

Abstract
Nano-sized FePt capsules with two types of ultrathin shell were fabricated using a template method for use in a nano-scale drug delivery system. One capsule was composed of an inorganic-organic hybrid shell of a water-soluble polymer and FePt nanoparticles, and the other capsule was composed of a network of fused FePt nanoparticles. We demonstrated that FePt nanoparticles selectively accumulated on the polymer molecules adsorbed on the template silica particles, and investigated the morphologies of the particle accumulation by changing the concentration of the polymer solution with which the template particles were treated. Capsular size was reduced from 340 to less than 90 nm by changing the size of the silica template particles, and the shell thickness was controlled by changing the amount of FePt nanoparticles adsorbed on the template particles. The hybrid shell was maintained by the connection of FePt nanoparticles and polymer molecules, and the shell thickness was 10 nm at the maximum. The FePt network shell was fabricated by hydrothermal treatment of the FePt/polymer-modified silica composite particles. The FePt network shell was produced from only the FePt alloy, and the shell thickness was 3 nm. Water-soluble anti-cancer drugs could be loaded into the hollow space of FePt network capsules, and lipid-coated FePt network capsules loaded with anti-cancer drugs showed cellular toxicity. The nano-sized capsular structure and the ultrathin shell suggest applicability as a drug carrier in magnetically guided drug delivery systems.
Keywords: hydrothermal treatment, magnetic hollow sphere, magnetically guided drug delivery system, nano-scale drug delivery system, organic-inorganic magnetic material, porous capsule, size-controllable
Introduction
Magnetic capsules are currently being developed in the field of biotechnology as carriers of therapeutic agents in magnetically guided drug delivery systems (MG-DDSs) because they have unique properties such as a hollow structure, low material density, and magnetism. The shells of these magnetic capsules are usually composed of magnetic inorganic and/or organic materials such as Fe3O4, CoFe2O3, block copolymers, silica or layer-by-layer assemblies of polyelectrolytes.1-13 In previous studies, the shells of these capsules have been too thick (> 20 nm) to afford sufficient internal space when applied to nano-scale drug delivery systems (nano-DDSs) targeting cancer lesions.14-17 In nano-DDSs targeting cancer tissue, the size of the carrier is limited because small carriers (< 5 nm) are eliminated by renal excretion through the kidney, and large size (> 200–500 nm) of foreign material are easily removed by the mononuclear phagocyte systems in the lung, spleen and liver. Therefore, to avoid the elimination of carriers after intravenous administration, the total size of nano-scale drug capsules must be between several tens of nanometers and 200 nm.14-17 When using a magnetic capsule with a diameter of 100 nm and a shell thickness of 20 nm, the loading capacity for therapeutic agents is limited to only 22% of the capsule volume. The loading capacity was calculated from their whole diameter and shell thickness. A thinner shell is therefore required to increase the loading capacity for biomedical agents and to decrease the amount of magnetic components administered to human bodies while still adhering to the size limitations applicable to nano-DDSs.
Our previous papers demonstrated two types of FePt magnetic capsules with thinner shells (d = 5–10 nm) compared with previously reported magnetic capsules.1-13,18-20Scheme 1 shows the fabrication process for these two types of FePt magnetic capsules and representative transmission electron microscopy (TEM) images. The first type of FePt magnetic capsule was an inorganic-organic hybrid capsule that was fabricated by growing ordered alloy FePt nanoparticles on silica template particles with an anionic surface that had been modified with a cationic polymer, poly(diallyldimethylammonium chloride) (PDDA), and then dissolving the silica template particles from the FePt-nanoparticle/PDDA/silica particles by using NaOH aqueous solution.18 This hybrid shell was composed of 3 to 5 nm of FePt nanoparticles and a single layer of PDDA. The second type of FePt magnetic capsule was a porous capsule composed of an ordered alloy FePt network fabricated by hydrothermally treating the FePt-nanoparticle/PDDA/silica particles.19,20 Through the hydrothermal treatment the PDDA layer and silica template particles were decomposed in supercritical water and the FePt nanoparticles were thermally fused to each other. These FePt capsules were designed for use as a magnetic carrier in a MG-DDS.21-24
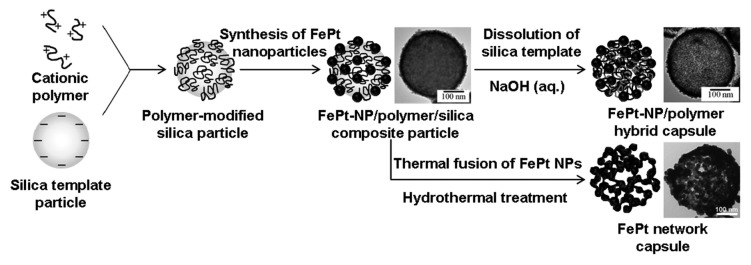
Scheme 1. Fabrication process for two types of FePt magnetic capsules and representative transmission electron microscopy images. FePt-NP, FePt nanoparticle.
An ordered FePt alloy was used for fabricating the nano-sized shells in our studies because it has a higher magnetocrystalline anisotropy constant (105–106 Jm−3) than iron oxides, which have also been used in the production of magnetic capsules developed for MG-DDSs.25 The higher magnetocrystalline anisotropy constant means that the thickness of the shells can be reduced without any deterioration of the magnetic response. Although the total sizes of the two types of FePt capsules in our previous work were too large (330–340 nm) for them to be used in a nano-DDS,18,19 capsular size could be controlled by varying the size of the silica template particles. In addition, shell thickness could be controlled by changing the amount of FePt nanoparticles accumulated on the surface of the PDDA-modified silica template particles because our FePt-nanoparticle/PDDA hybrid shell was composed of FePt nanoparticles and a single layer of PDDA, and the FePt network shell was composed of only FePt alloy. Therefore, the total size of the hollow sphere depends on the diameters of the silica template particles and the FePt nanoparticles.
In the present work, we fabricated smaller FePt-nanoparticle/PDDA hybrid capsules and FePt network capsules with a nanometer-thick shell using PDDA-modified silica template particles with an average diameter of 100 nm. In addition, we demonstrated a feasibility-for-drug-delivery of FePt network capsules by loading the capsules with water-soluble anti-cancer agents and cell assay. We revealed that FePt nanoparticles selectively accumulated on the PDDA molecules adsorbed on the surface of silica template particles by changing the concentration of the PDDA aqueous solution with which the silica template particles were treated. Shell thickness was controlled by changing the amount of FePt nanoparticles accumulated on the PDDA-modified silica template particles. We discussed the control of the amount of FePt nanoparticles and PDDA molecules is absolutely imperative to obtain the thin shell and the whole diameter suitable for nano-DDS and demonstrated sufficient amount of FePt nanoparticles and PDDA molecules to obtain 3D structure. Especially in the FePt-nanoparticle/PDDA hybrid shell, FePt nanoparticles anchor the PDDA molecules in an aqueous solution, therefore the amounts of PDDA molecules adsorbed on the silica particles and FePt nanoparticles accumulated on the PDDA layer were important to maintain the structure of the shell. FePt network capsules loaded with anti-cancer drugs and coated with lipid membrane to avoid leaks of that drugs showed cellular toxicity to gastric cancer.
Results and Discussion
Morphologies
Composite particles and hybrid capsules
FePt nanoparticles were synthesized at 503 K in the presence of silica particles treated with various concentrations of PDDA aqueous solution (1–7 wt%) by reduction of Fe(acetylacetonate)3 (0.106 mmol) and Pt(acetylacetonate)2 (0.096 mmol).26,27 The modification of negatively charged silica template particles with a cationic polymer resulted in the zeta potential of the silica template particles changing from negative to positive. The adsorption of PDDA molecules on the surface of silica particles was confirmed by measuring their zeta potentials. Increasing the concentration of the PDDA solution used to treat the silica particles, the morphologies of the composite particles fabricated with these PDDA-modified silica particles were slightly different (Fig. 1A–C). FePt nanoparticles accumulated on the surface of the PDDA-modified silica particles, and there was no difference in the size and shape of these FePt nanoparticles; the diameter of the FePt nanoparticles was between 3 and 5 nm. However, silica template particles treated with lower concentrations of PDDA solution (1 or 5 wt%) were only partially covered with FePt nanoparticles, whereas silica particles treated with 7 wt% PDDA solution were entirely covered with FePt nanoparticles (Fig. 1A–C). Our previous report demonstrated that FePt nanoparticles accumulate on organic compounds adsorbed on the surface of silica particles18 confirming that the surface of the silica template particles treated with 1 or 5 wt% PDDA solution were not entirely covered with PDDA molecules, and that FePt nanoparticles accumulated only at the PDDA molecules adsorbed on the silica surface. This means that the FePt nanoparticles were selectively grown on the PDDA layer. Figure 1D–F shows TEM images of FePt-nanoparticle/PDDA hybrid capsules fabricated by dissolution of the silica template particles from the composite particles shown in Figure 1A–C. Complete FePt-nanoparticle/PDDA hybrid capsules were successfully obtained by dissolving the silica template particles from the composite particles entirely covered with FePt nanoparticles (Fig. 1F). In contrast, most of the FePt nanoparticle/PDDA aggregates were broken and dispersed after dissolution of the silica template particles from the composite particles partially covered with FePt nanoparticles (Fig. 1D and E).

Figure 1. TEM images of FePt-nanoparticle/PDDA/silica composite particles fabricated with silica template particles treated with various concentrations of PDDA aqueous solutions, (A) 1 wt%, (B) 5 wt% and (C) 7 wt%, and FePt-nanoparticle/PDDA hybrid capsules fabricated by dissolution of the silica template particles from the composite particles. (D−F) are images of the hybrid capsules fabricated from A, B and C respectively.
Figure 2A–C shows TEM images of FePt-nanoparticle/PDDA/silica composite particles fabricated by using weight ratios of FePt precursors to silica particles of 4, 2 and 1, respectively, and by using silica template particles treated with 7 wt% of PDDA solution. The size of the FePt nanoparticles was not affected by changing the amount of precursors used in their synthesis; only the amount of FePt nanoparticles accumulated on the surface of the PDDA-modified silica particles changed, and a thinner shell was obtained by decreasing the weight ratio. Figure 2D–F shows TEM images of FePt-nanoparticle/PDDA hybrid capsules fabricated by dissolution of the silica template particles from the composite particles shown in Figure 2A–C. Decreasing the amount of FePt nanoparticles accumulated on the PDDA-modified silica particles resulted in increased rupturing of the capsules. Few magnetic capsules were obtained after dissolving the template particles from the composite particles fabricated by using a weight ratio of FePt precursors to silica particles of 1 (Fig. 2F). Using a weight ratio of FePt precursors to silica particles of 4, we obtained FePt-nanoparticle/PDDA hybrid capsules that had an average diameter of 120 nm. The thickness of the hybrid shell was 10 nm at the maximum. These hybrid shells had many pores (d < 6 nm) and were stable in water. These shells may be useful for nano-DDSs because of their controlled structures.14-17

Figure 2. TEM images of FePt-nanoparticle/PDDA/silica composite particles fabricated by changing the weight ratio of FePt precursors to silica particles to (A) 4, (B) 2 and (C) 1 using silica template particles treated with 7 wt% of PDDA solution, and FePt-nanoparticle/PDDA hybrid capsules fabricated by dissolution of silica template particles from the composite particles. (D−F) are images of hybrid capsules fabricated from A, B and C, respectively.
Scheme 2 shows a proposed model of the morphology of the FePt-nanoparticle/PDDA hybrid capsules in water. PDDA, a water-soluble polymer, adsorbed on the surface of the silica particles is dispersed again after dissolution of the silica template particles. Our results show that when the amount of PDDA molecules adsorbed on the silica particles, or the amount of FePt nanoparticles adsorbed on the PDDA molecules, is small, most of the hybrid capsules rupture. FePt nanoparticles and PDDA molecules were connected each other, and the amounts of FePt nanoparticles and PDDA molecules were effective to maintain the three-dimensional (3D) structure of the hybrid capsules. It is hypothesized that the FePt nanoparticles anchor the PDDA molecules so that they are not dispersed in water despite their water-solubility, and that this combination of FePt nanoparticles and PDDA molecules accounts for the 3D structure being retained.
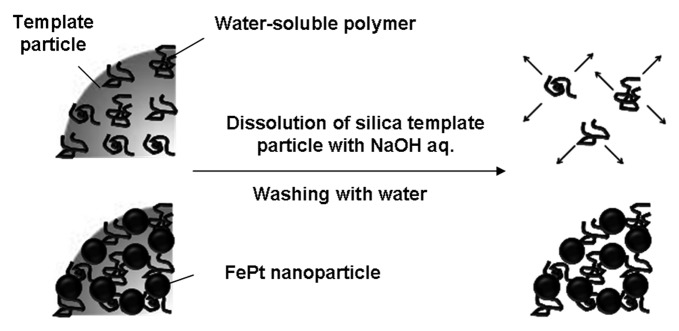
Scheme 2. Proposed model of the morphology of FePt-nanoparticle/PDDA hybrid capsules.
FePt network capsules
Figure 3 shows TEM images of FePt-nanoparticle/PDDA/silica composite particles and the FePt network capsules fabricated from them by hydrothermal treatment at 673K and 37 MPa for 3 h. The composite particles were fabricated with silica particles treated with 7 wt% of PDDA solution and a weight ratio of FePt precursors to silica particles of 1 (Fig. 3A). When the weight ratio of FePt precursors to silica particles was 1, FePt-nanoparticle/PDDA hybrid capsules could not be obtained (Fig. 2F); however, FePt network capsules could be successfully obtained (Fig. 3B). Despite the ratio of FePt precursors to silica particles being too low to obtain hybrid capsules after dissolution of the silica particles, and only a small amount of FePt nanoparticles being adsorbed on the PDDA-modified silica particles, hydrothermal treatment fused the FePt nanoparticles resulting in porous capsules being produced from the 3D network of the FePt alloy. The total diameter of the FePt network capsules was about 90 nm and the shell thickness was about 3 nm. In addition, there were large pores in the shell of the FePt network capsules that had a diameter up to 10 nm. A magnetic capsule with a diameter of 90 nm and a shell thickness of 3 nm has a loading capacity for therapeutic agents of 80% of the capsule volume. The larger internal capacity and larger pores in the network capsules are advantageous for carrying and releasing larger amounts anti-cancer drugs.1-13 Additionally, the large pores are useful for loading sterically-bulky molecules such as therapeutic agents.
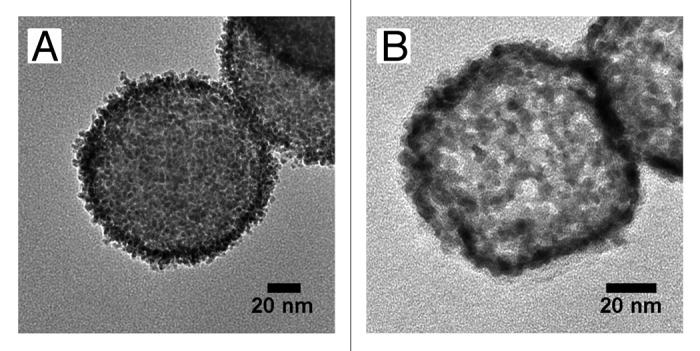
Figure 3. TEM images of (A) FePt-nanoparticle/PDDA/silica composite particles fabricated by using a weight ratio of FePt precursors to silica particles of 1, and (B) FePt network capsules fabricated by hydrothermal treatment of the composite particles.
Crystalline structure and magnetic properties of composite particles and FePt capsules
X-ray diffraction (XRD) patterns of the FePt-nanoparticle/PDDA/silica composite particles, FePt-nanoparticle/PDDA hybrid capsules, and FePt network capsules are shown in Figure 4. The composite particles were synthesized with 7 wt% PDDA solution and a weight ratio of FePt precursors to silica particles of 4 (Fig. 2A). Hybrid capsules were fabricated from these composite particles (Fig. 2D). The FePt network capsules were fabricated by hydrothermal treatment of composite particles fabricated with a weight ratio of FePt precursors to silica particles of 1 (Figs. 2C and 3A). The composite particles and hybrid capsules exhibited the typical XRD pattern of FePt, and there was no marked difference in their crystalline structure.25 Crystallite sizes were calculated by using Scherrer’s formula (Table 1). We found that crystallite size before hydrothermal treatment does not depend on the synthesis conditions except for reaction temperature (results not shown). Crystallite size increased from 2.0 to 5.5 nm for the FePt network capsules after hydrothermal treatment. Diffraction peaks from spinel-type iron oxide were observed for the FePt network capsules. Supercritical water has high oxidizability and is used to oxidize organic compounds. Therefore, either iron oxide was formed on the surface of the FePt network shell or Fe was dissolved out into the supercritical water and then oxidized during the hydrothermal treatment.
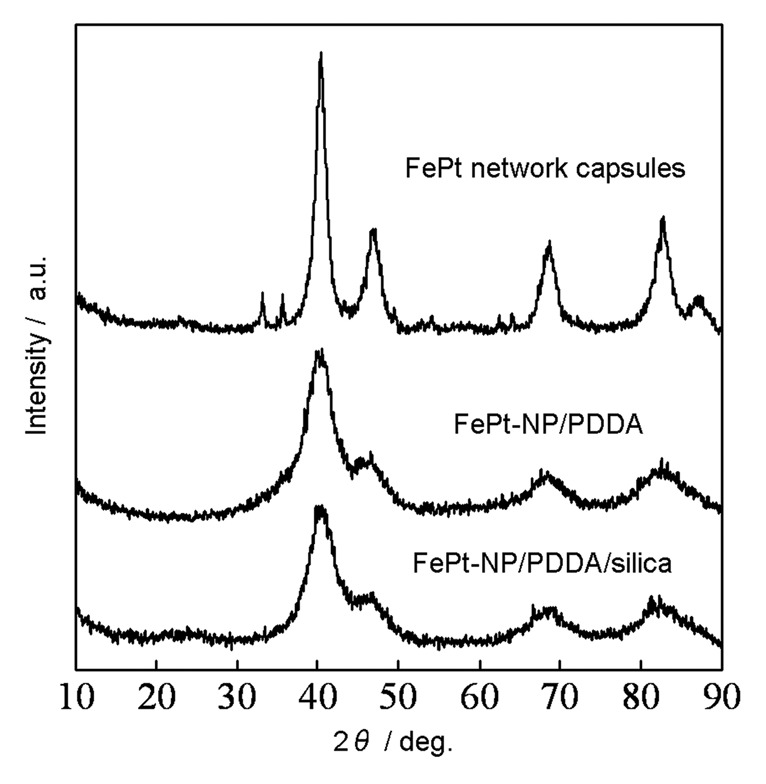
Figure 4. X-ray diffraction patterns of FePt-nanoparticle/PDDA/silica composite particles, FePt-nanoparticle/PDDA hybrid capsules, and FePt network capsules after hydrothermal treatment.
Table 1. Crystallite size and saturation magnetization of FePt-nanoparticle/PDDA/silica composite particles, FePt-nanoparticle/PDDA hybrid capsules and FePt network capsules after hydrothermal treatment.
| Sample | Crystallite size (nm) | Magnetization at 9 T (emu/g) |
|---|---|---|
|
FePt-nanoparticle/PDDA/silica |
2.0 |
2.6 |
|
FePt-nanoparticle/PDDA hybrid capsules |
2.0 |
7.6 |
| FePt network capsules | 5.5 | 12.4 |
Magnetization loops of the composite particles and the two types of FePt capsules were measured at 300 K in applied magnetic fields from −9 to 9 T, and their magnetization at 9 T is summarized in Table 1. Magnetization per weight at 9 T was increased after dissolution of the silica template particles from the composite particles due to the removal of the non-magnetic silica particles (7.6 emu/g). The magnetization at 9 T of the FePt network capsules after hydrothermal treatment was 12.4 emu/g. FePt network capsules exhibited higher magnetization than FePt-nanoparticle/PDDA hybrid capsules because the crystallite size was increased by the hydrothermal treatment. The magnetic characterization revealed that the two types of FePt magnetic capsules exhibited superparamagnetic behavior at 300 K. This property should help to avoid the embolism of blood vessels during delivery of therapeutic agents to cancer lesions because the capsules do not have spontaneous magnetization and do not coagulate without magnetic fields.22,28
Drug Delivery Experiment
Lipid-coated FePt network capsules loaded with doxorubicin (FePt-Dox) were obtained using the FePt network capsules fabricated by hydrothermal treatment of composite particles fabricated with a weight ratio of FePt precursors to silica particles of 1 (Figs. 2C and 3A). Figure 5 shows an optical micrograph and fluorescence micrograph of the cells incubated with FePt-Dox. The fluorescent micrograph was observed in the same field of the optical micrograph. Fluorescence microscopy revealed doxorubicin (Dox) was loaded into an internal space of FePt network capsules. Dox was translated through their pores to their hollow space by diffusion, leading to the formation of FePt-Dox. We found that the cells were dyed by FePt-Dox from the morphology change of the cells capturing FePt-Dox from the comparison between Figure 5A and B. FePt network capsules without Dox were not toxic to the cells reported in our previous work.19 These results showed FePt-Dox had cellular toxicity to gastric cancer cells.
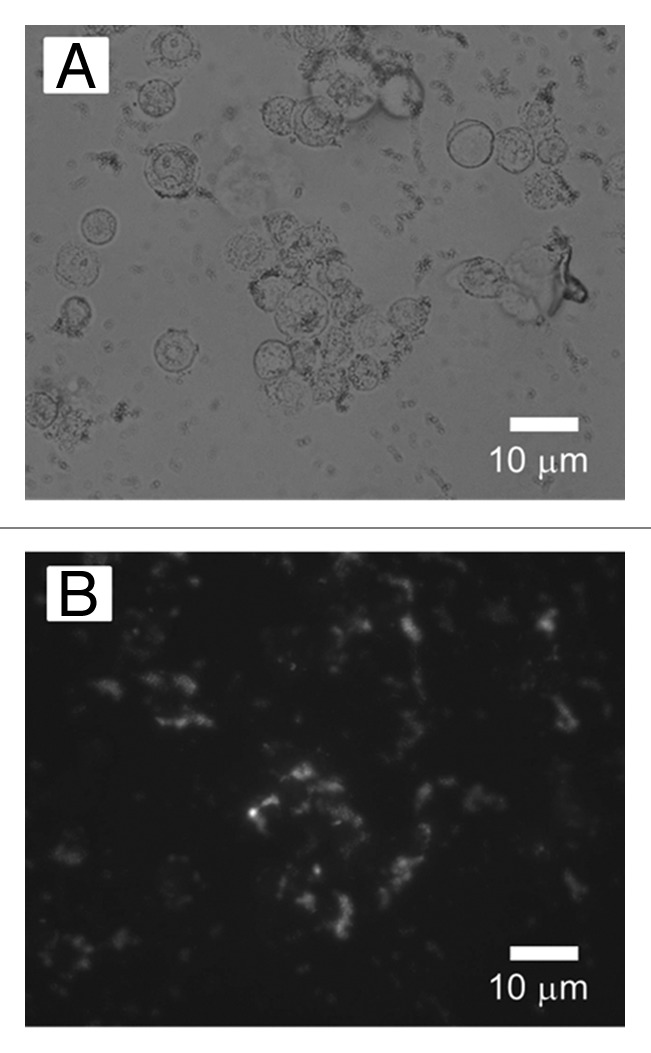
Figure 5. (A) Optical micrograph and (B) fluorescence micrograph of gastric cancer cells incubated with FePt- Dox. Fluorescent microscopy was investigated under the excitation/emission wavelength: 510/550 nm.
Conclusion
The present study demonstrates that the diameters of two types of FePt capsules can be reduced by changing the size of silica template particles used in their fabrication, and that shell thickness can be controlled by changing the amount of FePt nanoparticles accumulated on PDDA-modified silica template particles. FePt nanoparticles selectively accumulated onto PDDA molecules adsorbed on the surface of the silica template particles, and the amount of FePt nanoparticles could be easily controlled by changing the amount of precursors added to the synthesis of FePt-nanoparticle/PDDA/silica composite particles. FePt nanoparticles accumulated on the polymer molecules would anchor the polymer molecules resulting in the 3D structure being retained in the hybrid capsules. The shell thickness of the hybrid capsules was 10 nm at the maximum, and the hybrid capsules exhibited magnetization at 9 T of 7.6 emu/g. It may be possible for the hybrid capsules to be fabricated with different functional polymers, such as a thermo-responsive polymer, and these hybrid capsules should have wide applicability. FePt network capsules were fabricated by the hydrothermal treatment of FePt-nanoparticle/PDDA/silica composite particles, and their shell thickness was 3 nm. The network capsules exhibited magnetization at 9 T of 12.4 emu/g. Both of two types of the magnetic capsules will exhibit superparamagnetic behavior at approximate body temperature. Fluorescence microscopy showed water-soluble anti-cancer drugs could be loaded into the hollow space of FePt network capsules, and optical microscopy revealed lipid-coated FePt network capsules loaded with anti-cancer drugs showed cellular toxicity.
Materials and Methods
Materials
Amorphous silica particles (average diameter: 100 nm) were purchased from Nippon Shokubai (KE-P10). Fe(acetylacetonate)3 (517003-10G), Pt(acetylacetonate)2 (282782-5G), tetraethylene glycol (TEG) (110175–1KG), PDDA aqueous solution (522376-1L, weight average molecular weight: < 100 kg mol−1), and ethanol were purchased from Sigma-Aldrich. Doxorubicin hydrochloride (040–21521) was purchased from Wako pure chemical industries.
Synthesis
Modification of silica template particles with PDDA
PDDA-modified silica particles were fabricated by mixing an aqueous dispersion (5 mL) of amorphous silica particles (80 mg; average diameter: 100 nm) with various concentrations (1–7 wt%) of PDDA aqueous solution (25 mL).18 The PDDA-modified silica particles were purified to remove excess PDDA by washing with water. The PDDA-modified silica template particles (40 mg) were then dispersed in TEG (50 mL) after solvent exchange by centrifugation.
Synthesis of FePt-nanoparticle/PDDA/silica composite particles
FePt nanoparticles were synthesized by using the following polyol method.18 Fe(acetylacetonate)3 and Pt(acetylacetonate)2 were used as precursors of the FePt alloy. A mixture of Fe(acetylacetonate)3 (0.212, 0.106 or 0.053 mmol), Pt(acetylacetonate)2 (0.192, 0.096 or 0.048 mmol), silica template particles (40 mg) and TEG (50 mL) was added to a 100 mL three-necked, round-bottom flask and the mixture heated with stirring at 503 K by refluxing for 2 h in an inert gas (Ar/H2). After the reaction, the composite particles were precipitated by centrifugation and washing with ethanol.
Fabrication of FePt-nanoparticle/PDDA hybrid capsules
The FePt-nanoparticle/PDDA/silica composite particles were stirred with 3 mol dm−3 NaOH aqueous solution at 343 K for 1 h to dissolve the silica template particles. The FePt-nanoparticle/PDDA hybrid capsules were then purified several times by centrifugation and redispersion by using an ultrasonic bath in deionized water.18
Hydrothermal treatment of FePt-nanoparticle/PDDA/silica composite particles
An aqueous dispersion (5 mL) of FePt-nanoparticle/PDDA/silica composite particles was added to a reaction cell. The reaction cell (11 cm3) was heated and kept at 673 K and 37 MPa for 3 h to produce FePt network capsules.19
Preparation of lipid-coated FePt network capsule containing anti-cancer drug
FePt-Dox were fabricated by the following mehod.19 One milliliter of an aqueous solution of Dox (5 mg/ml) was mixed with the FePt network capsules (2 mg). Then this mixture was evaporated so as to remove the air in the FePt network capsules and fill them with Dox. The FePt network capsules containing Dox were mixed with 1 ml of egg phosphatidylcholine/chloroform (2 mg/ml). This mixture was stirred vigorously with a vortex-mixer, then, the mixture was stirred in an ultrasonic bath and evaporated. FePt-Dox were dispersed in Phosphate buffered saline (PBS).
Fluorescence image of magnetically guided FePt-Dox
The accumulation of the FePt-Dox was investigated in the cancer cell line KATO-III. Cells (5 × 103) were cultured for 12 h in a well of a 96 well plate and then culture medium was removed. PBS containing FePt-Doxs (the final concentration of DOX: 1 µg/ml) were introduced into a well of a 96 well plate. Cells were incubated with FePt-Dox and a NdFeB magnet (0.2 T) was directly attached under the well for 5 min. Six hours after the initiation of the treatment, cells were washed with PBS and fixed with 10% formaldehyde/PBS. Ultimately, the cellular uptake of Dox was evaluated with fluorescent microscopy (excitation/emission wavelength: 510/550 nm).
Measurements
The zeta potentials of the silica particles and PDDA-modified silica particles were measured with a nanoparticle analyzer (SZ-100; Horiba). The crystal structure of the FePt-nanoparticle/PDDA/silica composite particles and magnetic capsules was investigated by using a powder X-ray diffractometer (RINT 2100V; Rigaku). The size and morphology of the magnetic capsules was assessed with a transmission electron microscope (H-8100; Hitachi). Magnetic properties were measured using a Physical Property Measurement System (Quantum Design) at 300 K. Optical microscopy and fluorescence microscopy were investigated with fluorescence microscope (Axiovert 200; Zeiss).
Acknowledgments
This work was supported by an Industrial Technology Research Grant, Program 08C46049a, from the New Energy and Industrial Technology Development Organization of Japan, by a Funding Program for Next Generation World-Leading Researchers (LS114), and by a Grant-in Aid for Scientific Research (#20310077) from the Japan Society for the Promotion of Science.
Glossary
Abbreviations:
- MG-DDSs
magnetically guided drug delivery systems
- nano-DDSs
nano-scale drug delivery systems
- TEM
transmission electron microscopy
- PDDA
poly(diallyldimethylammonium chloride)
- FePt-NP
FePt nanoparticle
- TEG
tetraethylene glycol
- FePt-Dox
lipid-coated FePt network capsule loaded with doxorubicin
- Dox
doxorubicin
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/biomatter/article/22617
References
- 1.Zhang C, Zhang H, Du B, Hou R, Guo S. Facile organic solvent-free synthesis of size-controlled hierarchically structured magnetic hollow spheres and potential application in adsorption for bovine serum album. J Colloid Interface Sci. 2012;368:97–106. doi: 10.1016/j.jcis.2011.10.056. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Huang Z, Tang F, Ren J. Ferrite hollow spheres with tunable magnetic properties. Thin Solid Films. 2006;515:2555–61. doi: 10.1016/j.tsf.2006.04.049. [DOI] [Google Scholar]
- 3.Wang C, Yan J, Cui X, Wang H. Synthesis of raspberry-like monodisperse magnetic hollow hybrid nanospheres by coating polystyrene template with Fe(3)O(4)@SiO(2) particles. J Colloid Interface Sci. 2011;354:94–9. doi: 10.1016/j.jcis.2010.09.078. [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Zhang X, Qian H. A novel approach to fabrication of superparamagnetite hollow silica/magnetic composite spheres. J Magn Magn Mater. 2010;322:2172–6. doi: 10.1016/j.jmmm.2010.02.004. [DOI] [Google Scholar]
- 5.Lu Z, Qin Y, Fang J, Sun J, Li J, Liu F, et al. Monodisperse magnetizable silica composite particles from heteroaggregate of carboxylic polystyrene latex and Fe3O4 nanoparticles. Nanotech. 2008;19:055602. doi: 10.1088/0957-4484/19/05/055602. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura M, Katagiri K, Koumoto K. Preparation of hybrid hollow capsules formed with Fe3O4 and polyelectrolytes via the layer-by-layer assembly and the aqueous solution process. J Colloid Interface Sci. 2010;341:64–8. doi: 10.1016/j.jcis.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Abe M, Nishio N, Hatakeyama M, Hanyu N, Tanaka T, Tada M, et al. Preparation and medical application of magnetic beads conjugated with bioactive molecules. J Magn Magn Mater. 2009;321:645–9. doi: 10.1016/j.jmmm.2008.11.086. [DOI] [Google Scholar]
- 8.Liu J, Deng Y, Liu C, Sun Z, Zhao D. A simple approach to the synthesis of hollow microspheres with magnetite/silica hybrid walls. J Colloid Interface Sci. 2009;333:329–34. doi: 10.1016/j.jcis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Shen S, Wu W, Guo K, Meng H, Chen J. A novel process to synthesize magnetic hollow silicamicrospheres. Colloid Surf A. 2007;311:99–105. doi: 10.1016/j.colsurfa.2007.06.002. [DOI] [Google Scholar]
- 10.Xia H, Foo P, Yi J. Water-dispersible spherically hollow clusters of magnetic nanoparticles. Chem Mater. 2009;21:2442–51. doi: 10.1021/cm900268z. [DOI] [Google Scholar]
- 11.Lu X, Mao H, Zhang W. Fabrication of core-shell Fe3O4/polypyrrole and hollow polypyrrole microspheres. Polym Composites. 2009;30:847–54. doi: 10.1002/pc.20666. [DOI] [Google Scholar]
- 12.Wang C, Chen I, Lin C. Preparation and characterization of hollow magnetic silica (CoFe2O4–SiO2) microspheres. J Magn Magn Mater. 2006;304:451–3. doi: 10.1016/j.jmmm.2006.02.064. [DOI] [Google Scholar]
- 13.Yang S, Liu H, Huang H, Zhang Z. Fabrication of superparamagnetic magnetite/poly(styrene-co-12-acryloxy-9-octadecenoic acid) nanocomposite microspheres with controllable structure. J Colloid Interface Sci. 2009;338:584–90. doi: 10.1016/j.jcis.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71:227–34. doi: 10.1016/j.reactfunctpolym.2010.10.009. [DOI] [Google Scholar]
- 15.Yokoyama M, Satoh A, Sakurai Y, Okano T, Matsumura Y, Kakizoe T, et al. Incorporation of water-insoluble anticancer drug into polymeric micelles and control of their particle size. J Control Release. 1998;55:219–29. doi: 10.1016/S0168-3659(98)00054-6. [DOI] [PubMed] [Google Scholar]
- 16.Nishiyama N, Bae Y, Miyata K, Fukushima S, Kataoka K. Smart polymeric micelles for gene and drug delivery. Drug Discov Today. 2005;2:21–6. doi: 10.1016/j.ddtec.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Litzinger DC, Buiting AMJ, van Rooijen N, Huang L. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim Biophys Acta. 1994;1190:99–107. doi: 10.1016/0005-2736(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 18.Fuchigami T, Kawamura R, Kitamoto Y, Nakagawa M, Namiki Y. Ferromagnetic FePt-nanoparticles/polycation hybrid capsules designed for a magnetically guided drug delivery system. Langmuir. 2011;27:2923–8. doi: 10.1021/la1041019. [DOI] [PubMed] [Google Scholar]
- 19.Fuchigami T, Kawamura R, Kitamoto Y, Nakagawa M, Namiki Y. A magnetically guided anti-cancer drug delivery system using porous FePt capsules. Biomaterials. 2012;33:1682–7. doi: 10.1016/j.biomaterials.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Namiki Y, Fuchigami T, Tada N, Kawamura R, Matsunuma S, Kitamoto Y, et al. Nanomedicine for cancer: lipid-based nanostructures for drug delivery and monitoring. Acc Chem Res. 2011;44:1080–93. doi: 10.1021/ar200011r. [DOI] [PubMed] [Google Scholar]
- 21.Namiki Y, Namiki T, Yoshida H, Ishii Y, Tsubota A, Koido S, et al. A novel magnetic crystal-lipid nanostructure for magnetically guided in vivo gene delivery. Nat Nanotechnol. 2009;4:598–606. doi: 10.1038/nnano.2009.202. [DOI] [PubMed] [Google Scholar]
- 22.Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys. 2003;26:167–80. doi: 10.1088/0022-3727/36/13/201. [DOI] [Google Scholar]
- 23.Hirota Y, Akiyama Y, Izumi Y, Nishijima S. Fundamental study for development magnetic drug delivery system. Physica C. 2009;469:1853–6. doi: 10.1016/j.physc.2009.05.248. [DOI] [Google Scholar]
- 24.Vlaskou D, Mykhaylyk O, Krötz F, Hellwig N, Renner R, Schillinger U, et al. Magnetic and acoustically active lipospheres for magnetically targeted nucleic acid delivery. Adv Funct Mater. 2010;20:3881–94. doi: 10.1002/adfm.200902388. [DOI] [Google Scholar]
- 25.Sakuma H, Taniyama T, Ishii K, Kitamoto Y, Yamazaki Y. Analysis of atomic arrangement in magnetic Fe-Pt nanoparticles. J Magn Magn Mater. 2006;300:284–92. doi: 10.1016/j.jmmm.2005.05.016. [DOI] [Google Scholar]
- 26.Kitamoto Y, He J. Chemical synthesis of FePt nanoparticles with high alternate current magnetic susceptibility for biomedical applications. Electrochim Acta. 2009;54:5969–72. doi: 10.1016/j.electacta.2009.02.092. [DOI] [Google Scholar]
- 27.Iwamoto T, Matsumoto K, Kitamoto Y, Toshima N. Direct synthesis of fct-structured FePt nanoparticles at low temperature with assistance of poly(N-vinyl-2-pyrrolidone) J Colloid Interface Sci. 2007;308:564–7. doi: 10.1016/j.jcis.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 28.Neuberger T, Schöpf B, Hofmann H, Hofmann M, Rechenberg B. Superparamagnetic nanoparticles for biomedical applications: possibilities and limitations of a new drug delivery system. J Magn Magn Mater. 2005;293:483–96. doi: 10.1016/j.jmmm.2005.01.064. [DOI] [Google Scholar]