

Abstract
In vertebrates, the biosynthesis of steroid hormones is initiated by cytochrome P450 CYP11A1 which converts cholesterol to pregnenolone. We investigated whether some of the experimental and FDA-approved therapeutic agents alter the activity of CYP11A1 in the reconstituted system in vitro. We found that under the experimental conditions used and when phospholipids are included, ketoconazole, posaconazole, carbenoxolone, and selegiline inhibit CYP11A1-mediated production of pregnenolone by at least 67%. Conversely, pemirolast, clobenpropit, desogestrel, dexmedetomidine, and tizanidine stimulate the enzyme activity by up to 70%. We then evaluated the identified inhibitors and activators for spectral binding to CYP11A1 and their effect on enzyme activity in the absence of phospholipids. The data obtained provide insight into how different drugs interact with CYP11A1 and demonstrate that P450 association with the lipid bilayer determines, in many cases, a drug’s effect on enzyme activity.
Keywords: cytochrome P450, CYP11A1, CYP46A1, inhibition, activation, drug target, cholesterol, steroidogenesis
1. Introduction
Cytochrome P450 11A1 (CYP11A1) or cholesterol side chain cleavage enzyme, is a mitochondrial monooxygenase catalyzing the conversion of cholesterol to pregnenolone, the precursor of all steroid hormones. CYP11A1 has been intensively studied during the last 40 years (reviewed in (Guengerich, 2005; Pikuleva, 2006; Miller et al., 2011)), yet little is currently known about the effect of marketed drugs on activity of CYP11A1, both in vivo and in vitro. Our knowledge in this area is mainly limited to earlier studies in the field showing that CYP11A1, as well as some other steroidogenic P450s, are inhibited by the antifungal agent ketoconazole and anticonvulsant aminoglutethimide (withdrawn from the US market in 1986), whose administration leads to adrenocortical dysfunction (Bossche, 1992; Harvey et al., 2003). Recently, we determined the crystal structure of CYP11A1 (Mast et al., 2011) and compared it to the structure of CYP46A1, another P450 which utilizes cholesterol as the endogenous substrate. CYP46A1, or cholesterol 24-hydroxylase, is a microsomal enzyme expressed predominantly in the brain whether it initiates the major pathway of cholesterol removal (Lutjohann et al., 1996; Russell et al., 2009). We found that in CYPs 11A1 and 46A1, enzymes that share <25% of amino acid sequence identity, the shape of the active site is similar, a long curved tube, as is the positioning of cholesterol (Mast et al., 2011). The major difference is that the active site in CYP11A1 is longer and more narrow, providing an explanation for the more strict substrate specificity of CYP11A1 as compared to that of CYP46A1 which may bind compounds structurally unrelated to cholesterol (Mast et al., 2003; Mast et al., 2008; Mast et al., 2010). Similar architecture of the active sites in CYP11A1 and CYP46A1 gave impetus to the present work in which we investigated whether the pharmaceuticals that modulate CYP46A1 activity in vitro, also affect the activity of purified recombinant CYP11A1. We identified several strong CYP11A1 inhibitors, and unexpectedly found that CYP11A1 activity could also be stimulated. The latter is a novel finding which enhances our understanding of CYP11A1 and opens new directions in studies of this enzyme as a target for therapeutic agents.
2. Materials and methods
2.1. Materials
Pharmaceuticals for screening were purchased from one of the following sources: Sigma-Aldrich Co (St. Louis, MO), Cayman Chemical Company (Ann Arbor, MI), Alfa Aesar (Ward Hill, MA), Waterstone Technology LLC (Carmel, IN), and Toronto Research Chemicals Inc. (North York, Ontario, Canada). Cholesterol and [3H]cholesterol were from Steraloids Inc (Newport, RI) and American Radiolabeled Chemicals, Inc (St. Louis, MO), respectively. All other chemicals were from Sigma-Aldrich unless otherwise specified. Bovine recombinant CYP11A1 was purified as previously described (Mast et al., 2011), except that 22R-hydroxycholesterol was omitted from all the buffers, and detergent (CHAPS) was removed after the last purification step by repeated dilutions and concentrations of the enzyme preparation with detergent-free buffer, 50 mM potassium phosphate (KPi), pH 7.2, containing 1 mM EDTA and 20% glycerol. Bovine recombinant adrenodoxin reductase (Adr) and adrenodoxin (Adx) were purified as previously described (Sagara et al., 1992; Sagara et al., 1993).
2.2. Enzyme assay
Incubations were carried out as previously described (Heo et al., 2012). The reaction volume was 1 ml and the buffer was 40 mM KPi, pH 7.2, containing 1 mM EDTA. Liposomes (20 μg, prepared from dilauroylglycerol-3-phosphatidylcholine (DLPC)) were added to the buffer first followed by addition of cold cholesterol (1 μM), [3H]cholesterol (250,000 cpm), proteins (0.1 μM CYP11A1, 1.6 μM Adx and 0.4 μM Adr), and a drug (10 μM). The components were reconstituted for 20 min at room temperature and then for 2 min at 30°C followed by addition of 1 mM NADPH to initiate the enzymatic reaction. The enzymatic reaction was carried out for 2 min and terminated by the addition of CH2Cl2 (two 2 ml extractions). Organic layer was isolated, and evaporated, and the extract was dissolved in CH3CN and analyzed by HPLC (Pikuleva et al., 1997). The utilized concentration of cholesterol (added from 1 mM stock in 4.5% aqueous 2-hydroxypropyl-β-cyclodextrin (HPCD)) was equal to 0.5 Km of CYP11A1 for cholesterol. The concentration of test drug was equal to 5 Km for cholesterol. These conditions were selected based on our previous studies of CYP46A1 showing that if, under such a cholesterol concentration and drug-to-substrate ratio, a test drug inhibits P450-mediated cholesterol hydroxylation by ≥50%, this is usually an indication that the drug in question has nanomolar to low micromolar affinity for the P450 (Mast et al., 2008). The drugs were dissolved in solvents in which they had maximal solubility; methanol, DMSO, water or HPCD (Supplemental Table 1).
2.3. Spectral Binding Assay
Binding affinities of different drugs for CYP11A1 were estimated as described previously (Pikuleva et al., 1995), except the buffer (40 mM KPi, pH, 7.2, 1 mM EDTA) did not contain any detergent. The temperature was 18°C. When titrations were carried out in the presence of phospholipids (PLs), liposomes were added to the buffer first, at a final concentration of 40 μg/ml, followed by the addition of 0.4 μM CYP11A1. To test drug binding to cholesterol-bound CYP11A1, the reagents were placed in a tube in the following order: buffer, liposomes (40 μg/ml), cholesterol (4 μM), and CYP11A1 (0.4 μM). Apparent binding constants (Kd) were calculated using either the ΔA = (ΔAmax[L])/(Kd + [L]) or equations, in which ΔA is the spectral response at different ligand (drug) concentrations [L], and ΔAmax is the maximal amplitude of the spectral response.
3. Results
3.1. Effect of CYP46A1 inhibitors on activity of CYP11A1
Only compounds inhibiting CYP46A1 activity by more than 45% in our previous studies (Mast et al., 2012) were tested in this CYP11A1 enzyme assay. More than half of these pharmaceuticals did not significantly affect CYP11A1 activity under the experimental conditions used (Fig. 1, Supplemental Table 1). Yet, ketoconazole, posaconazole, carbenoxolone and selegeline decreased pregnenolone production by >65%, whereas clobenpropit and dexmedetomidine increased CYP11A1-mediated cholesterol metabolism by up to 170%. Pharmacologic stimulation of CYP11A1 activity prompted us to test desogestrel, pemirolast and tizanidine. In studies of CYP46A1, desogestrel had no effect, and pemirolast and tizanidine had a slight stimulatory effect on enzyme activity. These compounds increased CYP11A1 activity by 161%, 125%, and 170%, respectively.
Fig. 1.
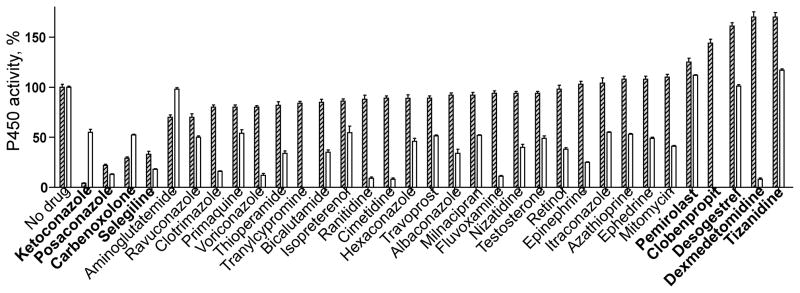
Effect of CYP46A1 inhibitors on activity of CYP11A1. Dashed and open bars correspond to CYP11A1 and CYP46A1, respectively. Data on the activity of CYP46A1 are taken from Mast et al., 2012, and shown for comparison. Enzyme assay was carried out as described under Section 2. The assay mixture contained PLs. The results are the mean ± S.D of three independent measurements. Drugs mentioned in Sections 3 and 4 are shown in bold.
3.2. Spectral changes in CYP11A1 elicited by inhibitors and activators
The identified strong CYP11A1 inhibitors and all of the enzyme activators were then evaluated in the spectral binding assay. Of them, only 4 elicited significant spectral shifts in CYP11A1 (Fig. 2–4). These were two inhibitors (ketoconazole and posaconazole) and two activators (clobenpropit and dexmedetomidine). At saturating concentrations, ketoconazole and posaconazole shifted λmax in the CYP11A1 absolute spectrum from 417 nm to 422 nm (Fig. 2A and C), the same wavelength as observed in previous studies with amine-containing steroids that bind to the CYP11A1 active site and serve as the enzyme inhibitors by coordinating the P450 heme iron with their nitrogen atom (Sheets et al., 1982; Sheets et al., 1983). Clobenpropit and dexmedetomidine also red-shifted the λmax of CYP11A1 (Fig. 3A, 4A), but the position of the Soret peak was at a shorter wavelength (420–421nm). The difference spectra of CYP11A1 in the presence of ketoconazole, posaconazole, clobenpropit and dexmedetomidine were all similar and of type II (Remmer et al., 1966; Schenkman et al., 1967) with the troughs at 412 nm and the peaks at 433–434 nm (Fig. 2B and D, 3B, 4B). Equilibrium binding constants were determined from the difference spectra (Table 1). The spectral Kd values were 1.0 μM and 1.5 μM for the inhibitors, and 7.0 μM and 18 μM for the activators.
Fig. 2.
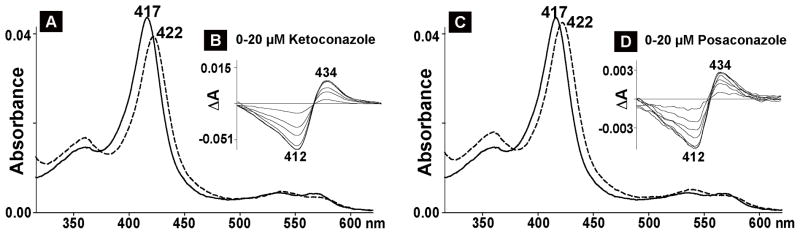
Spectral analysis of CYP11A interaction with inhibitory drugs. The concentration of CYP11A1 was 0.4 μM, and the buffer was 40 mM KPi, pH, 7.2, containing 1 mM EDTA. Numbers above or below the spectra indicate the wavelengths of absorption maxima or minima. A and C, absolute spectra; solid and dashed lines represent CYP11A1 spectrum in the absence and presence of ketoconazole (A) and posaconazole (B), respectively. Inhibitors’ concentrations were 15 μM (ketoconazole) and 10 μM (posaconazole), equal to 10 Kd of for the studied drug (1.5 μM and 1.0 μM, respectively) for substrate-free CYP11A1. B and D, difference spectra.
Fig. 4.

Spectral analysis of CYP11A interaction with dexmedetomidine. Numbers above or below the spectra indicate the wavelengths of absorption maxima or minima. A, absolute spectra; black and gray lines represent the spectra of cholesterol-free (black) and cholesterol-bound (gray) CYP11A1 in the absence (solid line) and presence (dashed line) of dexmedetomidine. The concentration of CYP11A1 was 0.4 μM; the concentrations of cholesterol and dexmedetomidine were 4 μM and 70 μM, respectively. These ligand concentrations are equal to 20 Kd of cholesterol (0.2 μM) and 10 Kd of dexmedetomidine (7.0 μM) for cholesterol-free CYP11A1. B–E, difference spectra of 0.4 μM CYP11A1 titrated under different assay conditions. B, titration with dexmedetomidine when no cholesterol or DLPC was added to the buffer (40 mM KPi, pH, 7.2, containing 1 mM EDTA). C, titration with cholesterol when no DLPC was added to the buffer. D, titration with dexmedetomidine in the presence of cholesterol when no DLPC was added to the buffer. E, titration with dexmedetomidine in the presence of cholesterol and DLPC.
Fig. 3.

Spectral analysis of CYP11A interaction with clobenpropit. Numbers above or below the spectra indicate the wavelengths of absorption maxima or minima. A, absolute spectra; black and gray lines represent the spectra of cholesterol-free (black) and cholesterol-bound (gray) CYP11A1 in the absence (solid line) and presence (dashed line) of clobenpropit. The concentration of CYP11A1 was 0.4 μM; the concentrations of cholesterol and clobenpropit were 4 μM and 150 μM, respectively. These ligand concentrations are equal to 20 Kd of cholesterol (0.2 μM) and 8 Kd of clobenpropit (18 μM) for cholesterol-free CYP11A1. B–E, difference spectra of 0.4 μM CYP11A1 titrated under different assay conditions. B, titration with clobenpropit when no cholesterol or DLPC was added to the buffer (40 mM KPi, pH, 7.2, containing 1 mM EDTA). C, titration with cholesterol when no DLPC was added to the buffer. D, titration with clobenpropit in the presence of cholesterol when no DLPC was added to the buffer. E, titration with clobenpropit in the presence of cholesterol and DLPC. Under the concentrations used, the absorption of clobenpropit in the visible region is up to 0.003 absorbance units.
Table 1.
Spectral changes in CYP11A1 elicited by inhibitors and activatorsa
| Drug | λmax in the absolute spectrum, nm | λmin and λmax in the difference spectrum, nm | ΔAmax in the difference spectrumb | Spectral Kdc |
|---|---|---|---|---|
| None | 417 | NAd | NA | NA |
| Inhibitors | ||||
| Ketoconazole | 422 | 412, 432 | 0.1 ± 0.01 | 1.5 ± 0.2 |
| Posaconazole | 422 | 411, 429 | 0.02 ± 0.001 | 1.0 ± 0.2 |
| Carbenoxolone | 417e | WSRf | <0.004 | NDg |
| Selegeline | 417e | NOh | NA | ND |
| Activators | ||||
| Pemirolast | 417e | NO | NA | ND |
| Clobenpropit | 420 | 412, 433 | 0.03 ± 0.008 | 18.0 ± 2.0 |
| Dexmedetomidine | 421 | 413, 434 | 0.08 ± 0.01 | 7.0 ± 1.0 |
| Desogestrel | 417e | WSR | <0.002 | ND |
| Tizanidine | 417e | NO | NA | ND |
The assay buffer was 40 mM KPi, pH, 7.2, containing 1 mM EDTA.
Normalized per nmol of CYP11A1, except cases with weak or no spectral response.
The results are the mean ± S.D of three independent titrations.
NA, non applicable.
The absorbance at λmax was decreased by 2–8%.
WSR, weak spectral response.
ND, not determined.
NO, not observed.
3.3. Effect of phospholipids on drug modulation of CYP11A1 activity
CYP11A1 is a membrane-bound enzyme residing in the inner membrane of the mitochondrion. To gain insight into whether association with the membrane contributes to pharmacologic modulation of CYP11A1 activity, a set of drugs investigated in section 3.1 was tested in the in vitro enzyme assay again, except that PLs were omitted from the reaction mixture. Lack of PLs affected CYP11A1 activity in a drug-dependent manner (Fig. 5). The extent of inhibition remained the same, as in the presence of PLs, in the case ketoconoazole and was increased in the case of posaconazole, selegeline, ravuconazole, clotrimazole and hexaconazole. In contrast, inhibitory effect of carbenoxolone was abolished as is the stimulatory effect of pemirolast, clobenpropit, desogestrel, dexmedetomidine and tizanidine.
Fig. 5.
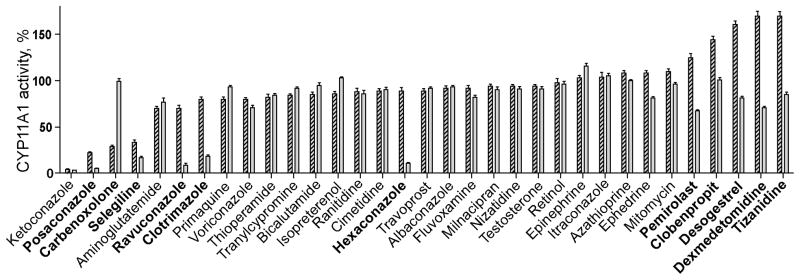
Effect of phospholipids on modulation of CYP11A1 activity by different drugs. Dashed and gray bars correspond to activity measurements in the presence or absence of DLPC, respectively, after normalization to basal CYP11A1 turnover: 2.6 min−1 in the presence of DLPC and 2.0 min−1 in the absence of DLPC. The results are the mean ± S.D of three independent measurements. Drugs mentioned in Sections 3 and 4 are shown in bold.
3.4. Effect of cholesterol and PLs on activator-induced spectral changes in CYP11A1
Since PLs seem to be essential for a stimulatory effect of drugs on CYP11A1, we carried out spectral titrations of this P450 under the conditions modeling those in the enzyme assay: 10 to 1 molar ratio of cholesterol to CYP11A1 but a 4-fold increased substrate and enzyme concentrations to obtain good quality spectra. Consistent with previous studies (Takikawa et al., 1978), cholesterol-induced spectral shifts in CYP11A1 were of type I (Fig. 3A and C) occurring when compound in question binds to the P450 active site and displaces water molecule coordinating the heme iron in substrate-free enzyme. Subsequent titration of cholesterol-bound CYP11A1 with either pemirolast, clobenpropit, desogestrel, dexmedetomidine or tizanidine revealed that, as in experiments with substrate-free CYP11A1, only clobenpropit and dexmedetomidine elicited spectral changes in substrate-bound CYP11A (Fig. 3A and D, 4A and D). At subsaturating clobenpropit or dexmedetomidine concentrations (we were never able to reach a plateau in these titrations), only a shoulder was observed in the 417–418 nm region in the absolute spectrum, and a trough at 391 nm and peak at 428 nm in the difference spectrum. This difference spectrum was neither of classical type II (e.g., Fig. 3B, 4B) nor reverse type I characterized by the trough at 380–390 nm and peak at 415–420 nm (Mailman et al., 1974).
Next, we evaluated the effect of PLs on CYP11A1 spectral response. The P450 was reconstituted first into PL vesicles followed by the saturation with cholesterol, and titration with clobenpropit or dexmedetomidine. Upon sequential additions of cholesterol and the activator, CYP11A1 displayed spectral shifts identical to those observed in the absence of PLs (not shown). The only detectable effect was on the magnitude of the P450 spectral response. At the same drug concentrations as in experiments with no PLs present, the spectral response was smaller in the case of clobenpropit and larger in the case of dexmedetomidine, as assessed by the shoulder at 417–418 nm in the absolute spectra (not shown) and the amplitude of ΔA in the difference spectrum (Fig. 3E vs D, and Fig. 4E vs D).
4. Discussion
The present work capitalized on our previous structural studies of CYP46A1 and CYP11A1 (Mast et al., 2008; Mast et al., 2010; Mast et al., 2011) and our recent screening of the experimental or approved by the FDA drugs for the inhibitory effect on CYP46A1 in vitro (Mast et al., 2012). We established that the shape of the active site is similar in the two enzymes and hypothesized that some of the compounds that inhibit CYP46A1 in vitro may also inhibit CYP11A1. We also identified pharmaceuticals to be tested on CYP11A1, most of which were inhibitors of CYP46A1. A total of 31 CYP46A1 inhibitors and three compounds with no inhibitory effect on CYP46A1 were evaluated (Fig. 1). Of these 34 medications, nine (27%) modulated the activity of CYP11A1 under the experimental conditions used: 4 negatively and 5 positively. The identified inhibitors were the antifungal azole posaconazole, the experimental antiulcerative agent carbenoxolone, and the antiparkinsonian drug selegiline (Fig. 1). Posaconazole is structurally similar to the known CYP11A1 inhibitor ketoconazole (Fig. 6), yet is longer because contains two additional rings, phenol and triazole, that enhance drug’s affinity to its primary target – the fungal CYP51 enzyme (Dodds Ashley et al., 2005). Apparently, posaconazole also fits nicely into the narrow and long active site of CYP11A1 and coordinates the heme iron with one of its nitrogen atoms as indicated by type II P450 spectral response (Fig. 2C and D). Type II spectral response is consistent with the inhibitory effect of posaconazole on CYP11A1 activity because type II ligands stabilize the P450 low reduction potential and are more often inhibitors than substrates (Gigon et al., 1968). In contrast, steroid-like carbenoxolone has no nitrogen atoms and elicits only a very weak spectral response in CYP11A1. Nevertheless, we suggest, on the basis of structural similarity to the CYP11A1 substrate cholesterol, that carbenoxolone also enters the active site of CYP11A1 and is retained there via interactions involving its oxygen atom(s). Finally, selegiline is the smallest of the three identified inhibitors. Its mode of CYP11A1 inhibition is currently difficult to explain. Additional investigation is required to determine whether selegiline binds to CYP11A1 and competes, like posaconazole and carbenoxolone, with cholesterol for the enzyme active site. Previous studies by others revealed that small molecules can modulate the interactions between the CYP11A1 redox partners Adr and Adx and thus affect the activity of this enzyme (Berwanger et al., 2010).
Fig. 6.

Chemical structures of some of the compounds evaluated in the present study.
A search of CYP11A1 inhibitors unexpectedly led to identification of CYP11A1 activators. These were: the antiallergic drug pemirolast and the muscle relaxant tizanidine (also slightly stimulatory for CYP46A1); the experimental anticonvulsant clobenpropit; the oral contraceptive component desogestrel; and the sedative dexmedetomidine (Fig. 1). Although these CYP11A1 activating compounds have different chemical structures, they share some similarities with the structures of the enzyme inhibitors (Fig. 6). Clobenpropit is similar to posaconazole in that it is long and can adopt a linear configuration. Dexmedetomidine, like selegiline, is small and has rotatable bonds. Desogestrel is synthetic steroid and can be viewed as an analog of carbenoxolone. Finally, the ring structures in pemirolast and tizanidine are partially or completely conjugated, as are in cholesterol and other steroid molecules. All of the activators, except desogestrel, contain imidazole/triazole heterocycles, but only clobenpropit and dexmedetomidine elicit type II spectral response in cholesterol-free CYP11A1, indicating that they coordinate the heme iron (Fig. 3B and 4B). This binding, however, seems to be weaker than that of the inhibitors as indicated by the 4.7-to 12-fold increase in the values of apparent binding constants (Table 1). Perhaps more important is that clobenpropit- and dexmedetomidine-induced type of the difference spectrum is changed when cholesterol is present in the active site and no longer represents the inhibitory type II response (Fig. 3D and 4D). To the best of our knowledge, the difference spectrum observed in Figs. 3D and 4D does not correspond to any of the previously reported types of the spectral response and could reflect a stimulatory effect of clobenpropit and dexmedetomidine on CYP11A1-mediated cholesterol metabolism.
Studies of CYP11A1 activity in the absence of PLs (Fig. 5) demonstrated that enzyme association with the lipid bilayer is an important contributor to the modulatory effect of drugs. In the case of posaconazole and selegeline, the extent of CYP11A1 inhibition was higher in the absence than in the presence of PLs suggesting that the effective inhibitor concentration is higher in PL-free buffer. Indeed, whlile lipophilic, posaconazole and selegeline probably have lower propensity toward the lipid bilayer than cholesterol which, due to its planarity and amphipathic nature, readily intercalates between the acyl chains of PLs. Therefore, in the presence of PLs, only a part of the added posaconazole or selegeline is likely incorporated in the liposomes, thus lowering the effective concentration of the inhibitor. This interpretation assumes that posaconazole and selegeline inhibit CYP11A1 competitively and enter the P450 through the membrane like cholesterol (Seybert et al., 1979). Consistent with the lower, as compared to cholesterol, drug partitioning into the lipid bilayer, ravuconazole, clotrimazole and hexaconazole showed only a minor inhibition of CYP11A1 activity in the presence of PLs and significant enzyme inhibition in the PL-free buffer. In contrast, carbenoxolone did not inhibit CYP11A1 the absence of PLs. This could be due to the increased drug’s affinity, as compared to that of cholesterol, to the vehicle in which it was delivered. Both cholesterol and carbenoxolone were dissolved in 4.5% HPCD, a cyclic oligosaccharide whose hydrophobic cavity can encapsulate lipophilic compounds to form a soluble inclusion complex (Pitha et al., 1988). Carbenoxolone has more hydrogen-bonding functionalities than cholesterol and should form a stronger complex with HPCD. If so, carbenoxolone might not be released from the vehicle in the absence of PLs, and might thus inhibit CYP11A1.
Lack of PLs also abolished a stimulatory effect of pemirolast, clobenpropit, desogestrel and dexmedetomidine (Fig. 5). The mechanism of CYP11A1 activation is currently unknown, therefore interpretation of the effects of PLs is difficult. We ascertained that clobenpropit and dexmedetomidine can bind to the enzyme heme iron in cholesterol-free CYP11A1 (Fig. 3B, 4B) and alter spectral properties of cholesterol-bound CYP11A1 (Fig. 3D, 4D). We also established that PLs affect the magnitude of the activator-induced spectral response in cholesterol-bound CYP11A1 (Fig. 3E vs D, 4E vs D). These findings lead to a question about the site(s) of the activators’ binding: inside or outside the CYP11A1 active site or on one of the redox partners. Also, do different activators bind to the same or different sites? To address these questions, we will conduct a separate investigation to delineate the mechanism of CYP11A1 activation by therapeutic drugs. Overall, studies of the effect of PLs on modulation of CYP11A1 activity by different drugs emphasize the importance of the P450 association with the lipid bilayer and show how this association may alter drug effect on the enzyme whose substrate enters the active site through the lipid bilayer (Seybert et al., 1979). Not only might PLs change the effective drug concentration through drug partitioning into the lipid bilayer, but also affect CYP11A1 by itself by altering the iron spin state as well as enzyme activity as shown in previous studies (Kido et al., 1979; Lambeth et al., 1980; Hsu et al., 1985). Since changes in the P450 spin state reflect changes in the active site, PLs effect on CYP11A1 active site might also alter enzyme interactions with both cholesterol and a drug and thus either enhance or abolish drug effect on enzyme activity.
In conclusion, we continue to take advantage of the unique opportunity provided to us by nature and compare the P450 enzymes that act on the same endogenous substrate (cholesterol). We show here that knowledge gained during studies of one of these enzymes (CYP46A1) could be used in studies of the other enzyme (CYP11A) leading to quick and relatively inexpensive identification of pharmaceuticals with a modulatory effect on enzyme activity. We found that CYP11A1 modulators might not only be antifungal azoles but also medications from other drug classes. We also discovered that, similar to CYP46A1, the effect on enzyme activity might not only be inhibitory but also stimulatory. The present work expands our knowledge of chemical structures that can bind to CYP11A1 and warrants future studies assessing the pharmacological relevance of our in vitro findings and evaluation of CYP11A1 as a drug target.
Supplementary Material
Highlights.
Structurally distinct pharmaceuticals were found to alter the activity of CYP11A1.
The effect on CYP11A1 activity was not only inhibitory but also stimulatory.
CYP11A1-membrane interactions contribute to a drug’s effect on enzyme activity.
Acknowledgments
This study was supported by United Public Health Service Grant GM62882. I.A.P. is a recipient of the Jules and Doris Stein Professorship from the Research to Prevent Blindness.
The abbreviations used
- Adx
adrenodoxin
- Adr
adrenodoxin reductase
- DLPC
dilauroylglycerol-3-phosphatidylcholine
- HPCD
2-hydroxypropyl-β-cyclodextrin
- KPi
potassium phosphate
- PL
phospholipid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berwanger A, Eyrisch S, Schuster I, Helms V, Bernhardt R. Polyamines: naturally occurring small molecule modulators of electrostatic protein-protein interactions. J Inorg Biochem. 2010;104:118–125. doi: 10.1016/j.jinorgbio.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Bossche HV. Inhibitors of P450-dependent steroid biosynthesis: from research to medical treatment. J Steroid Biochem Molec Biol. 1992;43:1003–1021. doi: 10.1016/0960-0760(92)90328-G. [DOI] [PubMed] [Google Scholar]
- Dodds Ashley ES, Alexander BD. Posaconazole. Drugs Today (Barc) 2005;41:393–400. doi: 10.1358/dot.2005.41.6.898145. [DOI] [PubMed] [Google Scholar]
- Gigon PL, Gram TE, Gillette JR. Effect of drug substrates on the reduction of hepatic microsomal cytochrome P-450 by NADPH. Biochem Biophys Res Commun. 1968;31:558–562. doi: 10.1016/0006-291x(68)90514-7. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450. Kluwer Academic/Plenum Publishers; New York, Boston, Dordrecht, London, Moscow: 2005. pp. 377–530. [Google Scholar]
- Harvey PW, Everett DJ. The adrenal cortex and steroidogenesis as cellular and molecular targets for toxicity: critical omissions from regulatory endocrine disrupter screening strategies for human health? J Appl Toxicol. 2003;23:81–87. doi: 10.1002/jat.896. [DOI] [PubMed] [Google Scholar]
- Heo GY, Liao WL, Turko IV, Pikuleva IA. Features of the retinal environment which affect the activities and product profile of cholesterol-metabolizing cytochromes P450 CYP27A1 and CYP11A1. Arch Biochem Biophys. 2012;518:119–126. doi: 10.1016/j.abb.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DK, Huang YY, Kimura T. Liposomal cholesterol binding to steroid-free cytochrome P450scc: effects of fatty acyl and head groups in phospholipids. Endocr Res. 1985;11:45–58. doi: 10.3109/07435808509035424. [DOI] [PubMed] [Google Scholar]
- Kido T, Arakawa M, Kimura T. Adrenal cortex mitochondrial cytochrome P-450 specific to cholesterol side chain cleavage reaction. Spectral changes induced by detergents, alcohols, amines, phospholipids, steroid hydroxylase inhibitors, and steroid substrates, and conditions for adrenodoxin binding to the cytochrome. J Biol Chem. 1979;254:8377–8385. [PubMed] [Google Scholar]
- Lambeth JD, Kamin H, Seybert DW. Phosphatidylcholine vesicle reconstituted cytochrome P-450scc. Role of the membrane in control of activity and spin state of the cytochrome. J Biol Chem. 1980;255:8282–8288. [PubMed] [Google Scholar]
- Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, Bjorkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman RB, Kulkarni AP, Baker RC, Hodgson E. Cytochrome P-450 difference spectra: effect of chemical structure on type II spectra in mouse hepatic microsomes. Drug Metab Dispos. 1974;2:301–308. [PubMed] [Google Scholar]
- Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, Pikuleva IA. Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J Biol Chem. 2011;286:5607–5613. doi: 10.1074/jbc.M110.188433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem. 2010;285:31783–31795. doi: 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Clark M, Wiseman J, Stout CD, Pikuleva IA. In Silico and Intuitive Predictions of CYP46A1 Inhibition by Marketed Drugs with Subsequent Enzyme Crystallization in Complex with Fluvoxamine. Mol Pharmacol. 2012 doi: 10.1124/mol.112.080424. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Norcross R, Andersson U, Shou M, Nakayama K, Bjorkhem I, Pikuleva IA. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 2003;42:14284–14292. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci U S A. 2008;105:9546–9551. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res. 2011;52:2111–2135. doi: 10.1194/jlr.R016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikuleva IA. Cytochrome P450s and cholesterol homeostasis. Pharmacol Ther. 2006;112:761–773. doi: 10.1016/j.pharmthera.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Pikuleva IA, Bjorkhem I, Waterman MR. Expression, purification, and enzymatic properties of recombinant human cytochrome P450c27 (CYP27) Arch Biochem Biophys. 1997;343:123–130. doi: 10.1006/abbi.1997.0142. [DOI] [PubMed] [Google Scholar]
- Pikuleva IA, Mackman RL, Kagawa N, Waterman MR, Ortiz de Montellano PR. Active-site topology of bovine cholesterol side-chain cleavage cytochrome P450 (P450scc) and evidence for interaction of tyrosine 94 with the side chain of cholesterol. Arch Biochem Biophys. 1995;322:189–197. doi: 10.1006/abbi.1995.1451. [DOI] [PubMed] [Google Scholar]
- Pitha J, Irie T, Sklar PB, Nye JS. Drug solubilizers to aid pharmacologists: amorphous cyclodextrin derivatives. Life Sci. 1988;43:493–502. doi: 10.1016/0024-3205(88)90150-6. [DOI] [PubMed] [Google Scholar]
- Remmer H, Schenkman J, Estabrook RW, Sasame H, Gillette J, Narasimhulu S, Cooper DY, Rosenthal O. Drug interaction with hepatic microsomal cytochrome. Mol Pharmacol. 1966;2:187–190. [PubMed] [Google Scholar]
- Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara Y, Hara T, Ariyasu Y, Ando F, Tokunaga N, Horiuchi T. Direct expression in Escherichia coli and characterization of bovine adrenodoxins with modified amino-terminal regions. FEBS Lett. 1992;300:208–212. doi: 10.1016/0014-5793(92)80847-a. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Wada A, Takata Y, Waterman MR, Sekimizu K, Horiuchi T. Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization. Biol Pharm Bull. 1993;16:627–630. doi: 10.1248/bpb.16.627. [DOI] [PubMed] [Google Scholar]
- Schenkman JB, Remmer H, Estabrook RW. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- Seybert DW, Lancaster JR, Jr, Lambeth JD, Kamin H. Participation of the membrane in the side chain cleavage of cholesterol. Reconstitution of cytochrome P-450scc into phospholipid vesicles. J Biol Chem. 1979;254:12088–12098. [PubMed] [Google Scholar]
- Sheets JJ, Vickery LE. Proximity of the substrate binding site and the heme-iron catalytic site in cytochrome P-450scc. Proc Natl Acad Sci U S A. 1982;79:5773–5777. doi: 10.1073/pnas.79.19.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets JJ, Vickery LE. Active site-directed inhibitors of cytochrome P-450scc. Structural and mechanistic implications of a side chain-substituted series of amino-steroids. J Biol Chem. 1983;258:11446–11452. [PubMed] [Google Scholar]
- Takikawa O, Gomi T, Suhara K, Itagaki E, Takemori S, Katagiri M. Properties of an adrenal cytochrome P-450 (P-450SCC) for the side chain cleavage of cholesterol. Arch Biochem Biophys. 1978;190:300–306. doi: 10.1016/0003-9861(78)90279-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.