

Abstract
The symptoms and signs constituting the congestive heart failure (CHF) syndrome have their pathophysiologic origins rooted in a salt-avid renal state mediated by effector hormones of the renin-angiotensin-aldosterone and adrenergic nervous systems. Controlled clinical trials, conducted over the past decade in patients having minimally to markedly severe symptomatic heart failure, have demonstrated the efficacy of a pharmacologic regimen that interferes with these hormones, including aldosterone receptor binding with either spironolactone or eplerenone. Potential pathophysiologic mechanisms which have not hitherto been considered involved for the salutary responses and cardioprotection provided by these mineralocorticoid receptor antagonists are reviewed herein. In particular, we focus on the less well-recognized impact of catecholamines and aldosterone on mono- and divalent cation dyshomeostasis which leads to hypokalemia, hypomagnesemia, ionized hypocalcemia with secondary hyperparathyroidism and hypozincemia. Attendant adverse cardiac consequences include a delay in myocardial repolarization with increased propensity for supra- and ventricular arrhythmias and compromised antioxidant defenses with increased susceptibility to nonischemic cardiomyocyte necrosis.
Keywords: mineralocorticoid receptor antagonists, congestive heart failure, potassium, magnesium, calcium, zinc, cardioprotection, mitochondria, oxidative stress, antioxidant defenses
Introduction
The congestive heart failure (CHF) syndrome with its disabling symptoms and signs is now the leading admitting diagnosis to U.S. hospitals. All too frequently CHF is recurrent. Its origins are rooted in a salt-avid state induced by an inappropriate and unwanted homeostatic response—neurohormonal activation—evoked by underperfused kidneys. It is this homeostatic response gone awry that causes dyshomeostasis and what has been characterized as a disorder of adaptation [1]. Effector hormones of activated renin-angiotensin-aldosterone (RAAS) and adrenergic nervous (ANS) systems mediate the retention of salt and water. The current medical management of patients with CHF therefore draws on interfering with these effector hormones by either disrupting their formation or receptor binding.
Large-scale controlled clinical trials, conducted over the past several decades, have demonstrated the efficacy of interfering with these hormones, especially when pharmacologic agents are administered in optimally tolerated oral doses. This regimen has included mineralocorticoid receptor antagonism (MCRa) with either spironolactone (Spiro) or eplerenone (Epler), given in combination with an angiotensin-converting enzyme inhibitor or AT1 receptor antagonist, beta adrenergic receptor antagonist, and loop diuretic. Significant benefits and risk reduction for mortal and morbid events were reported in symptomatic patients enrolled in either the RALES or EPHESUS trials with reduced ejection fraction [2, 3]. Pathophysiologic responses which could be favorably intercepted by MCRa in CHF were considered nearly a decade ago and reported in this Journal [4].
More recently, another favorable trial with MCRa in patients with comparable systolic dysfunction was reported [5••]. Of note, patients enrolled in the EMPHASIS-HF Study were minimally symptomatic and were receiving standard of care before their double-blind randomization to either Epler or placebo. Primary outcomes were met and included reduced risk of death from cardiovascular causes or hospitalization for heart failure. It is therefore timely to address potential pathophysiologic mechanisms which have not hitherto been considered responsible for the MCRa efficacy in patients with either compensated or decompensated heart failure. Hence, we present a fresh perspective on cardioprotection with MCRa in heart failure. We focus on less well-recognized impact of catecholamines and aldosterone on mono- and divalent cation dyshomeostasis which leads to hypokalemia, hypomagnesemia, ionized hypocalcemia with secondary hyperparathyroidism (SHPT) and hypozincemia.
PATHOPHYSIOLOGIC MECHANISMS IN HEART FAILURE
Neurohormonal Activation in Heart Failure: A Brief Overview
CHF is a systemic illness. What begins with a failing muscular pump having reduced EF will eventuate into the clinical CHF syndrome with its characteristic disabling symptoms and debilitating signs. At first and as seen in Figure 1, there is the release of a family of natriuretic peptides (ANP and BNP) from distended atria and ventricles; they maintain urinary Na+ excretion in proportion to dietary Na+ intake. Hence, despite reduced EF (<35%) patients are compensated and only become symptomatic upon carrying out marked workloads (NYHA Class I and II) or with excessive dietary Na+. This important dissociation between EF and clinical severity of CHF with RAAS activation was enforced by the SOLVD Study. Herein, asymptomatic patients with EF <35% and normal plasma renin activity were enrolled into the prevention arm of the study prior to receiving enalapril or placebo; symptomatic patients, whose EF was also <35%, had increased plasma renin activity and were enrolled in the treatment arm [6, 7]. Activation of the RAAS overwhelms the natriuretic peptides leading to salt and water retention and the appearance of decompensated heart failure. Exaggerated RAAS activity can occur intermittently leading to what is considered salt sensitivity [8].
Figure 1.

An overview of compensated and decompensated phases of heart failure relative to renal perfusion and urinary Na+/K+ ratio. Myocardial failure elicits an interplay between atrial (ANP) and brain (BNP, not shown) natriuretic peptides, elicited by atrial and/or ventricular distention, and effector hormones of the renin-angiotensin-aldosterone system (RAAS) on urinary sodium reabsorption. When the impairment in renal perfusion is mild to moderate, ANP and BNP dominate with urinary Na+/K+ ratio >1.0. There is no retention of Na+ and water with the patient compensated (minimally or asymptomatic). When renal perfusion is more markedly impaired, the RAAS dominates with urinary Na+ retention and a Na+/K+ ratio <1.0 leading to decompensated (symptomatic) failure. Patients can transition back and forth between compensated and decompensated phases of failure. From N Engl J Med, Weber KT, “Aldosterone in Congestive Heart Failure,” Volume 345, Pages 1689–97. Copyright ©2001 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
CHF: A Systemic Illness
Neurohormonal activation involving RAAS hormones and their endocrine properties creates a systemic illness whose collective features have been referred to as a proinflammatory phenotype. Its features include: i) oxidative stress, in which the rate of reactive oxygen species generation overwhelm their rate of detoxification largely rendered by Zn2+-based endogenous antioxidant defenses; ii) an immunostimulatory state in which activated peripheral blood mononuclear cells (PBMC:lymphocytes and monocytes) release proinflammatory cytokines (e.g., TNF-α and IL-6) and contribute to a vasculopathy of the intramural coronary, renal and mesenteric vasculatures; and iii) a wasting of soft tissues and bone, termed cardiac cachexia.
Neurohormonal Activation and Myocardial Remodeling
In addition to this systemic illness there is a progressive pathologic remodeling of the myocardium. This includes an ongoing loss of cardiomyocytes to necrotic and apoptotic forms of cell death. Elevations in plasma troponins, biomarkers of myocyte necrosis, are found in patients hospitalized because of their CHF [9–15•]. Each episode of decompensated failure is based on neurohormonal activation with recurrent necrosis. Most prominent features of this pathologic remodeling are the appearance of foci of microscopic scars, footprints indicative of cardiomyocyte necrosis, found widely scattered throughout both the failing right and left atria and ventricles, irrespective of the etiologic origins of heart failure [16–18]. Absent are inflammatory cell and fibroblast responses and hence apoptosis is not accompanied by tissue repair and therefore leaves behind no morphologic footprint in the form of scarring [19].
In both the explanted failing human heart and postmortem tissue obtained from the failing myocardium, a progressive fibrosis is recognized as the major component to adverse structural remodeling. The prevalence of fibrosis implicates a crucial pathophysiologic role of ongoing cardiomyocyte necrosis, irrespective of its etiologic origins [16]. The pathophysiologic mechanisms accounting for myocyte necrosis are therefore of fundamental importance.
Neurohormonal Activation and Polycation Dyshomeostasis
The relative importance of angiotensin (Ang) II and aldosterone (Aldo) on proximal and distal Na+ resorption in the nephron, as well as the colon, and K+ excretion at these sites of high-density aldosterone receptor binding has been well documented. Herein, we focus on less well-recognized impact of catecholamines and aldosterone on mono- and divalent cation dyshomeostasis which leads to hypokalemia, hypomagnesemia, ionized hypocalcemia and hypozincemia, together with delayed myocardial repolarization and increased propensity for cardiac arrhythmias, compromised antioxidant defenses and susceptibility to cardiomyocyte necrosis.
CHF: A HYPERADRENERGIC STRESSOR STATE WITH CATION DYSHOMEOSTASIS
Mg2+-Dependent Na/K ATPase
Na/K ATPase is a membrane-bound, energy-dependent pump whose activity contributes to the regulation of intracellular K+; it also has an obligatory dependence on Mg2+. These pumps are abundantly present in skeletal muscle, whose activity is regulated by catecholamines [20••]. Elevations in plasma epinephrine and norepinephrine accompany hyperadrenergic stressor states, such as CHF, where they activate these pumps leading to marked intracellular K+ uptake by muscle and the appearance of hypokalemia (see Figure 2). Reduction in myocardial K+ is accompanied by delayed repolarization and prolongation of the QTc interval of the electrocardiogram that predisposes to supra- and ventricular arrhythmias.
Figure 2.
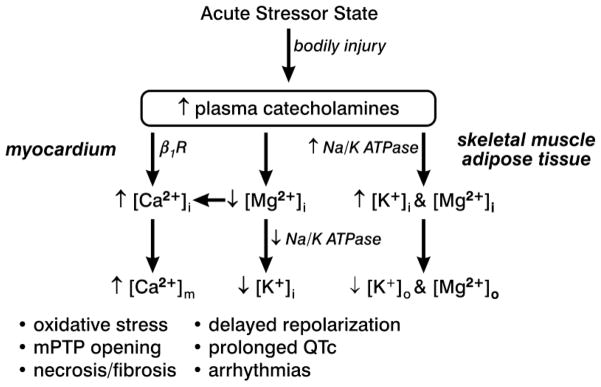
An acute stressor state, such as CHF or bodily injury, is associated with increased circulating concentrations of epinephrine and norepinephrine. These catecholamines have an impact on mono- and divalent cations involving skeletal muscle, adipose tissue and heart. See text for further discussion. Reprinted from Khan MU, et al., “Cation Interdependency in Acute Stressor States,” Am J Med Sci 2012 (In press), with permission.
Potassium balance preexisting an episode of CHF is a critical determinant of the severity of the ensuing hypokalemia that appears. Diuretic-induced excessive loss of K+ is also known to predispose to marked hypokalemia in response to the catecholamines [21]. Spironolactone, a K+-sparing aldosterone receptor antagonist, is protective against hypokalemia and ventricular arrhythmias [22]. Patients with CHF who are receiving long-term loop diuretic treatment may acquire marginal K+ and Mg2+ reserves. These limiting reserves are further compromised by the hyperadrenergic state that may readily lead to marked hypokalemia and hypomagnesemia with QTc prolongation, thus provoking a propensity for arrhythmias. Spironolactone, by attenuating urinary and fecal K+ losses, is therefore cardioprotective. Albuterol, a short-acting β2 adrenergic receptor agonist, predisposes to hypokalemia and hypomagnesemia in normal volunteers, and even more so when they are receiving a diuretic [23, 24]. Chronic excessive use of such a β2 receptor agonist may lead to marked hypokalemia and a greater propensity for arrhythmias. Antibiotics, antidepressants and antipsychotics can also lead to drug-induced prolongation of myocardial repolarization and QTc interval conducive of arrhythmogenicity.
Elevations in plasma catecholamines are accompanied by hypomagnesemia (see Figure 2). This is related to a cyclic AMP-mediated rise in intracellular Mg2+, together with increased lipolysis with Mg2+ binding to free fatty acids and subsequent sequestration in adipocytes causing hypomagnesemia. Predisposing risk factors include hypokalemia, hypocalcemia, thiazide and loop diuretic use, and sepsis. The preexisting hypomagnesemia at the time of admission in these patients may become more severe during their prolonged hospital stay due to ongoing excessive excretory losses and reduced Mg2+ intake [25]. Chronic Mg2+ deficiency contributes to systolic dysfunction and proinflammatory cardiac phenotype [26••]. By attenuating Mg2+ losses, spironolactone would be cardioprotective.
Atrial and ventricular arrhythmias appear when hypomagnesemia is of moderate to marked severity (1.68±0. 7 mg/dL). Mg2+-dependent Na/K ATPase activity is reduced with hypomagnesemia and further prolongs the QTc interval enhancing the propensity for arrhythmias. Pharmacologic reversal of impaired K+ balance and hypokalemia will prove difficult unless Mg2+ is first replaced. Abnormal prolongation of the QTc interval (>440 ms) suggests a deficiency of myocardial K+ and Mg2+. Daily monitoring of the QTc interval and its normalization via Mg2+ and K+ supplementation can be used to gauge the adequacy of their intracellular replacement in cardiomyocytes. Attainment of normal QTc with these supplements may require several more days than needed for the correction of hypokalemia and hypomagnesemia alone. Less than 1% of Mg2+ is extracellular and hence serum Mg2+ is not an accurate indicator of intracellular Mg2+ stores; therefore the QTc interval can serve as a more reliable surrogate. Digoxin, a Na/K ATPase inhibitor, accentuates the dyshomeostasis of intracellular K+ and Mg2+ predisposing to QTc prolongation and arrhythmias.
Concurrent hypokalemia and hypomagnesemia are common with interactions between K+ and Mg2+ which are diverse and complex, including the importance of Mg2+ deficiency, that interferes with K+ retention while raising urinary K+ excretion [27, 28]. Mg2+ deficiency contemporaneously begets K+ deficiency. The effective clinical resolution of hypokalemia mandates simultaneous reversal of hypomagnesemia [29].
In the absence of gastrointestinal losses or diuretic usage, hypomagnesemia and hypokalemia due to impaired renal tubular reabsorption and presenting as urinary K+ and Mg2+ wasting cannot be overlooked in patients with cardiac arrhythmias, including atrial fibrillation. Differential diagnostic evaluation of inheritable renal tubular disorders associated with such excessive urinary Mg2+ losses (e.g., Gitelman syndrome) should always be considered via determination of urinary Mg2+ excretion. Patients with Mg2+ deficiency will retain urinary Mg2+ and not waste it. This caveat is also applicable when the resolution of these cations, using standard oral Mg2+ and K+ supplements, proves difficult to achieve. Spironolactone may prove efficacious in many of these Mg2+-wasting disorders.
Mg2+ Efflux from Cardiomyocytes
Mg2+ is an endogenous antagonist to Ca2+ entry in cardiomyocytes and their mitochondria and vice versa [30]. Catecholamines promote the efflux of Mg2+ from cardiomyocytes which, in turn, augments Ca2+ entry and the potential for intracellular Ca2+ overloading [31]. A β1 adrenergic receptor antagonist prevents catecholamine-induced Mg2+ losses. Reduced myocardial Mg2+ content slows repolarization and prolongs QTc interval provoking arrhythmogenicity. The efficacy of β1 and β2 adrenergic receptor blockade includes their favorable impact on catecholamine-driven dyshomeostasis of K+ and Mg2+ that accompanies hyperadrenergic states.
Intracellular Ca2+ Overloading and Oxidative Stress
Reductions in plasma ionized [Ca2+]o, or ionized hypocalcemia, are commonly found in adults presenting to the emergency department or admitted to intensive care units with an acute hyperadrenergic stressor state [32, 33]. The extent to which [Ca2+]o falls correlates with the severity of the hyperadrenergic response (see Figure 3). Ionized hypocalcemia serves as an in-hospital marker of survival. Hypoalbuminemia can contribute to reduced total Ca2+ concentration since a lesser amount of serum proteins are available for Ca2+ binding.
Figure 3.

An acute stressor state with elevated circulating catecholamines is responsible for intracellular Ca2+ overloading with a subsequent fall in plasma ionized [Ca2+]o which, in turn, provokes the parathyroid glands to release parathyroid hormone (PTH), a calcitropic hormone. PTH likewise contributes to intracellular Ca2+ overloading. In cardiomyocytes Ca2+ overloading is accompanied by the induction of oxidative stress by subsarcolemmal mitochondria (SSM), which leads to the opening of their permeability transition pore (mPTP) with ensuing osmotic swelling and injury. The necrosis of cardiomyocytes follows accompanied by the leak of intracellular troponins into the interstitial space accounting for the ultimate rise in plasma troponins. Cardiac myocytes lost to necrosis are replaced by fibrous tissue, or scarring, which preserves the structural integrity of this hollow muscular organ. Reprinted from Khan MU, et al., “Cation Interdependency in Acute Stressor States,” Am J Med Sci 2012 (In press), with permission.
The appearance of ionized hypocalcemia in critically ill patients is based on a catecholamine-mediated shift in circulating Ca2+ into the intracellular compartment of various tissues that includes the heart, skeletal muscle and PBMC and accounts for intracellular Ca2+ overloading. Ionized hypocalcemia is followed by increased release of parathyroid hormone (PTH) and ensuing PTH-mediated excessive Ca2+ entry (see Figure 3). Collectively, catecholamine- and PTH-facilitated excessive intracellular Ca2+ accumulation in cardiomyocytes leads to Ca2+ overloading of their mitochondria accounting for the induction of oxidative stress by these organelles, where the rate of reactive oxygen and nitrogen species overwhelm their rate of detoxification by endogenous antioxidant defenses. The ensuing necrotic death of cardiomyocytes is followed by tissue repair with the resultant fibrous tissue response, or scarring. This replacement fibrosis preserves the structural integrity of the injured myocardium and ventricular function. It constitutes a morphologic footprint of previous necrotic cell death. Fibrosis, however, has adverse consequences. The addition of stiff fibrillar type I collagen in scar tissue compromises ventricular function in diastole and systole and serves as substrate for reentrant arrhythmias.
The catecholamine-induced, Ca2+ overload-initiated nonischemic necrosis of cardiomyocytes is accompanied by the release of troponins, an intracellular protein whose release plays a crucial role in discerning myocardial injury. A modest increase in plasma troponins has been reported in diverse stressor states, including decompensated heart failure, sepsis, hemorrhagic shock, subarachnoid hemorrhage, trauma, gastrointestinal bleeding, or pulmonary embolus [34]. The levels to which plasma troponins rise in such patients does not reach the more marked elevations seen with segmental loss of myocardium that accompanies ischemia and infarction or that which follows myocardial contusion.
CHF: A RAAS STRESSOR STATE WITH CATION DYSHOMEOSTASIS
Secondary Aldosteronism of CHF Accompanied by Secondary Hyperparathyroidism
Increased circulating PTH levels, coupled to a resorption of bone, are less well-recognized features of the secondary aldosteronism of CHF. SHPT arises because of the marked urinary and fecal excretory losses of Ca2+ and Mg2+ and the resultant appearance of ionized hypocalcemia and hypomagnesemia, each transmits stimuli to increased PTH secretion [35–37]. In man with autonomous adrenal aldosterone production, SHPT is corrected by either adrenal surgery or Spiro treatment [37]. In experimental animals receiving aldosterone/salt treatment, where plasma aldosterone levels are raised to those found in CHF, SHPT can be prevented by: cotreatment with Spiro, which prevents the heightened excretory losses of Ca2+ and Mg2+ in urine and feces [36]; cotreatment with a Ca2+- and Mg2+-supplemented diet, together with vitamin D, to prevent ionized hypocalcemia and hypomagnesemia [38]; parathyroidectomy [39]; or cotreatment with a calcimimetic, cinacalcet, which resets the threshold of the parathyroid glands’ Ca2+-sensing receptor to prevent heightened elaboration of PTH [40]. In preventing SHPT and its attendant pathophysiologic consequences, Spiro is cardioprotective.
A dyshomeostasis of serum K+ and Ca2+ is found in patients hospitalized with decompensated biventricular failure having a dilated cardiomyopathy of ischemic or nonischemic origins. This metabolic profile is also found in patients having low-renin or salt-sensitive hypertension [41–44] and in those with primary aldosteronism [45–47]. Furthermore, elevated PTH, evoked in response to ionized hypocalcemia, serves as a stimulus to adrenal aldosterone production producing contemporaneous elevations in plasma aldosterone [48–51]. In patients with primary hyperparathyroidism, preoperative PTH levels in excess of 00 ng/mL are independent predictors of abnormal elevations in plasma aldosterone [51]. Major pathogenic events accounting for cardiomyocyte necrosis in aldosteronism focus on the relative importance of PTH-mediated intracellular Ca2+ overloading and induction of oxidative stress [36, 39, 40]. The role of elevations in circulating aldosterone and which are inappropriate for dietary Na+ must also be considered [52].
Abnormal elevations in serum PTH (>65 pg/mL) serve as a potent mediator of Ca2+ overloading in cardiomyocytes and their mitochondria [36, 53, 54]. This may contribute to the increased cardiovascular morbidity and mortality associated with primary hyperparathyroidism [55, 56]. Elevations in serum PTH are likewise associated with increased mortality in frail elderly persons independent of their 25(OH)D status, bone mass or renal function [57, 58]. In patients having primary hyperparathyroidism, the increased incidence of left ventricular hypertrophy, Ca2+ deposits in the myocardium and heart valve leaflets, and intracellular Ca2+ overloading may further contribute to increased risk of cardiovascular mortality [55, 59–62].
Elevated PTH levels are found in patients hospitalized with decompensated heart failure and those awaiting cardiac transplantation [63–66], and serve as an independent predictor of CHF, the need for hospitalization and cardiovascular mortality [67–70]. Moreover, PTH levels have been shown to be an independent risk factor for mortality and cardiovascular events in community-dwelling individuals [71–73••]. SHPT is especially prevalent in African-Americans (AA) with protracted (>4 wks) decompensated biventricular failure, where chronic elevations in plasma aldosterone contribute to symptoms and signs of CHF and plasma ionized hypocalcemia [44, 64]. SHPT is also associated with the prevalence of hypovitaminosis D in AA, where the increased melanin content of dark skin serves as a natural sunscreen [64]. Accordingly, the prevalence of hypovitaminosis D, often of marked severity (<20 ng/mL), compromises Ca2+ homeostasis predisposing AA to ionized hypocalcemia and consequent SHPT [64, 74, 75]. Vitamin D deficiency is also reported in Caucasians and Asians with heart failure whose effort intolerance predisposes to an indoors lifestyle [67, 68, 76, 77]. Other factors which may be associated with compromised Ca2+ stores and contribute to the appearance of SHPT, especially in AA with CHF, have been reviewed elsewhere [78].
Osteopenia and osteoporosis accompany CHF and predispose elderly patients to atraumatic fractures, especially of the hip [79]. In cachectic patients, atrophy of proximal muscle groups reduce limb strength and predispose to increased falls. Patients with CHF treated with Spiro have a reduced incidence of atraumatic fractures, especially of the hip [80]. Osteopenia and osteoporosis are also accompanying adverse outcomes to chronic SHPT; they predispose to atraumatic bone fractures [81]. Patients with heart failure have reduced bone density, which is related to SHPT and vitamin D deficiency, coupled with effort intolerance due to symptomatic failure and consequent reduced physical activity [63, 65, 82–86]. The risk of such fractures is further increased in elderly patients with heart failure receiving a loop diuretic, where consequent hypercalciuria is also contributory, but preventable when given in combination with spironolactone [79, 80]. In elderly patients with hip fracture, elevated PTH levels are associated with perioperative myocardial injury with elevated serum troponins and all-cause mortality [87].
Mg2+-Dependent Secretion of Parathyroid Hormone
In response to catecholamine-driven ionized hypocalcemia, the Ca2+-sensing receptor of the parathyroid glands provokes the increased secretion of PTH. In turn, secondary hyperparathyroidism (SHPT) with increased plasma PTH seeks to restore extracellular Ca2+ homeostasis by promoting resorption of bone Ca2+ and increased Ca2+ absorption from the gut and kidneys by 25(OH)2D3 which is synthesized by the kidneys in response to PTH provocation. When hypocalcemia is associated with hypomagnesemia, PTH secretion is impaired and can be corrected by reversing hypomagnesemia [88, 89].
CHF: A DYSHOMEOSTASIS OF ZINC AS ANTIOXIDANT
Excretory Zn2+ Losses in Aldosteronism
Deficiency in antioxidant reserves is also an important contributor to the imbalance in prooxidant:antioxidant equilibrium leading to cardiomyocyte necrosis that accompanies neurohormonal activation [90, 91]. Zinc is integral to antioxidant defenses, as well as wound healing [92]. Upregulation of metallothionein, a Zn2+-binding protein, occurs at sites of tissue injury, including the heart, where it promotes local accumulation of Zn2+ and its involvement in gene transcription and cell replication [93, 94]. Zn2+ deficiency, evident with reduced Zn2+ levels in bone and lymphocytes [95, 96], will compromise these antioxidant reserves and healing after cardiomyocyte necrosis.
In the secondary aldosteronism of CHF, increased urinary and fecal losses of Zn2+ result in hypozincemia with simultaneous cellular and subcellular dyshomeostasis of Zn2+ [94, 95]. Accompanying Zn2+ deficiency compromises the activity of Cu/Zn superoxide dismutase, an important endogenous antioxidant. Urinary Zn2+ excretion is increased in response to angiotensin-converting enzyme inhibitor or angiotensin receptor antagonist, commonly used in the management of CHF [97, 98]. Serum and myocardial Zn2+ levels are reduced in patients with a dilated cardiomyopathy and individuals with arterial hypertension [44, 96, 99–104••]. Underlying causes for Zn2+ deficiency, including inadequate dietary intake and excess urinary excretion, remain to be elucidated.
Intricate interactions between Zn2+ with Ca2+ have long been recognized [54, 92, 105, 106]. The prooxidant effect representing intracellular Ca2+ overloading that accompanies elevations in either plasma catecholamines or PTH is intrinsically coupled to increased Zn2+ entry in cardiomyocytes acting as an antioxidant [53, 54, 107, 108]. Zn2+ entry is known to occur via L-type Ca2+ channels, however, it enters predominantly via Zn2+ transporters activated by oxidative stress. Increased cytosolic free [Zn2+]i may also occur via release of inactive Zn2+ bound to metallothionein (MT)-1 and can be induced by nitric oxide (NO) derived from endothelial NO synthase [109]. Elevations in [Zn2+]i can also be achieved by a ZnSO4 supplement [53, 108, 110–115]. Increased cytosolic free [Zn2+]i activates its sensor, metal-responsive transcription factor (MTF)-1 which, upon its translocation to the nucleus, upregulates antioxidant defense genes [107]. These observations raise the therapeutic prospect that cation-modulating nutriceutical supplementation capable of favorably influencing the extra- and intracellular Ca2+ and Zn2+ equilibrium enhancing overall antioxidant capacity, could prove pivotal to combating mitochondria-based oxidative injury and cardiomyocyte necrosis, while promoting Zn2+-based endogenous cardioprotective potential.
SUMMARY AND CONCLUSIONS
A dyshomeostasis of extra- and intracellular K+, Mg2+, ionized Ca2+ and Zn2+ accompanies CHF—a coupled hyperadrenergic stressor state with RAAS activation. This includes catecholamine- and aldosterone-driven translocation of these cations: i) from the intravascular compartment to intracellular sites of storage in such organs as muscle, adipose tissue and liver, ii) to sites of injury, where they participate in tissue repair, and iii) increased urinary and fecal excretory losses. Hypokalemia, hypomagnesemia, ionized hypocalcemia and hypozincemia are consequences of this neurohormonally-driven dyshomeostasis of cations. The myocardium is particularly vulnerable to these pathophysiologic events. This includes a delay in repolarization, as reflected in QTc interval prolongation on the ECG, with increased propensity for supra- and ventricular arrhythmias. Intracellular Ca2+ overloading of its cardiomyocytes and mitochondria, mediated by catecholamines and/or PTH, leads to the induction of oxidative stress by these organelles and opening of their inner membrane permeability transition pore. There ensues mitochondrial degeneration followed by cell necrosis and consequent tissue repair with reparative fibrosis, or scarring. The cardioprotective potential of a MCRa therefore includes the rescue of these mono- and divalent cations and their attendant adverse consequences with protection from arrhythmias and pathologic remodeling of myocardium.
Acknowledgments
The authors are supported by NIH grants R01-HL73043 and R01-HL90867. The contents of this submission are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Disclosure
No potential conflicts of interest relevant to this article were reported.
References
- 1.Kamalov G, Bhattacharya SK, Weber KT. Congestive heart failure: where homeostasis begets dyshomeostasis. J Cardiovasc Pharmacol. 2010;56:320–328. doi: 10.1097/FJC.0b013e3181ed064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 3.Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 4.Weber KT. Efficacy of aldosterone receptor antagonism in heart failure: potential mechanisms. Curr Heart Fail Rep. 2004;1:51–56. doi: 10.1007/s11897-004-0025-4. [DOI] [PubMed] [Google Scholar]
- 5••.Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21. doi: 10.1056/NEJMoa1009492. The efficacy of a mineralocorticoid receptor antagonist was evaluated in the EMPHASIS-HF trial which consisted of patients having mild heart failure symptoms. A reduced risk of death from cardiovascular causes or hospitalization for heart failure was found. [DOI] [PubMed] [Google Scholar]
- 6.Francis GS, Benedict C, Johnstone DE, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure: a substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- 7.The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 8.Volpe M, Tritto C, DeLuca N, et al. Abnormalities of sodium handling and of cardiovascular adaptations during high salt diet in patients with mild heart failure. Circulation. 1993;88:1620–1627. doi: 10.1161/01.cir.88.4.1620. [DOI] [PubMed] [Google Scholar]
- 9.Ishii J, Nomura M, Nakamura Y, et al. Risk stratification using a combination of cardiac troponin T and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol. 2002;89:691–695. doi: 10.1016/s0002-9149(01)02341-4. [DOI] [PubMed] [Google Scholar]
- 10.Peacock WF, 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–2126. doi: 10.1056/NEJMoa0706824. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi R, Sato Y, Nishio Y, Kimura T, Kita T. Measurements of baseline and follow-up concentrations of cardiac troponin-T and brain natriuretic peptide in patients with heart failure from various etiologies. Heart Vessels. 2006;21:344–349. doi: 10.1007/s00380-006-0909-1. [DOI] [PubMed] [Google Scholar]
- 12.Sukova J, Ostadal P, Widimsky P. Profile of patients with acute heart failure and elevated troponin I levels. Exp Clin Cardiol. 2007;12:153–156. [PMC free article] [PubMed] [Google Scholar]
- 13.Ilva T, Lassus J, Siirilä-Waris K, et al. Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail. 2008;10:772–779. doi: 10.1016/j.ejheart.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Sato Y, Nishi K, Taniguchi R, et al. In patients with heart failure and non-ischemic heart disease, cardiac troponin T is a reliable predictor of long-term echocardiographic changes and adverse cardiac events. J Cardiol. 2009;54:221–230. doi: 10.1016/j.jjcc.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 15•.Miller WL, Hartman KA, Burritt MF, Grill DE, Jaffe AS. Profiles of serial changes in cardiac troponin T concentrations and outcome in ambulatory patients with chronic heart failure. J Am Coll Cardiol. 2009;54:1715–1721. doi: 10.1016/j.jacc.2009.07.025. Patients with decompensated heart failure having neurohormonal activation can have nonischemic cardiomyocyte necrosis with elevated serum troponins which contributes to an ongoing adverse remodeling of myocardium, including ensuing reparative fibrosis. [DOI] [PubMed] [Google Scholar]
- 16.Beltrami CA, Finato N, Rocco M, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- 17.Cotran RS, Kumar V, Robbins SL. Robbins Pathologic Basis of Disease. Philadelphia: W B Saunders; 1989. [Google Scholar]
- 18.Sun Y, Ramires FJA, Weber KT. Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc Res. 1997;35:138–147. doi: 10.1016/s0008-6363(97)00097-7. [DOI] [PubMed] [Google Scholar]
- 19.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 20••.Kjeldsen K. Hypokalemia and sudden cardiac death. Exp Clin Cardiol. 2010;15:e96–e99. The underappreciated importance of hypokalemia in clinical practice and its relationship to cardiac arrhythmias, both supraventricular and ventricular. [PMC free article] [PubMed] [Google Scholar]
- 21.Struthers AD, Whitesmith R, Reid JL. Prior thiazide diuretic treatment increases adrenaline-induced hypokalaemia. Lancet. 1983;1(8338):1358–1361. doi: 10.1016/s0140-6736(83)92140-2. [DOI] [PubMed] [Google Scholar]
- 22.Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol. 2000;85:1207–1211. doi: 10.1016/s0002-9149(00)00729-3. [DOI] [PubMed] [Google Scholar]
- 23.Reid JL, Whyte KF, Struthers AD. Epinephrine-induced hypokalemia: the role of beta adrenoceptors. Am J Cardiol. 1986;57:23F–27F. doi: 10.1016/0002-9149(86)90884-2. [DOI] [PubMed] [Google Scholar]
- 24.Ahrens RC, Smith GD. Albuterol: an adrenergic agent for use in the treatment of asthma pharmacology, pharmacokinetics and clinical use. Pharmacotherapy. 1984;4:105–121. doi: 10.1002/j.1875-9114.1984.tb03330.x. [DOI] [PubMed] [Google Scholar]
- 25.Ryzen E. Magnesium homeostasis in critically ill patients. Magnesium. 1989;8:201–212. [PubMed] [Google Scholar]
- 26••.Weglicki WB. Hypomagnesemia and inflammation: clinical and basic aspects. Annu Rev Nutr. 2012;32:55–71. doi: 10.1146/annurev-nutr-071811-150656. The underappreciated importance of chronic hypomagnesemia in contributing to pathologic remodeling of myocardium and ventricular dysfunction. [DOI] [PubMed] [Google Scholar]
- 27.Sheehan JP, Seelig MS. Interactions of magnesium and potassium in the pathogenesis of cardiovascular disease. Magnesium. 1984;3:301–314. [PubMed] [Google Scholar]
- 28.Solomon R. The relationship between disorders of K+ and Mg+ homeostasis. Semin Nephrol. 1987;7:253–262. [PubMed] [Google Scholar]
- 29.Leier CV, Dei Cas L, Metra M. Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia, and hypomagnesemia. Am Heart J. 1994;128:564–574. doi: 10.1016/0002-8703(94)90633-5. [DOI] [PubMed] [Google Scholar]
- 30.Szanda G, Rajki A, Gallego-Sandin S, Garcia-Sancho J, Spät A. Effect of cytosolic Mg2+ on mitochondrial Ca2+ signaling. Pflugers Arch. 2009;457:941–954. doi: 10.1007/s00424-008-0551-0. [DOI] [PubMed] [Google Scholar]
- 31.Romani A, Marfella C, Scarpa A. Regulation of magnesium uptake and release in the heart and in isolated ventricular myocytes. Circ Res. 1993;72:1139–1148. doi: 10.1161/01.res.72.6.1139. [DOI] [PubMed] [Google Scholar]
- 32.Carlstedt F, Lind L, Rastad J, et al. Parathyroid hormone and ionized calcium levels are related to the severity of illness and survival in critically ill patients. Eur J Clin Invest. 1998;28:898–903. doi: 10.1046/j.1365-2362.1998.00391.x. [DOI] [PubMed] [Google Scholar]
- 33.Carlstedt F, Lind L, Wide L, et al. Serum levels of parathyroid hormone are related to the mortality and severity of illness in patients in the emergency department. Eur J Clin Invest. 1997;27:977–981. doi: 10.1046/j.1365-2362.1997.2310778.x. [DOI] [PubMed] [Google Scholar]
- 34.Gunnewiek JM, Van Der Hoeven JG. Cardiac troponin elevations among critically ill patients. Curr Opin Crit Care. 2004;10:342–346. doi: 10.1097/01.ccx.0000135514.20538.44. [DOI] [PubMed] [Google Scholar]
- 35.Chhokar VS, Sun Y, Bhattacharya SK, et al. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol. 2004;287:H2023–H2026. doi: 10.1152/ajpheart.00477.2004. [DOI] [PubMed] [Google Scholar]
- 36.Chhokar VS, Sun Y, Bhattacharya SK, et al. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation. 2005;111:871–878. doi: 10.1161/01.CIR.0000155621.10213.06. [DOI] [PubMed] [Google Scholar]
- 37.Rossi E, Perazzoli F, Negro A, et al. Acute effects of intravenous sodium chloride load on calcium metabolism and on parathyroid function in patients with primary aldosteronism compared with subjects with essential hypertension. Am J Hypertens. 1998;11:8–13. doi: 10.1016/s0895-7061(97)00366-x. [DOI] [PubMed] [Google Scholar]
- 38.Goodwin KD, Ahokas RA, Bhattacharya SK, et al. Preventing oxidative stress in rats with aldosteronism by calcitriol and dietary calcium and magnesium supplements. Am J Med Sci. 2006;332:73–78. doi: 10.1097/00000441-200608000-00004. [DOI] [PubMed] [Google Scholar]
- 39.Vidal A, Sun Y, Bhattacharya SK, et al. Calcium paradox of aldosteronism and the role of the parathyroid glands. Am J Physiol Heart Circ Physiol. 2006;290:H286–H294. doi: 10.1152/ajpheart.00535.2005. [DOI] [PubMed] [Google Scholar]
- 40.Selektor Y, Ahokas RA, Bhattacharya SK, et al. Cinacalcet and the prevention of secondary hyperparathyroidism in rats with aldosteronism. Am J Med Sci. 2008;335:105–110. doi: 10.1097/MAJ.0b013e318134f013. [DOI] [PubMed] [Google Scholar]
- 41.Resnick LM, Nicholson JP, Laragh JH. Calcium metabolism and the renin-aldosterone system in essential hypertension. J Cardiovasc Pharmacol. 1985;7(Suppl 6):S187–S193. doi: 10.1097/00005344-198500076-00033. [DOI] [PubMed] [Google Scholar]
- 42.Resnick LM, Nicholson JP, Laragh JH. Calcium, the renin-aldosterone system, and the hypotensive response to nifedipine. Hypertension. 1987;10:254–258. doi: 10.1161/01.hyp.10.3.254. [DOI] [PubMed] [Google Scholar]
- 43.Resnick LM. Calciotropic hormones in salt-sensitive essential hypertension: 1,25-dihydroxyvitamin D and parathyroid hypertensive factor. J Hypertens Suppl. 1994;12:S3–S9. [PubMed] [Google Scholar]
- 44.LaGuardia SP, Dockery BK, Bhattacharya SK, et al. Secondary hyperparathyroidism and hypovitaminosis D in African-Americans with decompensated heart failure. Am J Med Sci. 2006;332:112–118. doi: 10.1097/00000441-200609000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Hellman DE, Kartchner M, Komar N, Mayes D, Pitt M. Hyperaldosteronism, hyperparathyroidism, medullary sponge kidneys, and hypertension. JAMA. 1980;244:1351–1353. [PubMed] [Google Scholar]
- 46.Resnick LM, Laragh JH. Calcium metabolism and parathyroid function in primary aldosteronism. Am J Med. 1985;78:385–390. doi: 10.1016/0002-9343(85)90328-6. [DOI] [PubMed] [Google Scholar]
- 47.Rossi E, Sani C, Perazzoli F, et al. Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens. 1995;8:884–893. doi: 10.1016/0895-7061(95)00182-O. [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg J, Pines M, Hurwitz S. Response of adrenal cells to parathyroid hormone stimulation. J Endocrinol. 1987;112:431–437. doi: 10.1677/joe.0.1120431. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberg J, Pines M, Hurwitz S. Stimulation of chick adrenal steroidogenesis by avian parathyroid hormone. J Endocrinol. 1988;116:91–95. doi: 10.1677/joe.0.1160091. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg J, Pines M, Hurwitz S. Inhibition of aldosterone secretion by atrial natriuretic peptide in chicken adrenocortical cells. Biochim Biophys Acta. 1989;1014:189–194. doi: 10.1016/0167-4889(89)90033-5. [DOI] [PubMed] [Google Scholar]
- 51.Brunaud L, Germain A, Zarnegar R, et al. Serum aldosterone is correlated positively to parathyroid hormone (PTH) levels in patients with primary hyperparathyroidism. Surgery. 2009;146:1035–1041. doi: 10.1016/j.surg.2009.09.041. [DOI] [PubMed] [Google Scholar]
- 52.Marney AM, Brown NJ. Aldosterone and end-organ damage. Clin Sci (Lond) 2007;113:267–278. doi: 10.1042/CS20070123. [DOI] [PubMed] [Google Scholar]
- 53.Gandhi MS, Deshmukh PA, Kamalov G, et al. Causes and consequences of zinc dyshomeostasis in rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2008;52:245–252. doi: 10.1097/FJC.0b013e3181833eb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamalov G, Deshmukh PA, Baburyan NY, et al. Coupled calcium and zinc dyshomeostasis and oxidative stress in cardiac myocytes and mitochondria of rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2009;53:414–423. doi: 10.1097/FJC.0b013e3181a15e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersson P, Rydberg E, Willenheimer R. Primary hyperparathyroidism and heart disease--a review. Eur Heart J. 2004;25:1776–1787. doi: 10.1016/j.ehj.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 56.Yu N, Donnan PT, Flynn RW, et al. Increased mortality and morbidity in mild primary hyperparathyroid patients. The Parathyroid Epidemiology and Audit Research Study (PEARS) Clin Endocrinol (Oxf) 2010;73:30–34. doi: 10.1111/j.1365-2265.2009.03766.x. [DOI] [PubMed] [Google Scholar]
- 57.Sambrook PN, Chen JS, March LM, et al. Serum parathyroid hormone is associated with increased mortality independent of 25-hydroxy vitamin D status, bone mass, and renal function in the frail and very old: a cohort study. J Clin Endocrinol Metab. 2004;89:5477–5481. doi: 10.1210/jc.2004-0307. [DOI] [PubMed] [Google Scholar]
- 58.Bjorkman M, Sorva A, Tilvis R. Parathyroid hormone as a mortality predictor in frail aged inpatients. Gerontology. 2009;55:601–606. doi: 10.1159/000239757. [DOI] [PubMed] [Google Scholar]
- 59.Stefenelli T, Abela C, Frank H, et al. Cardiac abnormalities in patients with primary hyperparathyroidism: implications for follow-up. J Clin Endocrinol Metab. 1997;82:106–112. doi: 10.1210/jcem.82.1.3666. [DOI] [PubMed] [Google Scholar]
- 60.Stefenelli T, Mayr H, Bergler-Klein J, et al. Primary hyperparathyroidism: incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med. 1993;95:197–202. doi: 10.1016/0002-9343(93)90260-v. [DOI] [PubMed] [Google Scholar]
- 61.Kiernan TJ, O’Flynn AM, McDermott JH, Kearney P. Primary hyperparathyroidism and the cardiovascular system. Int J Cardiol. 2006;113:E89–E92. doi: 10.1016/j.ijcard.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 62.Walker MD, Fleischer JB, Di Tullio MR, et al. Cardiac structure and diastolic function in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2010;95:2172–2179. doi: 10.1210/jc.2009-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shane E, Mancini D, Aaronson K, et al. Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med. 1997;103:197–207. doi: 10.1016/s0002-9343(97)00142-3. [DOI] [PubMed] [Google Scholar]
- 64.Alsafwah S, LaGuardia SP, Nelson MD, et al. Hypovitaminosis D in African Americans residing in Memphis, Tennessee with and without heart failure. Am J Med Sci. 2008;335:292–297. doi: 10.1097/MAJ.0b013e318167b0bd. [DOI] [PubMed] [Google Scholar]
- 65.Lee AH, Mull RL, Keenan GF, et al. Osteoporosis and bone morbidity in cardiac transplant recipients. Am J Med. 1994;96:35–41. doi: 10.1016/0002-9343(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 66.Schmid C, Kiowski W. Hyperparathyroidism in congestive heart failure. Am J Med. 1998;104:508–509. [PubMed] [Google Scholar]
- 67.Ogino K, Ogura K, Kinugasa Y, et al. Parathyroid hormone-related protein is produced in the myocardium and increased in patients with congestive heart failure. J Clin Endocrinol Metab. 2002;87:4722–4727. doi: 10.1210/jc.2002-020314. [DOI] [PubMed] [Google Scholar]
- 68.Zittermann A, Schleithoff SS, Tenderich G, et al. Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol. 2003;41:105–112. doi: 10.1016/s0735-1097(02)02624-4. [DOI] [PubMed] [Google Scholar]
- 69.Sugimoto T, Tanigawa T, Onishi K, et al. Serum intact parathyroid hormone levels predict hospitalisation for heart failure. Heart. 2009;95:395–398. doi: 10.1136/hrt.2008.147652. [DOI] [PubMed] [Google Scholar]
- 70.Schierbeck LL, Jensen TS, Bang U, et al. Parathyroid hormone and vitamin D--markers for cardiovascular and all cause mortality in heart failure. Eur J Heart Fail. 2011;13:626–632. doi: 10.1093/eurjhf/hfr016. [DOI] [PubMed] [Google Scholar]
- 71.Pilz S, Tomaschitz A, Drechsler C, et al. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J. 2010;31:1591–1598. doi: 10.1093/eurheartj/ehq109. [DOI] [PubMed] [Google Scholar]
- 72••.Hagström E, Hellman P, Larsson TE, et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation. 2009;119:2765–2771. doi: 10.1161/CIRCULATIONAHA.108.808733. Elevations in parathyroid hormone and the presence of secondary hyperparathyroidism as contributing to increased risk of cardiovascular mortality in a large elderly male cohort followed in Sweden. [DOI] [PubMed] [Google Scholar]
- 73••.Hagström E, Ingelsson E, Sundström J, et al. Plasma parathyroid hormone and risk of congestive heart failure in the community. Eur J Heart Fail. 2010;12:1186–1192. doi: 10.1093/eurjhf/hfq134. Elevations in parathyroid hormone as predictive of risk for congestive heart failure in the large male cohort from Sweden emphasizing the value of monitoring serum parathyroid hormone. [DOI] [PubMed] [Google Scholar]
- 74.Bell NH, Greene A, Epstein S, et al. Evidence for alteration of the vitamin D-endocrine system in blacks. J Clin Invest. 1985;76:470–473. doi: 10.1172/JCI111995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sawaya BP, Monier-Faugere MC, Ratanapanichkich P, et al. Racial differences in parathyroid hormone levels in patients with secondary hyperparathyroidism. Clin Nephrol. 2002;57:51–55. doi: 10.5414/cnp57051. [DOI] [PubMed] [Google Scholar]
- 76.Zittermann A, Fischer J, Schleithoff SS, et al. Patients with congestive heart failure and healthy controls differ in vitamin D-associated lifestyle factors. Int J Vitam Nutr Res. 2007;77:280–288. doi: 10.1024/0300-9831.77.4.280. [DOI] [PubMed] [Google Scholar]
- 77.Zittermann A, Schleithoff SS, Gotting C, et al. Poor outcome in end-stage heart failure patients with low circulating calcitriol levels. Eur J Heart Fail. 2008;10:321–327. doi: 10.1016/j.ejheart.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 78.Borkowski BJ, Cheema Y, Shahbaz AU, Bhattacharya SK, Weber KT. Cation dyshomeostasis and cardiomyocyte necrosis. The Fleckenstein hypothesis revisited. Eur Heart J. 2011;32:1846–1853. doi: 10.1093/eurheartj/ehr063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Diepen S, Majumdar SR, Bakal JA, McAlister FA, Ezekowitz JA. Heart failure is a risk factor for orthopedic fracture: a population-based analysis of 16,294 patients. Circulation. 2008;118:1946–1952. doi: 10.1161/CIRCULATIONAHA.108.784009. [DOI] [PubMed] [Google Scholar]
- 80.Carbone LD, Cross JD, Raza SH, et al. Fracture risk in men with congestive heart failure. Risk reduction with spironolactone. J Am Coll Cardiol. 2008;52:135–138. doi: 10.1016/j.jacc.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 81.Teucher B, Dainty JR, Spinks CA, et al. Sodium and bone health: impact of moderately high and low salt intakes on calcium metabolism in postmenopausal women. J Bone Miner Res. 2008;23:1477–1485. doi: 10.1359/jbmr.080408. [DOI] [PubMed] [Google Scholar]
- 82.Kerschan-Schindl K, Strametz-Juranek J, Heinze G, et al. Pathogenesis of bone loss in heart transplant candidates and recipients. J Heart Lung Transplant. 2003;22:843–850. doi: 10.1016/s1053-2498(02)00806-9. [DOI] [PubMed] [Google Scholar]
- 83.Nishio K, Mukae S, Aoki S, et al. Congestive heart failure is associated with the rate of bone loss. J Intern Med. 2003;253:439–446. doi: 10.1046/j.1365-2796.2003.01130.x. [DOI] [PubMed] [Google Scholar]
- 84.Kenny AM, Boxer R, Walsh S, Hager WD, Raisz LG. Femoral bone mineral density in patients with heart failure. Osteoporos Int. 2006;17:1420–1427. doi: 10.1007/s00198-006-0148-4. [DOI] [PubMed] [Google Scholar]
- 85.Frost RJ, Sonne C, Wehr U, Stempfle HU. Effects of calcium supplementation on bone loss and fractures in congestive heart failure. Eur J Endocrinol. 2007;156:309–314. doi: 10.1530/EJE-06-0614. [DOI] [PubMed] [Google Scholar]
- 86.Abou-Raya S, Abou-Raya A. Osteoporosis and congestive heart failure (CHF) in the elderly patient: double disease burden. Arch Gerontol Geriatr. 2009;49:250–254. doi: 10.1016/j.archger.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 87.Fisher AA, Southcott EK, Srikusalanukul W, et al. Relationships between myocardial injury, all-cause mortality, vitamin D, PTH, and biochemical bone turnover markers in older patients with hip fractures. Ann Clin Lab Sci. 2007;37:222–232. [PubMed] [Google Scholar]
- 88.Anast CS, Winnacker JL, Forte LR, Burns TW. Impaired release of parathyroid hormone in magnesium deficiency. J Clin Endocrinol Metab. 1976;42:707–717. doi: 10.1210/jcem-42-4-707. [DOI] [PubMed] [Google Scholar]
- 89.Rude RK, Oldham SB, Singer FR. Functional hypoparathyroidism and parathyroid hormone end-organ resistance in human magnesium deficiency. Clin Endocrinol (Oxf) 1976;5:209–224. doi: 10.1111/j.1365-2265.1976.tb01947.x. [DOI] [PubMed] [Google Scholar]
- 90.Singal PK, Kirshenbaum LA. A relative deficit in antioxidant reserve may contribute in cardiac failure. Can J Cardiol. 1990;6:47–49. [PubMed] [Google Scholar]
- 91.Kirshenbaum LA, Singal PK. Antioxidant changes in heart hypertrophy: significance during hypoxia-reoxygenation injury. Can J Physiol Pharmacol. 1992;70:1330–1335. doi: 10.1139/y92-186. [DOI] [PubMed] [Google Scholar]
- 92.Sharir H, Zinger A, Nevo A, Sekler I, Hershfinkel M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J Biol Chem. 2010;285:26097–26106. doi: 10.1074/jbc.M110.107490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iwata M, Takebayashi T, Ohta H, et al. Zinc accumulation and metallothionein gene expression in the proliferating epidermis during wound healing in mouse skin. Histochem Cell Biol. 1999;112:283–290. doi: 10.1007/s004180050449. [DOI] [PubMed] [Google Scholar]
- 94.Thomas M, Vidal A, Bhattacharya SK, et al. Zinc dyshomeostasis in rats with aldosteronism. Response to spironolactone. Am J Physiol Heart Circ Physiol. 2007;293:H2361–H2366. doi: 10.1152/ajpheart.00200.2007. [DOI] [PubMed] [Google Scholar]
- 95.Selektor Y, Parker RB, Sun Y, et al. Tissue 65zinc translocation in a rat model of chronic aldosteronism. J Cardiovasc Pharmacol. 2008;51:359–364. doi: 10.1097/FJC.0b013e318165b96e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tubek S. Role of zinc in regulation of arterial blood pressure and in the etiopathogenesis of arterial hypertension. Biol Trace Elem Res. 2007;117:39–51. doi: 10.1007/BF02698082. [DOI] [PubMed] [Google Scholar]
- 97.Golik A, Modai D, Averbukh Z, et al. Zinc metabolism in patients treated with captopril versus enalapril. Metabolism. 1990;39:665–667. doi: 10.1016/0026-0495(90)90098-w. [DOI] [PubMed] [Google Scholar]
- 98.Golik A, Zaidenstein R, Dishi V, et al. Effects of captopril and enalapril on zinc metabolism in hypertensive patients. J Am Coll Nutr. 1998;17:75–78. doi: 10.1080/07315724.1998.10720459. [DOI] [PubMed] [Google Scholar]
- 99.Arroyo M, LaGuardia SP, Bhattacharya SK, et al. Micronutrients in African-Americans with decompensated and compensated heart failure. Transl Res. 2006;148:301–308. doi: 10.1016/j.trsl.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 100.Oster O. Trace element concentrations (Cu, Zn, Fe) in sera from patients with dilated cardiomyopathy. Clin Chim Acta. 1993;214:209–218. doi: 10.1016/0009-8981(93)90112-h. [DOI] [PubMed] [Google Scholar]
- 101.Topuzoglu G, Erbay AR, Karul AB, Yensel N. Concentrations of copper, zinc, and magnesium in sera from patients with idiopathic dilated cardiomyopathy. Biol Trace Elem Res. 2003;95:11–17. doi: 10.1385/bter:95:1:11. [DOI] [PubMed] [Google Scholar]
- 102.Salehifar E, Shokrzadeh M, Ghaemian A, Aliakbari S, Saeedi Saravi SS. The study of Cu and Zn serum levels in idiopathic dilated cardiomyopathy (IDCMP) patients and its comparison with healthy volunteers. Biol Trace Elem Res. 2008;125:97–108. doi: 10.1007/s12011-008-8151-6. [DOI] [PubMed] [Google Scholar]
- 103.Kosar F, Sahin I, Taskapan C, et al. Trace element status (Se, Zn, Cu) in heart failure. Anadolu Kardiyol Derg. 2006;6:216–220. [PubMed] [Google Scholar]
- 104••.Frustaci A, Sabbioni E, Fortaner S, et al. Selenium- and zinc-deficient cardiomyopathy in human intestinal malabsorption: preliminary results of selenium/zinc infusion. Eur J Heart Fail. 2012;14:202–210. doi: 10.1093/eurjhf/hfr167. The underappreciated role of hypozincemia and hyposelenemia in leading to a dilated cardiomyopathy in patients with intestinal malabsorption and its reversibility with Zn2+ and Se2+ supplementation. [DOI] [PubMed] [Google Scholar]
- 105.Tuncay E, Bilginoglu A, Sozmen NN, et al. Intracellular free zinc during cardiac excitation-contraction cycle: calcium and redox dependencies. Cardiovasc Res. 2011;89:634–642. doi: 10.1093/cvr/cvq352. [DOI] [PubMed] [Google Scholar]
- 106.Turan B. Zinc-induced changes in ionic currents of cardiomyocytes. Biol Trace Elem Res. 2003;94:49–60. doi: 10.1385/BTER:94:1:49. [DOI] [PubMed] [Google Scholar]
- 107.Kamalov G, Ahokas RA, Zhao W, et al. Temporal responses to intrinsically coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria during aldosteronism. Am J Physiol Heart Circ Physiol. 2010;298:H385–H394. doi: 10.1152/ajpheart.00593.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kamalov G, Ahokas RA, Zhao W, et al. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol. 2010;55:248–254. doi: 10.1097/FJC.0b013e3181cf0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schulz R, Rassaf T, Massion PB, Kelm M, Balligand JL. Recent advances in the understanding of the role of nitric oxide in cardiovascular homeostasis. Pharmacol Ther. 2005;108:225–256. doi: 10.1016/j.pharmthera.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 110.Wang J, Song Y, Elsherif L, et al. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation. 2006;113:544–554. doi: 10.1161/CIRCULATIONAHA.105.537894. [DOI] [PubMed] [Google Scholar]
- 111.Chung MJ, Hogstrand C, Lee SJ. Cytotoxicity of nitric oxide is alleviated by zinc-mediated expression of antioxidant genes. Exp Biol Med (Maywood) 2006;231:1555–1563. doi: 10.1177/153537020623100916. [DOI] [PubMed] [Google Scholar]
- 112.Chung MJ, Walker PA, Brown RW, Hogstrand C. ZINC-mediated gene expression offers protection against H2O2-induced cytotoxicity. Toxicol Appl Pharmacol. 2005;205:225–236. doi: 10.1016/j.taap.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 113.Singal PK, Dhillon KS, Beamish RE, Dhalla NS. Protective effect of zinc against catecholamine-induced myocardial changes electrocardiographic and ultrastructural studies. Lab Invest. 1981;44:426–433. [PubMed] [Google Scholar]
- 114.Singal PK, Kapur N, Dhillon KS, Beamish RE, Dhalla NS. Role of free radicals in catecholamine-induced cardiomyopathy. Can J Physiol Pharmacol. 1982;60:1390–1397. doi: 10.1139/y82-207. [DOI] [PubMed] [Google Scholar]
- 115.Chvapil M, Owen JA. Effect of zinc on acute and chronic isoproterenol induced heart injury. J Mol Cell Cardiol. 1977;9:151–159. doi: 10.1016/0022-2828(77)90046-3. [DOI] [PubMed] [Google Scholar]