.1016/j.biomaterials.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 54.Sherwood JK, Riley SL, Palazzolo R, Brown SC, Monkhouse DC, Coates M, et al. A three-dimensional osteochondral composite scaffold for articular cartilage repair. Biomaterials. 2002;23:4739–51. doi: 10.1016/S0142-9612(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 55.Bose S, Darsell J, Hosick HL, Yang L, Sarkar DK, Bandyopadhyay A. Processing and characterization of porous alumina scaffolds. J Mater Sci Mater Med. 2002;13:23–8. doi: 10.1023/A:1013622216071. [DOI] [PubMed] [Google Scholar]
- 56.Boland T, Mironov V, Gutowska A, Roth EA, Markwald RR. Cell and organ printing 2: fusion of cell aggregates in three-dimensional gels. Anat Rec A Discov Mol Cell Evol Biol. 2003;272:497–502. doi: 10.1002/ar.a.10059. [DOI] [PubMed] [Google Scholar]
- 57.Bibette J, Leal-Calderon F, Schmitt V, Poulin P. Emulsion science: basic principles: an overview. In, Fukuyama H, Kuhn M, Muller T, Ruckenstein A, Steiner F, Trumper J et al., eds. Germany: Springer tracts in modern physics. Berlin Germany: Springer, 2002:5-42. [Google Scholar]
- 58.Kitchener JA, Mussellwhite PA. The theory of stability of emulsions. In: Sherman P, ed. Emulsion science. 1. London: Academic Press, 1968. [Google Scholar]
- 59.Forgiarini A, Esquena J, Gonzalez C, Solans C. Formation of Nano-emulsions by Low-Energy. Emulsification Methods at Constant Temperature Langmuir. 2001;17:2076–83. [Google Scholar]
- 60.Whang K, Goldstick TK, Healy KE. A biodegradable polymer scaffold for delivery of osteotropic factors. Biomaterials. 2000;21:2545–51. doi: 10.1016/S0142-9612(00)00122-8. [DOI] [PubMed] [Google Scholar]
- 61.Tcholakova S, Denkov ND, Ivanov IB, Campbell B. Coalescence stability of emulsions containing globular milk proteins. Adv Colloid Interface Sci. 2006;123-126:259–93. doi: 10.1016/j.cis.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 62.Sarker DK. Engineering of nanoemulsions for drug delivery. Curr Drug Deliv. 2005;2:297–310. doi: 10.2174/156720105774370267. [DOI] [PubMed] [Google Scholar]
- 63.Yang YY, Chung TS, Ng NP. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–41. doi: 10.1016/S0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y, Deng X. Influences of preparation conditions on particle size and DNA-loading efficiency for poly(DL-lactic acid-polyethylene glycol) microspheres entrapping free DNA. J Control Release. 2002;83:147–55. doi: 10.1016/S0168-3659(02)00176-1. [DOI] [PubMed] [Google Scholar]
- 65.Castellanos IJ, Flores G, Griebenow K. Effect of the molecular weight of poly(ethylene glycol) used as emulsifier on alpha-chymotrypsin stability upon encapsulation in PLGA microspheres. J Pharm Pharmacol. 2005;57:1261–9. doi: 10.1211/jpp.57.10.0004. [DOI] [PubMed] [Google Scholar]
- 66.Delgado A, Soriano I, Sánchez E, Oliva M, Evora C. Radiolabelled biodegradable microspheres for lung imaging. Eur J Pharm Biopharm. 2000;50:227–36. doi: 10.1016/S0939-6411(00)00109-0. [DOI] [PubMed] [Google Scholar]
- 67.Grinberg O, Binderman I, Bahar H, Zilberman M. Highly porous bioresorbable scaffolds with controlled release of bioactive agents for tissue-regeneration applications. Acta Biomater. 2010;6:1278–87. doi: 10.1016/j.actbio.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 68.Levy Y, Zilberman M. Novel bioresorbabale composite fiber structures loaded with proteins for tissue regeneration applications: microstructure and protein release. J Biomed Mater Res A. 2006;79:779–87. doi: 10.1002/jbm.a.30825. [DOI] [PubMed] [Google Scholar]
- 69.Elsner JJ, Berdicevsky I, Zilberman M. In vitro microbial inhibition and cellular response to novel biodegradable composite wound dressings with controlled release of antibiotics. Acta Biomater. 2011;7:325–36. doi: 10.1016/j.actbio.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 70.Elsner JJ, Shefy-Peleg A, Zilberman M. Novel biodegradable composite wound dressings with controlled release of antibiotics: microstructure, mechanical and physical properties. J Biomed Mater Res B Appl Biomater. 2010;93:425–35. doi: 10.1002/jbm.b.31599. [DOI] [PubMed] [Google Scholar]
- 71.Kraitzer A, Ofek L, Schreiber R, Zilberman M. Long-term in vitro study of paclitaxel-eluting bioresorbable core/shell fiber structures. J Control Release. 2008;126:139–48. doi: 10.1016/j.jconrel.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 72.Kraitzer A, Kloog Y, Zilberman M. Novel farnesylthiosalicylate (FTS)-eluting composite structures. Eur J Pharm Sci. 2009;37:351–62. doi: 10.1016/j.ejps.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 73.Jones SA, Bowler PG, Walker M, Parsons D. Controlling wound bioburden with a novel silver-containing Hydrofiber dressing. Wound Repair Regen. 2004;12:288–94. doi: 10.1111/j.1067-1927.2004.012304.x. [DOI] [PubMed] [Google Scholar]
- 74.Field FK, Kerstein MD. Overview of wound healing in a moist environment. Am J Surg. 1994;167(1A):2S–6S. doi: 10.1016/0002-9610(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 75.Lamke LO, Nilsson GE, Reithner HL. The evaporative water loss from burns and the water-vapour permeability of grafts and artificial membranes used in the treatment of burns. Burns. 1977;3:159–65. doi: 10.1016/0305-4179(77)90004-3. [DOI] [Google Scholar]
- 76.Boateng JS, Matthews KH, Stevens HN, Eccleston GM. Wound healing dressings and drug delivery systems: a review. J Pharm Sci. 2008;97:2892–923. doi: 10.1002/jps.21210. [DOI] [PubMed] [Google Scholar]
- 77.Sussman C, Bates-Jensen BM. Wound care: a collaborative practice manual for physical therapists and nurses. In, Sussman C, Bates-Jensen BM eds. Wound care: a collaborative practice manual for physical therapists and nurses (2nd Edn). Gaithersburg, MD; Aspen Publishers, 2001: 162-220. [Google Scholar]
- 78.Xu RX. Experimental and Clinical Study on Burns Regenerative Medicine and Therapy with MEBT/MEBO (Part 2). In, Xu RX, Sun X, Weeks BS Eds. Burns regenerative medicine and therapy. Basel, Switzerland, 2004:63-87. [Google Scholar]
- 79.Revathi G, Puri J, Jain BK. Bacteriology of burns. Burns. 1998;24:347–9. doi: 10.1016/S0305-4179(98)00009-6. [DOI] [PubMed] [Google Scholar]
- 80.Harrison-Balestra C, Cazzaniga AL, Davis SC, Mertz PM. A wound-isolated Pseudomonas aeruginosa grows a biofilm in vitro within 10 hours and is visualized by light microscopy. Dermatol Surg. 2003;29:631–5. doi: 10.1046/j.1524-4725.2003.29146.x. [DOI] [PubMed] [Google Scholar]
- 81.Pruitt BA, Jr., Levine NS. Characteristics and uses of biologic dressings and skin substitutes. Arch Surg. 1984;119:312–22. doi: 10.1001/archsurg.1984.01390150050013. [DOI] [PubMed] [Google Scholar]
- 82.Chung LY, Schmidt RJ, Hamlyn PF, Sagar BF, Andrews AM, Turner TD. Biocompatibility of potential wound management products: fungal mycelia as a source of chitin/chitosan and their effect on the proliferation of human F1000 fibroblasts in culture. J Biomed Mater Res. 1994;28:463–9. doi: 10.1002/jbm.820280409. [DOI] [PubMed] [Google Scholar]
- 83.Ruszczak Z, Friess W. Collagen as a carrier for on-site delivery of antibacterial drugs. Adv Drug Deliv Rev. 2003;55:1679–98. doi: 10.1016/j.addr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 84.Galdbart JO, Branger C, Andreassian B, Lambert-Zechovsky N, Kitzis M. Elution of six antibiotics bonded to polyethylene vascular grafts sealed with three proteins. J Surg Res. 1996;66:174–8. doi: 10.1006/jsre.1996.0391. [DOI] [PubMed] [Google Scholar]
- 85.Wu P, Grainger DW. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials. 2006;27:2450–67. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 86.Gold HS, Moellering RC., Jr. Antimicrobial-drug resistance. N Engl J Med. 1996;335:1445–53. doi: 10.1056/NEJM199611073351907. [DOI] [PubMed] [Google Scholar]
- 87.Gransden WR. Antibiotic resistance. Nosocomial gram-negative infection. J Med Microbiol. 1997;46:436–9. [PubMed] [Google Scholar]
- 88.Zilberman M. Novel composite fiber structures to provide drug/protein delivery for medical implants and tissue regeneration. Acta Biomater. 2007;3:51–7. doi: 10.1016/j.actbio.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 89.Zilberman M, Golerkansky E, Elsner JJ, Berdicevsky I. Gentamicin-eluting bioresorbable composite fibers for wound healing applications. J Biomed Mater Res A. 2009;89:654–66. doi: 10.1002/jbm.a.32013. [DOI] [PubMed] [Google Scholar]
- 90.Mi FL, Wu YB, Shyu SS, Schoung JY, Huang YB, Tsai YH, et al. Control of wound infections using a bilayer chitosan wound dressing with sustainable antibiotic delivery. J Biomed Mater Res. 2002;59:438–49. doi: 10.1002/jbm.1260. [DOI] [PubMed] [Google Scholar]
- 91.Sripriya R, Kumar MS, Sehgal PK. Improved collagen bilayer dressing for the controlled release of drugs. J Biomed Mater Res B Appl Biomater. 2004;70:389–96. doi: 10.1002/jbm.b.30051. [DOI] [PubMed] [Google Scholar]
- 92.Kim HW, Knowles JC, Kim HE. Porous scaffolds of gelatin-hydroxyapatite nanocomposites obtained by biomimetic approach: characterization and antibiotic drug release. J Biomed Mater Res B Appl Biomater. 2005;74:686–98. doi: 10.1002/jbm.b.30236. [DOI] [PubMed] [Google Scholar]
- 93.Queen D, Gaylor JD, Evans JH, Courtney JM, Reid WH. The preclinical evaluation of the water vapour transmission rate through burn wound dressings. Biomaterials. 1987;8:367–71. doi: 10.1016/0142-9612(87)90007-X. [DOI] [PubMed] [Google Scholar]
- 94.Boateng JS, Matthews KH, Stevens HN, Eccleston GM. Wound healing dressings and drug delivery systems: a review. J Pharm Sci. 2008;97:2892–923. doi: 10.1002/jps.21210. [DOI] [PubMed] [Google Scholar]
- 95.Lamke LO. The influence of different “skin grafts” on the evaporative water loss from burns. Scand J Plast Reconstr Surg. 1971;5:82–6. doi: 10.3109/02844317109042943. [DOI] [PubMed] [Google Scholar]
- 96.Rho KS, Jeong L, Lee G, Seo BM, Park YJ, Hong SD, et al. Electrospinning of collagen nanofibers: effects on the behavior of normal human keratinocytes and early-stage wound healing. Biomaterials. 2006;27:1452–61. doi: 10.1016/j.biomaterials.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 97.Lee SB, Kim YH, Chong MS, Hong SH, Lee YM. Study of gelatin-containing artificial skin V: fabrication of gelatin scaffolds using a salt-leaching method. Biomaterials. 2005;26:1961–8. doi: 10.1016/j.biomaterials.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 98.Gilbert P, Collier PJ, Brown MR. Influence of growth rate on susceptibility to antimicrobial agents: biofilms, cell cycle, dormancy, and stringent response. Antimicrob Agents Chemother. 1990;34:1865–8. doi: 10.1128/AAC.34.10.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–22. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 100.Dover R, Otto WR, Nanchahal J, Riches DJ. Toxicity testing of wound dressing materials in vitro. Briti J plast surg 1995; 48(4):230-235. [DOI] [PubMed]
- 101.Paddle-Ledinek JE, Nasa Z, Cleland HJ. Effect of different wound dressings on cell viability and proliferation. Plast Reconstr Surg. 2006;117(Suppl):110S–8S, discussion 119S-20S. doi: 10.1097/01.prs.0000225439.39352.ce. [DOI] [PubMed] [Google Scholar]
- 102.Hamid R, Rotshteyn Y, Rabadi L, Parikh R, Bullock P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol In Vitro. 2004;18:703–10. doi: 10.1016/j.tiv.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 103.Burd A, Kwok CH, Hung SC, Chan HS, Gu H, Lam WK, et al. A comparative study of the cytotoxicity of silver-based dressings in monolayer cell, tissue explant, and animal models. Wound Repair Regen. 2007;15:94–104. doi: 10.1111/j.1524-475X.2006.00190.x. [DOI] [PubMed] [Google Scholar]
- 104.Yannas IV, Burke JF, Orgill DP, Skrabut EM. Wound tissue can utilize a polymeric template to synthesize a functional extension of skin. Science. 1982;215:174–6. doi: 10.1126/science.7031899. [DOI] [PubMed] [Google Scholar]
- 105.Bjornson AB, Bjornson HS, Lincoln NA, Altemeier WA. Relative roles of burn injury, wound colonization, and wound infection in induction of alterations of complement function in a guinea pig model of burn injury. J Trauma. 1984;24:106–15. doi: 10.1097/00005373-198402000-00003. [DOI] [PubMed] [Google Scholar]
- 106.Kaufman T, Lusthaus SN, Sagher U, Wexler MR. Deep partial skin thickness burns: a reproducible animal model to study burn wound healing. Burns. 1990;16:13–6. doi: 10.1016/0305-4179(90)90199-7. [DOI] [PubMed] [Google Scholar]
- 107.Orenstein A, Klein D, Kopolovic J, Winkler E, Malik Z, Keller N, et al. The use of porphyrins for eradication of Staphylococcus aureus in burn wound infections. FEMS Immunol Med Microbiol. 1997;19:307–14. doi: 10.1016/S0928-8244(97)00097-7. [DOI] [PubMed] [Google Scholar]
- 108.Boon RJ, Beale AS, Sutherland R. Efficacy of topical mupirocin against an experimental Staphylococcus aureus surgical wound infection. J Antimicrob Chemother. 1985;16:519–26. doi: 10.1093/jac/16.4.519. [DOI] [PubMed] [Google Scholar]
- 109.Galandiuk S, Wrightson WR, Young S, Myers S, Polk HC., Jr. Absorbable, delayed-release antibiotic beads reduce surgical wound infection. Am Surg. 1997;63:831–5. [PubMed] [Google Scholar]
- 110.Kawai K, Suzuki S, Tabata Y, Taira T, Ikada Y, Nishimura Y. Development of an artificial dermis preparation capable of silver sulfadiazine release. J Biomed Mater Res. 2001;57:346–56. doi: 10.1002/1097-4636(20011205)57:3<346::AID-JBM1177>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 111.Mazurak VC, Burrell RE, Tredget EE, Clandinin MT, Field CJ. The effect of treating infected skin grafts with Acticoat on immune cells. Burns. 2007;33:52–8. doi: 10.1016/j.burns.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 112.Herndon DN, Wilmore DW, Mason AD., Jr. Development and analysis of a small animal model simulating the human postburn hypermetabolic response. J Surg Res. 1978;25:394–403. doi: 10.1016/S0022-4804(78)80003-1. [DOI] [PubMed] [Google Scholar]
- 113.Elsner JJ, Egozi D, Ullmann Y, Berdicevsky I, Shefy-Peleg A, Zilberman M. Novel biodegradable composite wound dressings with controlled release of antibiotics: results in a guinea pig burn model. Burns. 2011;37:896–904. doi: 10.1016/j.burns.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 114.Poon VKM, Burd A. In vitro cytotoxity of silver: implication for clinical wound care. Burns. 2004;30:140–7. doi: 10.1016/j.burns.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 115.Su SH, Chao RY, Landau CL, Nelson KD, Timmons RB, Meidell RS, et al. Expandable bioresorbable endovascular stent. I. Fabrication and properties. Ann Biomed Eng. 2003;31:667–77. doi: 10.1114/1.1575756. [DOI] [PubMed] [Google Scholar]
- 116.Alikacem N, Yoshizawa T, Nelson KD, Wilson CA. Quantitative MR imaging study of intravitreal sustained release of VEGF in rabbits. Invest Ophthalmol Vis Sci. 2000;41:1561–9. [PubMed] [Google Scholar]
- 117.Dunn RL, Lewis DH. J.M. G. Monolithic fibers for controlled delivery of tetracycline. Proc Int Symp Control Rel Bioact Mater, 1982:157-63. [Google Scholar]
- 118.Dunn RL, English JP, Stoner WC, Potter AG, Perkins BH. Biodegradable fibers for the controlled release of tetracycline in treatment of peridontal disease. Proc Int Symp Control Rel Bioact Mater, 1987:289-94. [Google Scholar]
- 119.Dunn RL, Lewis DH, Beck LR. Fibrous polymer for the delivery of contraceptive steroids to the female reproductive track. In: Lewis DH, ed. Controlled release of pesticides and pharmaceuticals. New York: Plenum press, 1981:125-46. [Google Scholar]
- 120.Eenink MJD, Feijen J, Oligslanger J, Albers JHM, Rieke JC. P.J. G. Biodegradable hollow fibers for the controlled release of hormones. J Control Release. 1987;6:225–37. doi: 10.1016/0168-3659(87)90079-4. [DOI] [Google Scholar]
- 121.Lazzeri L, Cascone MG, Quiriconi L, Morabito L, Giusti P. Biodegradable hollow microfibers to produce bioactive scaffolds. Polym Int. 2005;54:101–7. doi: 10.1002/pi.1648. [DOI] [Google Scholar]
- 122.Polacco G, Cascone MG, Lazzeri L, Ferrara S, Giusti P. Biodegradable hollow fibers containing drug-loaded nanoparticles as controlled release systems. Polym Int. 2002;51:1464–72. doi: 10.1002/pi.1086. [DOI] [Google Scholar]
- 123.Heldman AW, Cheng L, Jenkins GM, Heller PF, Kim DW, Ware M, Jr., et al. Paclitaxel stent coating inhibits neointimal hyperplasia at 4 weeks in a porcine model of coronary restenosis. Circulation. 2001;103:2289–95. doi: 10.1161/01.CIR.103.18.2289. [DOI] [PubMed] [Google Scholar]
- 124.Dhanikula AB, Panchagnula R. Localized paclitaxel delivery. Int J Pharm. 1999;183:85–100. doi: 10.1016/S0378-5173(99)00087-3. [DOI] [PubMed] [Google Scholar]
- 125.Kraitzer A. Paclitaxel-loaded composite fiber structures used in vascular stents. Material Science and Engineering. Tel Aviv: Tel Aviv University, 2006. [Google Scholar]
- 126.Feng S, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymers. J Control Release. 2001;71:53–69. doi: 10.1016/S0168-3659(00)00364-3. [DOI] [PubMed] [Google Scholar]
- 127.Farb A, Heller PF, Shroff S, Cheng L, Kolodgie FD, Carter AJ, et al. Pathological analysis of local delivery of paclitaxel via a polymer-coated stent. Circulation. 2001;104:473–9. doi: 10.1161/hc3001.092037. [DOI] [PubMed] [Google Scholar]
- 128.Marom M, Haklai R, Ben-Baruch G, Marciano D, Egozi Y, Kloog Y. Selective inhibition of Ras-dependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270:22263–70. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 129.George J, Sack J, Barshack I, Keren P, Goldberg I, Haklai R, et al. Inhibition of intimal thickening in the rat carotid artery injury model by a nontoxic Ras inhibitor. Arterioscler Thromb Vasc Biol. 2004;24:363–8. doi: 10.1161/01.ATV.0000112021.98971.f0. [DOI] [PubMed] [Google Scholar]
- 130.Kloog Y, Cox AD. Prenyl-binding domains: potential targets for Ras inhibitors and anti-cancer drugs. Semin Cancer Biol. 2004;14:253–61. doi: 10.1016/j.semcancer.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 131.Chitkara D, Shikanov A, Kumar N, Domb AJ. Biodegradable injectable in situ depot-forming drug delivery systems. Macromol Biosci. 2006;6:977–90. doi: 10.1002/mabi.200600129. [DOI] [PubMed] [Google Scholar]
- 132.Shikanov A, Vaisman B, Krasko MY, Nyska A, Domb AJ. Poly(sebacic acid-co-ricinoleic acid) biodegradable carrier for paclitaxel: in vitro release and in vivo toxicity. J Biomed Mater Res A. 2004;69:47–54. doi: 10.1002/jbm.a.20101. [DOI] [PubMed] [Google Scholar]
- 133.Kraitzer A, Kloog Y, Haklai R, Zilberman M. Composite fiber structures with antiproliferative agents exhibit advantageous drug delivery and cell growth inhibition in vitro. J Pharm Sci. 2011;100:133–49. doi: 10.1002/jps.22238. [DOI] [PubMed] [Google Scholar]
- 134.Burke J. Hildebrand Solubility Parameter. In: Jensen C, ed. The AIC Book and Paper Group Annual, 3. The Oakland Museum of California, 1984: 13-58 [Google Scholar]
- 135.Kraitzer A, Alperstein D, Kloog Y, Zilberman M. Mechanisms of antiproliferative drug release from bioresorbable porous structures. J Biomed Mater Res A. 2012 doi: 10.1002/jbm.a.34436. Accepted. [DOI] [PubMed] [Google Scholar]
- 136.Haklai R, Weisz MG, Elad G, Paz A, Marciano D, Egozi Y, et al. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37:1306–14. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 137.Zundelevich A, Elad-Sfadia G, Haklai R, Kloog Y. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid) Mol Cancer Ther. 2007;6:1765–73. doi: 10.1158/1535-7163.MCT-06-0706. [DOI] [PubMed] [Google Scholar]
- 138.Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005;65:999–1006. [PubMed] [Google Scholar]
- 139.Goldberg L, Kloog Y. A Ras inhibitor tilts the balance between Rac and Rho and blocks phosphatidylinositol 3-kinase-dependent glioblastoma cell migration. Cancer Res. 2006;66:11709–17. doi: 10.1158/0008-5472.CAN-06-1878. [DOI] [PubMed] [Google Scholar]
- 140.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920–6. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 141.Howard D, Buttery LD, Shakesheff KM, Roberts SJ. Tissue engineering: strategies, stem cells and scaffolds. J Anat. 2008;213:66–72. doi: 10.1111/j.1469-7580.2008.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Babensee JE, McIntire LV, Mikos AG. Growth factor delivery for tissue engineering. Pharm Res. 2000;17:497–504. doi: 10.1023/A:1007502828372. [DOI] [PubMed] [Google Scholar]
- 143.Chen RR, Mooney DJ. Polymeric growth factor delivery strategies for tissue engineering. Pharm Res. 2003;20:1103–12. doi: 10.1023/A:1025034925152. [DOI] [PubMed] [Google Scholar]
- 144.Basmanav FB, Kose GT, Hasirci V. Sequential growth factor delivery from complexed microspheres for bone tissue engineering. Biomaterials. 2008;29:4195–204. doi: 10.1016/j.biomaterials.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 145.Zhu XH, Wang CH, Tong YW. In vitro characterization of hepatocyte growth factor release from PHBV/PLGA microsphere scaffold. J Biomed Mater Res A. 2009;89:411–23. doi: 10.1002/jbm.a.31978. [DOI] [PubMed] [Google Scholar]
- 146.Elcin AE, Elcin YM. Localized angiogenesis induced by human vascular endothelial growth factor-activated PLGA sponge. Tissue Eng. 2006;12:959–68. doi: 10.1089/ten.2006.12.959. [DOI] [PubMed] [Google Scholar]
- 147.Wei G, Jin Q, Giannobile WV, Ma PX. Nano-fibrous scaffold for controlled delivery of recombinant human PDGF-BB. J Control Release. 2006;112:103–10. doi: 10.1016/j.jconrel.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Piazza R. Protein science and association: an open challenge for colloid science. Curr Opin Colloid Interface Sci. 2004;8:515–22. doi: 10.1016/j.cocis.2004.01.008. [DOI] [Google Scholar]
- 149.Tadros TF, Vandamme A, Levecke B, Booten K, Stevens CV. Stabilization of emulsions using polymeric surfactants based on inulin. Adv Colloid Interface Sci. 2004;108-109:207–26. doi: 10.1016/j.cis.2003.10.024. [DOI] [PubMed] [Google Scholar]