

Background: DyP-type peroxidases catalyze biotechnologically important reactions.
Results: Based on the crystal structure of a fungal DyP, the conformational flexibility of Asp-168 is elucidated. Tyr-337 is identified as a surface-exposed substrate interaction site.
Conclusion: Asp-168 and Tyr-337 are key residues directly involved in AauDyPI-catalysis.
Significance: Peroxidases are biocatalysts, much sought after and ubiquitous enzymes in nature.
Keywords: Crystal Structure, Enzyme Catalysis, Glycoprotein, Heme, X-ray Crystallography, Acid-Base Pair, Dye-decolorizing Peroxidases (DyPs), Lignin, Long Range Electron Transfer (LRET), Redox Enzyme
Abstract
Dye-decolorizing peroxidases (DyPs) belong to the large group of heme peroxidases. They utilize hydrogen peroxide to catalyze oxidations of various organic compounds. AauDyPI from Auricularia auricula-judae (fungi) was crystallized, and its crystal structure was determined at 2.1 Å resolution. The mostly helical structure also shows a β-sheet motif typical for DyPs and Cld (chlorite dismutase)-related structures and includes the complete polypeptide chain. At the distal side of the heme molecule, a flexible aspartate residue (Asp-168) plays a key role in catalysis. It guides incoming hydrogen peroxide toward the heme iron and mediates proton rearrangement in the process of Compound I formation. Afterward, its side chain changes its conformation, now pointing toward the protein backbone. We propose an extended functionality of Asp-168, which acts like a gatekeeper by altering the width of the heme cavity access channel. Chemical modifications of potentially redox-active amino acids show that a tyrosine is involved in substrate interaction. Using spin-trapping experiments, a transient radical on the surface-exposed Tyr-337 was identified as the oxidation site for bulky substrates. A possible long-range electron transfer pathway from the surface of the enzyme to the redox cofactor (heme) is discussed.
Introduction
Peroxidases (EC 1.11.1) are ubiquitous enzymes that catalyze the oxidative conversion of various compounds utilizing hydrogen peroxide (H2O2) as electron acceptor. As a common trait, most peroxidases, although phylogenetically unrelated, contain a heme B (iron protoporphyrin IX) molecule as the redox cofactor.
A division of this heterogenic group led to the formation of four families, one of which consist of the dye-decolorizing peroxidases or DyPs3 (EC 1.11.1.19) (1). According to Welinder's systematics, which were later on extended (2, 3), DyPs were initially grouped in the class of secretory fungal peroxidases viz. class II of the peroxidase-catalase superfamily. This class further comprises lignin peroxidase, manganese peroxidase, and versatile peroxidase. However, DyPs turned out to be phylogenetically as well as structurally unrelated to all hitherto described peroxidase families. As a result, a new family of enzymes was established to accommodate these unusual peroxidases (4). From a structural viewpoint, DyPs are best considered members of a highly diverse superfamily of proteins comprising, among others, Clds and DyPs, sharing a ferredoxin-like core of β-sheets as a common feature (5). Actually, even the term “DyP” circumscribes a polyphyletic group that can be roughly divided into four different entities. The groups DyPA–C comprise predominantly bacterial enzymes, whereas DyPD is a fungal group (6). The first DyP was discovered in 1995 (7). A mixture of extracellular enzymes secreted by the basidiomycete Bjerkandera adusta (the strain was back then misidentified as Thanatephorus cucumeris) was reported to efficiently oxidize anthraquinone dyes such as Reactive Blue 5 (7, 8). The enzyme responsible for the decolorization process was characterized as a heme peroxidase of unusual chemical properties. Despite the general versatility of peroxidase chemistry, no fungal peroxidases had been known to efficiently oxidize synthetic anthraquinone dyes so far.
DyPs catalyze many reactions, among them several conversions that are biotechnologically desirable. Enzymatic assays on a fungal DyP from the basidiomycete Auricularia auricula-judae (AauDyP, formerly labeled “AjP”) have been reported in a previous publication (9). The isolated enzyme exhibited the typical range of DyP-type peroxidase reactions. Interestingly, this reaction range also encompassed a low-pH activity to oxidize nonphenolic compounds such as 3,4-dimethoxybenzylalcohol (veratryl alcohol), like lignin peroxidase, a fungal peroxidase involved in lignin degradation. Lignin is considered one of the most abundant and recalcitrant biopolymers on earth. This suggests that at least this DyP-type peroxidase might take part in the decomposition of lignin. Lately, other reactions catalyzed by DyPs have been discovered, e.g. the oxidative cleavage of carotenoids (10) and sulfoxidation of aromatic sulfides (11). Although the mentioned and yet to be fully unraveled chemical properties of DyPs demonstrate their obvious biotechnological potential, their actual role in nature remains obscure.
The putative oxidative cycle of fungal DyPs is largely equivalent to that in other peroxidases, but was recently extended by the proposal of a swinging mechanism of a distal aspartate residue during Compound I formation (12). In Scheme 1, the formation of Compound I is depicted. In a first step, H2O2 enters the heme cavity of the enzyme in resting state where it displaces a water molecule that occupies the sixth ferric iron coordination site of the protoporphyrin IX system. A distal basic amino acid residue mediates the rearrangement of a proton in H2O2. In peroxidases, this base typically is a histidine, whereas in DyPs, this key residue is substituted by an aspartate (see “Results and Discussion”). The heme molecule is then oxidized to the radical-cationic oxoferryl species Compound I by two-fold single electron transfer, releasing a water molecule. Two electrons are successively drawn from substrate molecules, leading to their oxidized counterparts. Concomitantly, the heme is stepwise reduced back to its initial oxidation state, leading to the enzyme resting state in this process.
SCHEME 1.

General formation of Compound I in peroxidases. This is the key step in heme peroxidase catalysis. After proton rearrangement by the distal base-catalytic residue, two electrons are transferred from heme to hydrogen peroxide, leading to the active state of the enzyme, called Compound I (13). B indicates a basic residue, e.g. a histidine or aspartate/glutamate in peroxidases. L indicates the proximal heme ligand, usually a histidine residue.
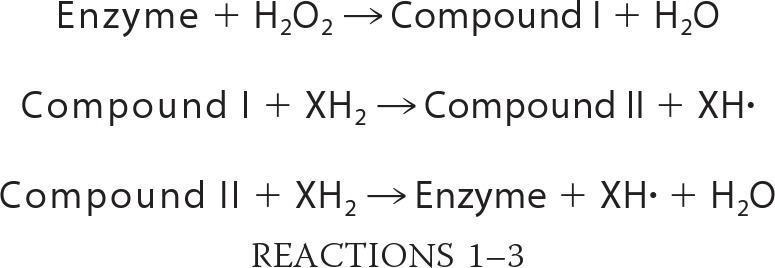 |
The above reactions summarize the general catalytic cycle of peroxidases. The term “Enzyme” indicates the peroxidase resting state, and XH2 and XH• are a substrate molecule and the corresponding radical species, respectively.
Up to the present, only a few DyP-like enzymes from fungi have been described biochemically in detail, and only one crystal structure has been published so far (14). Research on bacterial members has been comparatively more extensive. A native crystal structure was needed to substantiate recent results involving fungal as well as bacterial DyP-type peroxidases (5, 12). To gain further insight into the structural properties of the eucaryotic (in this case fungal) DyPs, AauDyPI from the jelly fungus A. auricula-judae (Auriculariales, Basidiomycota) was purified and crystallized, and the crystal structure was determined at 2.1 Å resolution (Protein Data Bank (PDB) code 4AU9).
EXPERIMENTAL PROCEDURES
Isolation of Nucleic Acids, PCR, and mRNA Sequencing
For detailed methodology, see elsewhere (49). In short, mRNA was extracted from mycelia of A. auricula-judae grown in agitated cultures. For cDNA synthesis, the total mRNA (1 μg) was primed and subsequently reverse-transcribed. Afterward the obtained cDNA was amplified following a customized PCR protocol.
Primers were designed to amplify fragments of a DyP-type peroxidase gene from A. auricula-judae. The obtained PCR products were purified and sequenced. Strain DSMZ 11236 (from the Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany) was checked for species identity by internal transcribed spacer PCR (ITS-PCR) on genomic DNA with primers ITS1 and ITS4 (15).
Protein Production and Purification of AauDyPI
Production and purification of AauDyPI were carried out as described in a previous publication (9). To summarize, A. auricula-judae was grown in agitated tomato juice medium at 24 °C. At maximum peroxidase activity level, the cultures were harvested and directly filtrated. The enzyme was purified using Q-Sepharose chromatography columns. Purified enzyme was stored in 5 mm sodium acetate buffer at 4 °C.
Crystallization
Purified AauDyPI was crystallized using protein concentrations of 10.7 mg ml−1 in 5 mm sodium acetate, pH 6.8. Various crystallization kits (Hampton Research, Aliso Viejo, CA and Jena Bioscience, Jena, Germany) were used for initial screens. Crystals were grown at 19 °C by the hanging drop vapor diffusion method with a 1:1 (v/v) ratio of protein-to-precipitant in 4-μl drops. Intergrown plate-like crystals were obtained with a precipitant solution consisting of 0.17 m sodium acetate trihydrate, 0.085 m Tris hydrochloride, pH 8.5, 25.5% (w/v) polyethylene glycol 4.000, and 15% (v/v) glycerol. From these crystal clusters, very small single fragments of about 0.2 × 0.1 × >0.001 mm were retrieved that seemed suitable for x-ray crystallographic experiments. To verify the results of structure determination, we examined the purified protein via LC-MS.
Data Collection and Processing
AauDyPI crystals were directly flash-cooled in liquid nitrogen. X-ray diffraction data were collected on the macromolecular crystallographic beamline ID14-4 at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France) (16). Data were indexed, processed, and scaled with XDS (17, 18). Data collection and processing statistics are given in Table 1.
TABLE 1.
Summary of data collection and refinement statistics of AauDyPI
Values in parentheses belong to the highest resolution shell.
| Data collection | |
| Space group | P21 |
| Unit-cell parameters (Å/°) | a = 66.60; b = 46.69; c = 141.20; β = 91.35 |
| Beamline | ID14-4, ESRF |
| Wavelength (Å) | 0.9395 |
| Temperature (K) | 100 |
| No. of crystals | 1 |
| Resolution range (Å) | 49.01–2.10 (2.23–2.10) |
| Total no. of reflections | 131,951 (19,754) |
| No. of unique reflections | 49,651 (7661) |
| Completeness (%) | 96.9 (93.4) |
| I/σ(I) | 11.02 (3.35) |
| Rmerge | 0.094 (0.437) |
| Refinement | |
| Rwork/Rfreea | 0.180/0.261 |
| Molecules per asymmetric unit | 2 |
| Solvent content (%) | 42 |
| No. of amino acid residues | 896 |
| No. of heme molecules | 2 |
| No. of carbohydrates | 8 |
| No. of ligands | 6 glycerol, 1 acetate, 1 Tris |
| No. of water molecules | 679 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.25 |
| Ramachandran plot (%) | |
| Most favored | 93.9 |
| Allowed | 5.4 |
| Disallowed | 0.7 |
| Average B factors (Å2) | |
| Main chain atoms | 24.3 |
| Side chain atoms | 25.2 |
| Water molecules | 37.1 |
| Ligands/hemes/carbohydrates | 36.1 |
| Overall | 26.2 |
a Rfree values are calculated with 5% of the data.
Structure Determination
The structure of AauDyPI was determined by molecular replacement using MOLREP (19) and Phaser (20). An ad interim structure of the B. adusta DyP (PDB code 2D3Q) was used as search model. This structure will be referred to as BadDyP in the following. Side chains of nonconserved amino acids were truncated at the last carbon atom common to the target and model residues with the CCP4-program CHAINSAW (21). Because there are two molecules per asymmetric unit, density modification including noncrystallographic symmetry averaging was performed to improve the initial phases using the CCP4 program PARROT (22). An initial model was built with Coot (23) and refined in REFMAC5 (24). Further improvement was achieved by successive cycles of model building and refinement.
Identification of a Redox-active Surface-exposed Tyrosine
Chemical modification of AauDyPI was carried out as described by Inokuchi et al. (25) and Miki et al. (26). In brief, AauDyPI was incubated with 5–150-fold excess of N-bromosuccinimide or tetranitromethane, respectively. The modified proteins were directly used in an enzyme assay monitoring the oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) at 420 nm. Spin trapping was performed using the protocol of Zhao et al. (27). At first AauDyPI was deglycosylated using 10 units of peptide-N-glycosidase F (Aldrich) at room temperature for 1.5 days. The thus prepared enzyme was mixed with excess proline nitric oxide after incubation with peroxyacetic acid. This mixture was incubated for 15 min at 37 °C and then rebuffered in 20 mm K3PO4 buffer, pH 7.2. The enzyme was recovered from the reaction mixture by SDS-PAGE and subsequent Coomassie Blue staining. The bands were cut from the gel using a sterile scalpel. Prior to mass spectrometry, the protein was digested in-gel with elastase and thermolysin, respectively. The peptide containing the spin-trapped amino acid residue was identified using an Agilent 6520 quadrupole-TOF mass spectrometer coupled to a reverse-phase HPLC unit and a nano-electrospray ionization source.
Electron Transfer Calculations, Probing of the Heme Access Channel, and Imaging
The calculation of electron transfer pathways was performed with the program HARLEM (as described in Ref. 28) using the PATHWAY module. Volume and shape of the heme access channel were calculated with HOLLOW (29).
Two coordinate files of AauDyPI were created from the original file, each one containing one of the two conformers of Asp-168. Solvent molecules were removed from the coordinate files, and a simulation of the Compound I state containing a randomly placed guaiacol (2-methoxyphenol) molecule was built using Coot. An oxygen atom was thus placed distally perpendicular to the heme plane at 1.7 Å distance from iron as found in the Compound I structure of horseradish peroxidase (PDB entry 1HCH (30)). Ligand docking was then performed with the Molegro Virtual Docker (MVD) (31). The ligand binding cavities were identified using the Expanded van der Waals algorithm with a grid size of 0.5 Å. 50 docking runs were performed using the guaiacol molecule as ligand. The population size was set to 200 over a radius of 12 Å around the predicted binding cavity with a grid size of 0.2 Å. 10,000 iterations per position were performed. Clustering of similar positions was enabled using an r.m.s. deviation of 1.5 Å. The ligand positioning was constrained to the predicted binding site, i.e. the distal heme cavity. Images were created in PyMOL (32) and CCP4mg (33).
RESULTS AND DISCUSSION
Structure Solution and Refinement
Improvement of crystal size and quality of AauDyPI proved unexpectedly arduous. Despite numerous variations of the initial crystallization condition, only very thin plate-like crystals were obtained. Crystal growth was exclusively observed in solutions with a high concentration of polyethylene glycol (PEG) 4000. The widely used ammonium sulfate yielded only amorphous precipitate. The presence of cryoprotectants such as glycerol or 2-methyl-2,4-pentanediol ab initio was beneficial to crystal quality. Additives such as various amino acids did not show any positive effects. Bearing in mind the size and morphology of the crystals, surprisingly good x-ray diffraction data were collected; the best crystals diffracted to a maximum resolution of about 2.1 Å (Table 1).
The relatively high sequence identity between AauDyPI and BadDyP (PDB code 2D3Q) suggested molecular replacement as a promising approach to solve the structure. Indeed, this technique was successful. Phase modification techniques gave a further improvement, resulting in a high-quality electron density map. This allowed us to build an initial model of the complete protein, which was subsequently refined in REFMAC5. The final refinement statistics are supplied in Table 1.
Overall Structure of AauDyPI
AauDyPI is a globular glycoprotein with a helical basic architecture and a prominent β-sheet motif spanning the distal side of the heme plane. A proximal N- and a distal C-terminal domain embed the heme molecule, which itself is partly flanked by the β-sheet of the ferredoxin-like fold. Like other DyPs, it is structurally very distinct from the classical heme peroxidases because the latter are practically completely helical. The monomeric AauDyPI has an ellipsoid-like shape with dimensions of 70 × 42 × 40 Å3. The refined model of AauDyPI consists of the complete polypeptide chain of 448 amino acids as predicted by gene sequencing.4 Even the C and N terminus could be modeled as well as some rather large flexible loop regions (Fig. 1A), where electron density was mostly poorly defined. In the crystal, there are two molecules per asymmetric unit. The Cα-r.m.s. between the two subunits is 0.375 Å, which is in the order of the estimated coordinate error of 0.257 Å. Only in the flexible surface loop regions were larger deviations between the two molecules observed. Given the consensus sequence Asn-X-Ser/Thr with X being any amino acid except proline or aspartic acid, there are four potential glycosylation sites. Three of them could be verified crystallographically, whereas closer inspection of Asn-322 reveals that this fourth site is inaccessible for glycosyltransferases and consequently is not glycosylated. The glycosylation sites are exclusively distributed on the “lower” end of the proximal side with respect to the heme molecule (Fig. 1A), especially at large, protruding loops that are susceptible to proteolytic attacks. Therefore, the carbohydrate moieties might have a protective function against proteases.
FIGURE 1.

Overview of the AauDyPI structure. A, ribbon diagram of AauDyPI. The ferredoxin-like β-sheet fold is highlighted in green, the protoporphyrin IX system is in dark violet with the central iron atom as a tan sphere, and the carbohydrate chains with their corresponding N-linked asparagine residues are in purple. The chain termini are labeled as well. B, superposition of BadDyP (mauve and purple) onto AauDyPI (green shading). The Cα-trace is shown with helices as tubes. C, superposition of Cld (pink) onto AauDyPI (whitish and green shading). The Cα-trace is shown with helices as tubes.
In AauDyPI, some carbohydrate chains are definitely glycosylated to a higher extent than outlined here. Residual electron density, albeit weak and difficult to interpret, strongly indicates the presence of branched N-glycans of the high-mannose type (34). Further model building was considered to be unrewarding.
AauDyPI shows an overall structure characteristic for the DyP-type family, which in turn is somewhat related to that of the chlorite detoxifying Clds (chlorite dismutases). The fold of these heme enzymes also includes the particular ferredoxin-like β-sheet, similar to that found in DyPs (35). However, a sequence alignment indicated only 8% identity between the Cld of Dechloromonas aromatica (a β-proteobacterium) and BadDyP, which is essentially nonhomologous (5). Consequently, a secondary structure matching superposition of AauDyP and Cld results in a large Cα-r.m.s. of 2.502 Å (Fig. 1C). The Cα-r.m.s. was calculated after secondary structure matching superposition with the CCP4-program SUPERPOSE (36).
Structurally, AauDyPI is most similar to BadDyP, as expected from the molecular replacement results, correlating with a Cα-r.m.s. of 1.14 Å for 427 common atoms (Fig. 1B). This value is relatively high in view of the sequence identity of 53% (66% similarity) and the unique molecular replacement solution obtained with PHASER. Helices and β-sheets are mostly conserved, whereas the loop regions are more diverse. DyP structures of procaryotic origin differ significantly from fungal enzymes because they lack the heme. In addition, their polypeptides are considerably (by one-third) smaller and form oligomers (dimers to hexamers) (37).
As in most peroxidases, a histidine residue coordinates to the central heme iron as the fifth ligand, with a distance of 2.1 Å (Fig. 2A). Usually heme-Fe(III) is coordinated six-fold, the distal ligand site occupied by a water molecule. Here, the water molecule closest to the iron atom is 3.11 Å apart. This distance is too large for a coordination bond. Probably the iron in the crystal structure represents a Fe(II) species, which usually coordinates five-fold. Most likely, the heme-Fe(III) was reduced by the high-intensity x-ray beam under cryocondition. From the distal side cavity, a channel leads to the exterior, which most likely serves as the substrate access channel (see below) (12).
FIGURE 2.

Heme and its environment in AauDyP. A, H-bonding pattern of propionate D. B, H-bonding pattern of propionate C.
A characteristic trait of the DyP-type and Cld-like enzymes is the unusual conformation of the heme propionate residue at pyrrole C (5). Generally, both propionate groups in peroxidases display a conformation like the one of the propionate at pyrrole ring D (Fig. 2A). In DyPs, the propionate at pyrrole C is tilted into an unusual conformation due to the formation of strong hydrogen bonds to the protein (Fig. 2B). Propionate D of AauDyPI forms a long H-bond with Nη2 of Arg-311 and additionally with four water molecules. This hydrogen-bonding pattern allows propionate D to maintain an unstrained conformation. In contrast, propionate C is much more confined by five hydrogen bonds. Among them, there are three strong H-bonds to the main chain N of Ile-170 and Ala-171 and to Nη2 of Arg-332, respectively. Additionally, two more H-bonds to Nϵ of Arg-255 and to one water molecule are formed. This strong hydrogen-bonding network obviously forces propionate C into a high-energy conformation, hence enforcing a rather close distance of 3.6 Å between one of the carboxyl oxygens and a methine group of the porphyrin ring. As a result, the sp2-plane of the propionate lies almost orthogonally to the heme plane. The oxygen sensor FixL might be another example where interactions with arginine side chains constrain the propionate chains into unusual geometries upon O2 binding (38).
Geometry of the Distal Heme Cavity and Implications for Catalysis
A first partial catalytic cycle for DyPs has been proposed by Sugano et al. (4). Fundamentally, the catalytic cycle in DyPs is the same as in other peroxidases. The heme molecule is oxidized by peroxides to the radical-cationic oxoferryl species Compound I, which is two electrons deficient from the resting state. One electron is abstracted from the iron, resulting in Fe(IV), and another one is abstracted from the porphyrin ring. It became apparent that a distal acid-base pair is essential for Compound I formation in peroxidases to rearrange a proton in the peroxide substrate. When compared with classical peroxidases, proton rearrangement or abstraction in DyPs is accomplished in a different way. Because binding to the heme iron lowers the pKa of hydrogen peroxide (39), deprotonation does not require any strong bases. Most peroxidases use a histidine residue, whereas in DyPs, an even weaker base, a deprotonated aspartate residue, is employed. An arginine is paired with the histidine or aspartate, respectively. It does not take part in the rearrangement process directly, but is essential for coordinating H2O2 at the sixth ligand site of heme-Fe(III) and stabilizing Compound I. Surprisingly, the aspartate residue is not essential for catalysis in procaryotic DyPs. Although Compound I stability is reduced in mutants lacking said aspartate, they retain a good portion of their enzymatic activity. Mutation of the distal arginine, however, results in complete failure to form Compound I and, concomitantly, loss of any peroxidase activity (40).
The mechanism of Compound I formation in DyPs has been amended recently by the concept of a swinging mechanism of the distal aspartate residue during this process (12). Said aspartate changes the location of its side chain upon the presence of cyanide, a structural analog of hydrogen peroxide, in the distal heme cavity when compared with the cyanide-free enzyme. It was proposed that this aspartate swings toward the heme molecule in the presence of H2O2 and thereupon mediates the rearrangement of a proton. Once Compound I formation is finished, it swings back to the initial position.
In AauDyPI, Asp-168 plays the part of the mediating residue. During model building, some issues arose concerning this key amino acid. Electron density clearly indicated two distinct conformations of Asp-168 as shown in Fig. 3.
FIGURE 3.

Multiple conformations of Asp-168. The two conformations of Asp-168 are shown in their corresponding electron density drawn as a light blue mesh. In this and all subsequent figures, distances are given in Å.
The Oδ1 and Oδ2 atoms of Asp-168 form hydrogen bonds to Nη1 of Arg-332, thus establishing the catalytic distal acid-base pair. This is the conformation necessary for Compound I formation. In the alternative conformation (refined with 0.5 occupancy), the side chain of Asp-168 has flipped toward a loop region in the opposite direction. The flipping angle is about 130°. Now both Oδ1 and Oδ2 are able to form hydrogen bonds to the protein backbone, more precisely to the carbonyl oxygen of Gly-160. Additionally, each carboxyl oxygen is bridged to a water molecule by a hydrogen bond of 3 Å. Average temperature factors of <30 Å2 for the atoms of Asp-168 in each conformation in both subunits create further confidence in the probability of our model. These data are consistent with two distinct conformations of the Asp-168 side chain, indicating flexibility in a peptide, which is otherwise rather rigid and crystallographically well defined. The two conformations might be indicative for a functionality related to the swinging mechanism proposed by Yoshida et al. (12). Moreover, this allows us to discuss yet another issue, the accessibility of the distal side cavity.
The heme access channel features an overall funnel-like shape that significantly narrows toward the distal site of the heme (see Fig. 5). It is largely hydrophobic with the exception of the distal Asp-168/Arg-332 pair and several hydrophilic residues directly at the channel entry. In BadDyP, the opening to the binding pocket was reported to measure about 3 Å, having a roughly circular appearance (Fig. 4A). The above mentioned Asp-168 is part of a strictly conserved tetrade that defines the distal substrate binding pocket. In one conformation, the side chain of Asp-168 coincides with that of Asp-171 in BadDyP. Consequently, AauDyPI features a very similar opening with a slightly ellipsoid appearance (Fig. 4B). These are the general conformations involved in forming Compound I. Fig. 4C shows the opening to the substrate binding pocket with Asp-168 in its alternative conformation facing toward the peripheral protein backbone. In this situation, the opening is enlarged significantly, measuring about 6 Å in one dimension and about 3 Å in the other. Switching the side chain toward the carbonyl oxygen of Gly-160, possibly after Compound I formation, Asp-168 might work like a gatekeeper that allows passage of substrate molecules bigger than hydrogen peroxide (i.e. small organic compounds). Consequently, this would enable the enzyme to oxidize substrates directly inside the heme cavity.
FIGURE 5.

Distribution of Trp and Tyr residues in AauDyPI and visualization of the heme access channel calculated with HOLLOW. The proposed LRET from Tyr-337 to the heme is shown in the inset.
FIGURE 4.
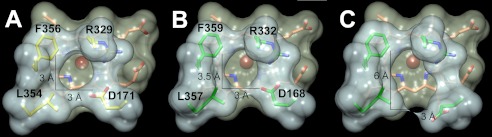
Side chain flipping of Asp-168 and its effects on the accession channel toward the heme plane. A and B, substrate accession channel in BadDyP (A) and AauDyPI (B) with Asp-168 facing to Arg-332. C, AauDyPI with Asp-168 facing to Gly-160.
Identification of Substrate Oxidation Sites
To explore the possibility of small substrates being oxidized directly in the heme cavity, we tentatively modeled a Compound I state of AauDyPI. As described above, Compound I was modeled based on the parameters of the oxoferryl species reported for horseradish peroxidase (PDB entry 1HCH 27). A guaiacol molecule was included into this hypothetical structure, a standard substrate for peroxidases. The MolDock algorithm of MOLEGRO was used to examine whether the distal substrate binding pocket is sufficiently spacious to contain the model compound. The calculations confirmed that molecules of this size narrowly fit in the heme access channel of the enzyme.
However, the majority of DyP substrates are considerably larger than guaiacol. Keeping in mind the nature of Compound I (involving formation of a radical cation) and widening the perspective on other peroxidases, we gathered an alternative: a surface-exposed substrate interaction site. Depending on the type of peroxidase, a one-electron hole migrates, resulting in a protein-based radical cation (41). Thus, in such a case, a novel protein-based substrate interaction site is introduced. This requires a long-range electron transfer (LRET) pathway (42) from the porphyrin ring to an appropriate redox active amino acid residue at the surface of the enzyme. An exposed oxidation site has been demonstrated extensively in a related heme enzyme viz. lignin peroxidase, where a hydroxylated tryptophan residue carries out the electron withdrawal from substrate molecules (43–45). In an electron density map of lignin peroxidase from Phanerochaete chrysosporium, strong electron density showed a hydroxylation at the Cβ position of the surface-exposed Trp-171. It was assumed that this covalent modification was the result of a reaction of water or oxygen with a radical cation intermediate at Trp-171. Because the Cβ-hydroxylation was absent in a crystal structure of pristine lignin peroxidase (i.e. enzyme before its first enzymatic turnover), it must have originated from an autocatalytic process during the first catalytic cycle. Trp-171 mutants (W171F, W171S) were shown to be completely inactive, confirming Trp-171 as the peripheral substrate oxidation site (44, 45) for substrates like veratryl alcohol. In a similar way, a tyrosine on the enzyme surface acts as the substrate interaction site in lignin peroxidase from Trametes cervina (26).
Because AauDyPI is able to degrade bulky substrates such as Reactive Blue 5 (46), it seems obvious to identify those redox-active residues (i.e. Trp, Tyr) in AauDyPI that might act as surface-exposed oxidation sites (Fig. 5). Because no post-translational changes that would directly indicate such redox-active residues could be detected during model building, we could not identify a surface-exposed substrate interaction site from the crystal structure. To address this issue experimentally, in a first step, we measured enzyme activities after chemically modifying tyrosine and tryptophan residues. For specific modification of these amino acids, tetranitromethane and N-bromosuccinimide were used, respectively. Although there was no effect on the activity using N-bromosuccinimide, with tetranitromethane, a significant decrease to roughly 20% of the original activity with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) as substrate was detected, strongly indicating the involvement of a tyrosine residue in the catalytic process. To locate this key residue (in AauDyPI, there are seven tyrosines in total), spin-trapping experiments were performed followed by mass spectrometric analysis. Excess proline nitric oxide was used to yield nitrotyrosine from transient tyrosyl radicals (27); it generates NO, which acts as an in situ radical scavenger trapping the expected tyrosyl radicals that were generated by adding peroxyacetic acid to an AauDyPI solution. Nitrosotyrosine thus formed is subsequently oxidized to the stable nitrotyrosine. The modified enzyme was finally digested proteolytically and subjected to LC-MS analysis. Nitrotyrosine was solely detected at residue 337.
Tyr-337 is surface-exposed and located at the beginning of a large protruding loop comprising 20 amino acids. Its side chain is partially embedded in a hollow formed by side chains of residues Leu-149 and Gln-218 and the main chain of residues 354–358 with the edge of the aromatic ring pointing toward the solvent. The latter peptide is the end of the aforementioned loop and extends into the inner β-sheet, which is closest to the distal site of the heme. Tyr-337 forms hydrogen bonds via backbone to Gly-356 and a van der Waals contact of its side chain to the nitrogen atom of Leu-357. Interestingly, the side chain of Leu-357 is in van der Waals contact (∼3.8 Å) with the heme, suggesting a possible LRET pathway. Therefore, potential electron transfer pathways from Tyr-337 to the heme molecule were analyzed with HARLEM. The best pathway is shown in the inset of Fig. 5. It proceeds from the benzene ring of Tyr-337 to the backbone amide nitrogen of Leu-357 involving a through-space jump of 3.3 Å followed by the leucine side chain and another jump (3.8 Å) from Cδ1 of Leu-357 to one of the pyrrole rings. The effective length of this pathway is about 13 Å. The hydroxyl group Tyr-337 forms a strong hydrogen bond of 2.6 Å with the carboxylate of Glu-354. It has been reported that acidic side chains in the vicinity of a tryptophan or a tyrosine stabilize an emerging radical cation (26, 47, 48). Alignments with other DyPs show that Tyr-337 is a conserved residue in fungal as well as bacterial DyPs and the related enzymes TyrA from Shewanella oneidensis and EfeB from Escherichia coli but is missing in Cld-like proteins (5).
Our experiments provide the first unambiguous identification of a surface-exposed oxidation site in the large group of dye-decolorizing peroxidases. Further investigations including crystallization of AauDyPI-substrate complexes and site-directed mutagenesis to further substantiate the significance of Tyr-337 and the suggested double responsibility of Asp-168 for the catalytic cycle are in progress.
Acknowledgments
We gratefully acknowledge the opportunity to collect diffraction data on the synchrotron beamline ID14-4 at the ESRF. We thank the staff for technical support. Dr. Eric Haaf of the proteomics unit of the Zentrum für Biosystemanalyse (ZBSA) in Freiburg, Germany is thanked for LC-MS experiments.
This work was partially financed by a grant from the Commission of the European Communities within the Sixth European Framework Programme (BIORENEW contract NMP2-CT-2006-026456) (to K. P. and M. H.) and by funding from the International Research Training Group Grant IRTG 1038 “Catalysts and Catalytic Reactions for Organic Synthesis” (CCROS) of the Deutsche Forschungsgemeinschaft (DFG) (to D. A. P.).
The atomic coordinates and structure factors (code 4AU9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DyP
- dye-decolorizing peroxidase
- Aau
- Auricularia auricula-judae
- Bad
- Bjerkandera adusta
- Cld
- chlorite dismutase
- r.m.s.
- root mean square
- LRET
- long-range electron transfer.
REFERENCES
- 1. Zámocký M., Obinger C. (2010) Molecular phylogeny of heme peroxidases. In: Biocatalysis Based on Heme Peroxidases. (Torres E., Ayala M., eds) pp. 7–35, Springer Verlag, Berlin, Germany [Google Scholar]
- 2. Welinder K. G. (1992) Superfamily of plant, fungal and bacterial peroxidases. Curr. Opin. Struct. Biol. 2, 388–393 [Google Scholar]
- 3. Martínez A. T. (2002) Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzyme Microb. Technol. 30, 425–444 [Google Scholar]
- 4. Sugano Y., Muramatsu R., Ichiyanagi A., Sato T., Shoda M. (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family. J. Biol. Chem. 282, 36652–36658 [DOI] [PubMed] [Google Scholar]
- 5. Goblirsch B., Kurker R. C., Streit B. R., Wilmot C. M., DuBois J. L. (2011) Chlorite dismutases, DyPs, and EfeB: 3 microbial heme enzyme families comprise the CDE structural superfamily. J. Mol. Biol. 408, 379–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmad M., Roberts J. N., Hardiman E. M., Singh R., Eltis L. D., Bugg T. D. H. (2011) Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 50, 5096–5107 [DOI] [PubMed] [Google Scholar]
- 7. Kim S. J., Ishikawa K., Hirai M., Shoda M. (1995) Characteristics of a newly isolated fungus, Geotrichum candidum Dec 1, which decolorizes various dyes. J. Ferment. Bioeng. 79, 601–607 [Google Scholar]
- 8. Kim S. J., Shoda M. (1999) Purification and characterization of a novel peroxidase from Geotrichum candidum Dec 1 involved in decolorization of dyes. Appl. Environ. Microbiol. 65, 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liers C., Bobeth C., Pecyna M., Ullrich R., Hofrichter M. (2010) DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl. Microbiol. Biotechnol. 85, 1869–1879 [DOI] [PubMed] [Google Scholar]
- 10. Scheibner M., Hülsdau B., Zelena K., Nimtz M., de Boer L., Berger R. G., Zorn H. (2008) Novel peroxidases of Marasmius scorodonius degrade β-carotene. Appl. Microbiol. Biotechnol. 77, 1241–1250 [DOI] [PubMed] [Google Scholar]
- 11. van Bloois E., Torres Pazmiño D. E., Winter R. T., Fraaije M. W. (2010) A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl. Microbiol. Biotechnol. 86, 1419–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshida T., Tsuge H., Konno H., Hisabori T., Sugano Y. (2011) The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J. 278, 2387–2394 [DOI] [PubMed] [Google Scholar]
- 13. Poulos T. L., Kraut J. (1980) The stereochemistry of peroxidase catalysis. J. Biol. Chem. 255, 8199–8205 [PubMed] [Google Scholar]
- 14. Sato T., Hara S., Matsui T., Sazaki G., Saijo S., Ganbe T., Tanaka N., Sugano Y., Shoda M. (2004) A unique dye-decolorizing peroxidase, DyP, from Thanatephorus cucumeris Dec 1: heterologous expression, crystallization and preliminary X-ray analysis. Acta Crystallogr. D 60, 149–152 [DOI] [PubMed] [Google Scholar]
- 15. White T. J., Bruns T., Lee S., Taylor J. (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols – a Guide to Methods and Applications (Innis M. A., Gelfand D. H., Sninsky J. J., White T. J., eds) pp. 315–322, Academic Press, San Diego, CA [Google Scholar]
- 16. McCarthy A. A., Brockhauser S., Nurizzo D., Theveneau P., Mairs T., Spruce D., Guijarro M., Lesourd M., Ravelli R. B. G., McSweeney S. (2009) A decade of user operation on the macromolecular crystallography MAD beamline ID14–4 at the ESRF. J. Synchrotron Radiat. 16, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kabsch W. (2010) Integration, scaling, space-group assignment, and post-refinement. Acta Crystallogr. D 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vagin A., Teplyakow A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 20. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 21. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 22. Cowtan K. (2010) Recent development in classical density modification. Acta Crystallogr. D 66, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Coot: model-building tool for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Skubák P., Murshudov G. N., Pannu N. S. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D 60, 2196–2201 [DOI] [PubMed] [Google Scholar]
- 25. Inokuchi N., Takahashi T., Yoshimoto A., Irie M. (1982) N-Bromosuccinimide oxidation of a glucoamylase from Aspergillus saitoi. J. Biochem. 91, 1661–1668 [DOI] [PubMed] [Google Scholar]
- 26. Miki Y., Calviño F. R., Pogni R., Giansanti S., Ruiz-Dueñas F. J., Martínez M. J., Basosi R., Romero A., Martínez A. T. (2011) Crystallographic, kinetic, and spectroscopic study of the first ligninolytic peroxidase presenting a catalytic tyrosine. J. Biol. Chem. 286, 15525–15534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao X., Girotto S., Yu S., Magliozzo R. S. (2004) Evidence for radical formation at Tyr-353 in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 279, 7606–7612 [DOI] [PubMed] [Google Scholar]
- 28. Prytkova T. R., Kurnikov I. V., Beratan D. N. (2005) Ab initio based calculations of electron-transfer rates in metalloproteins. J. Phys. Chem. B 109, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 29. Ho B. K., Gruswitz F. (2008) HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berglund G. I., Carlsson G. H., Smith A. T., Szöke H., Henriksen A., Hajdu J. (2002) The catalytic pathway of horseradish peroxidase at high resolution. Nature 417, 463–468 [DOI] [PubMed] [Google Scholar]
- 31. Thomsen R., Christensen M. H. (2006) MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 49, 3315–3321 [DOI] [PubMed] [Google Scholar]
- 32. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 33. Potterton E., McNicholas S., Krissinel E., Cowtan K., Noble M. (2002) The CCP4 molecular-graphics project. Acta Crystallogr. D 58, 1955–1957 [DOI] [PubMed] [Google Scholar]
- 34. Lerouge P., Cabanes-Macheteau M., Rayon C., Fischette-Lainé A. C., Gomord V., Faye L. (1998) N-Glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol. Biol. 38, 31–48 [PubMed] [Google Scholar]
- 35. Zubieta C., Krishna S. S., Kapoor M., Kozbial P., McMullan D., Axelrod H. L., Miller M. D., Abdubek P., Ambing E., Astakhova T., Carlton D., Chiu H. J., Clayton T., Deller M. C., Duan L., Elsliger M. A., Feuerhelm J., Grzechnik S. K., Hale J., Hampton E., Han G. W., Jaroszewski L., Jin K. K., Klock H. E., Knuth M. W., Kumar A., Marciano D., Morse A. T., Nigoghossian E., Okach L., Oommachen S., Reyes R., Rife C. L., Schimmel P., van den Bedem H., Weekes D., White A., Xu Q., Hodgson K. O., Wooley J., Deacon A. M., Godzik A., Lesley S. A., Wilson I. A. (2007) Crystal structures of two novel dye-decolorizing peroxidases reveal a β-barrel fold with a conserved heme-binding motif. Proteins 69, 223–233 [DOI] [PubMed] [Google Scholar]
- 36. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 37. Sugano Y. (2009) DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol. Life Sci. 66, 1387–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balland V., Bouzhir-Sima L., Anxolabéhère-Mallart E., Boussac A., Vos M. H., Liebl U., Mattioli T. A. (2006) Functional implications of the propionate 7-arginine 220 interaction in the FixLH oxygen sensor from Bradyrhizobium japonicum. Biochemistry 45, 2072–2084 [DOI] [PubMed] [Google Scholar]
- 39. Jones P., Dunford H. B. (2005) The mechanism of Compound I formation revisited. J. Inorg. Biochem. 99, 2292–2298 [DOI] [PubMed] [Google Scholar]
- 40. Singh R., Grigg J. C., Armstrong Z., Murphy M. E. P., Eltis L. D. (2012) Distal heme pocket residues of B-type dye-decolorizing peroxidases. J. Biol. Chem. 287, 10623–10630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mauro J. M., Fishel L. A., Hazzard J. T., Meyer T. E., Tollin G., Cusanovich M. A., Kraut J. (1988) Tryptophan-191 → phenylalanine, a proximal-side mutation in yeast cytochrome c peroxidase that strongly affects the kinetics of ferrocytochrome c oxidation. Biochemistry 27, 6243–6256 [DOI] [PubMed] [Google Scholar]
- 42. Klapper M. H., Faraggi M. (1979) Applications of pulse-radiolysis to protein chemistry. Q. Rev. Biophys. 12, 465–519 [DOI] [PubMed] [Google Scholar]
- 43. Doyle W. A., Blodig W., Veitch N. C., Piontek K., Smith A. T. (1998) Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry 37, 15097–15105 [DOI] [PubMed] [Google Scholar]
- 44. Blodig W., Doyle W. A., Smith A. T., Winterhalter K., Choinowski T., Piontek K. (1998) Autocatalytic formation of a hydroxy group at Cβ of Trp171 in lignin peroxidase. Biochemistry 37, 8832–8838 [DOI] [PubMed] [Google Scholar]
- 45. Blodig W., Smith A. T., Winterhalter K., Piontek K. (1999) Evidence from spin-trapping for a transient radical on tryptophan residue 171 of lignin peroxidase. Arch. Biochem. Biophys. 370, 86–92 [DOI] [PubMed] [Google Scholar]
- 46. Sugano Y., Matsushima Y., Shoda M. (2006) Complete decolorization of the anthraquinone dye Reactive blue 5 by the concerted action of two peroxidases from Thanatephorus cucumeris Dec 1. Appl. Microbiol. Biotechnol. 73, 862–871 [DOI] [PubMed] [Google Scholar]
- 47. Piontek K., Smith A. T., Blodig W. (2001) Lignin peroxidase structure and function. Biochem. Soc. Trans. 29, 111–116 [DOI] [PubMed] [Google Scholar]
- 48. Smith A. T., Doyle W. A., Dorlet P., Ivancich A. (2009) Spectroscopic evidence for an engineered, catalytically active Trp radical that creates the unique reactivity of lignin peroxidase. Proc. Natl. Acad. Sci. U.S.A. 106, 16084–16089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liers C., Pecyna M. J., Kellner H., Worrich A., Zorn H., Steffen K. T., Hofrichter M., Ullrich R. (2012) Appl. Microbiol. Biotechnol., DOI 10.1007/s00253-012-4521-2 [DOI] [PubMed] [Google Scholar]