

Abstract
To elucidate the functional roles of mitochondrial dynamics in vivo, we identified genes that become essential in cells lacking the dynamin-related proteins Fzo1 and Dnm1, which are required for mitochondrial fusion and division, respectively. The screen identified Num1, a cortical protein implicated in mitochondrial division and distribution that also functions in nuclear migration. Our data indicate that Num1, together with Mdm36, forms a physical tether that robustly anchors mitochondria to the cell cortex but plays no direct role in mitochondrial division. Our analysis indicates that Num1-dependent anchoring is essential for distribution of the static mitochondrial network in fzo1 dnm1 cells. Consistently, expression of a synthetic mitochondria–cortex tether rescues the viability of fzo1 dnm1 num1 cells. We find that the cortical endoplasmic reticulum (ER) also is a constituent of the Num1 mitochondria–cortex tether, suggesting an active role for the ER in mitochondrial positioning in cells. Thus, taken together, our findings identify Num1 as a key component of a mitochondria–ER–cortex anchor, which we termed “MECA,” that functions in parallel with mitochondrial dynamics to distribute and position the essential mitochondrial network.
The shape and cellular distribution of mitochondria depend on the integrated and regulated activities of mitochondrial division and fusion, motility, and tethering (1). A key question is how these mitochondrial behaviors are coordinated to shape and position mitochondria properly in response to the changing needs of the cell. To begin to address this question, an understanding of the molecular basis of mitochondrial behaviors is essential.
The molecular mechanisms underlying mitochondrial division and fusion are best understood in terms of mitochondrial behaviors (1, 2). At the heart of the molecular machines that mediate mitochondrial division and fusion are dynamin-related proteins (DRPs) that function via GTP-dependent self-assembly and GTP hydrolysis-mediated conformational changes to remodel membranes. The DRP Dnm1/DRP1 (in yeast and mammals, respectively) drives the scission of mitochondrial membranes, and the DRPs Fzo1/MFN1/2 and Mgm1/OPA1 mediate fusion of the outer and inner mitochondrial membranes, respectively. The relative rates of mitochondrial division and fusion are major determinants of the steady-state structure of the organelle and greatly influence its distribution. Attenuation of mitochondrial division leads to a more interconnected, collapsed, and less distributable mitochondrial network, and attenuation of mitochondrial fusion results in mitochondrial fragmentation and pronounced defects in the transmission and distribution of mtDNA.
Although mitochondrial division and fusion are important, additional parallel pathways also are likely to be important for mitochondrial distribution. For example, the stable positioning of mitochondria at specific cellular locations, an indication of active tethering mechanisms, has been observed in many different cell types. In yeast, mitochondria are tethered at both the bud tip and the distal end of the mother cell, presumably to ensure that daughter cells receive and mother cells retain the essential mitochondrial compartment (3, 4). In neurons, mitochondria are stably positioned at synapses, where there is a high demand for energy and calcium buffering (5–7). EM analysis of cell types such as neurons and myocytes has revealed physical structures thought to act as tethers that stably position mitochondria at the plasma membrane (PM) and/or endoplasmic/sarcoplasmic reticulum (8–11). Thus, although there is good evidence for mitochondrial-specific tethers, the molecular basis and regulation of these structures are poorly understood.
To identify pathways that act in parallel with mitochondrial fusion and division in the regulation of mitochondrial behavior and to explore the functional roles of mitochondrial dynamics, we screened for genes that become essential in Δfzo1 Δdnm1 cells. The screen identified Num1, which our analysis indicates is a core component of a mitochondria–endoplasmic reticulum (ER)–cell cortex tether that positions mitochondria at the cortex to retain mitochondria in mother cells and actively distribute the mitochondrial network.
Results
NUM1 Is Essential in Δfzo1 Δdnm1 Cells.
In a screen of the Saccharomyces cerevisiae deletion collection of nonessential genes for factors that become essential for growth on rich dextrose medium in the absence of FZO1 and DNM1, which encode the mitochondrial division and outer membrane fusion DRPs, respectively, we identified NUM1 as essential. Num1 encodes a cortical protein implicated in mitochondrial division and distribution that also functions, in nuclear migration during cell division, as an anchor for the microtubule motor dynein (12–14). We confirmed that NUM1 is essential in Δfzo1 Δdnm1 cells by tetrad analysis (Fig. 1A). The Δfzo1 Δdnm1 Δnum1 triple mutant exhibited a severe growth defect in comparison with each single mutant and double-mutant combination. Furthermore, unlike the single and double mutants, the Δfzo1 Δdnm1 Δnum1 triple mutant could not be propagated after the germinated spores were patched onto rich dextrose medium. Thus, our results indicate that NUM1 becomes essential in the absence of mitochondrial division and mitochondrial outer membrane fusion.
Fig. 1.

NUM1 is essential in fzo1 dnm1 cells. (A) Heterozygous Δfzo1 Δdnm1 Δnum1 diploid cells were sporulated, and spores from individual tetrads were arranged in a column on YPD medium. Growth on selective plates was used to score the deletion marker and determine the genotypes of haploid cells, which are indicated. (B) Construction of a temperature-sensitive fzo1 dnm1 num1 strain using fzo1-1. Serial dilutions of the indicated strains were plated on YPD medium and grown at the permissive (23 °C) or nonpermissive (37 °C) temperature. (C) The morphology of mitochondria in the indicated strains expressing mito-dsRED was analyzed by fluorescence microscopy. Wild-type, Δdnm1, Δnum1, and Δdnm1Δnum1 cells were grown at 30 °C before imaging, and fzo1-1 and fzo1-1 Δnum1 cells were imaged after the cells were shifted to the nonpermissive temperature (37 °C) for 5 h. (D) The distribution of mitochondria in the indicated cells expressing mito-dsRED was analyzed by fluorescence microscopy before and after a temperature shift from 23 °C to 37 °C for 5 h. Cells were stained with calcofluor to image septa and bud scars to distinguish mother (marked with an “M”) and daughter cells. Whole-cell projections of cells imaged after the temperature shift to 37 °C are shown. Data are shown as the mean ± SE of three independent experiments, in each of which >75 cells were counted. (Scale bars, 2 μm.)
To address the basis for the synthetic lethality observed in the Δfzo1 Δdnm1 Δnum1 triple mutant, we constructed a conditional fzo1 dnm1 num1 triple-mutant strain using a previously characterized temperature-sensitive allele of FZO1, fzo1-1 (15). As expected, at the nonpermissive temperature (37 °C), growth of the fzo1-1 Δdnm1 Δnum1 triple mutant was severely compromised in comparison with all possible double-mutant combinations (Fig. 1B). A growth defect, albeit more subtle, also was observed for the conditional triple-mutant strain at the permissive temperature (23 °C), likely because the fzo1-1 allele is hypomorphic.
To gain insight into the cellular basis for lethality in fzo1-1 Δdnm1 Δnum1 cells, we analyzed mitochondrial morphology in single, double, and triple mutants by expressing mitochondrial matrix-targeted dsRED (mito-dsRED). A comparison of the mitochondrial morphology in single fzo1-1, Δdnm1, and Δnum1 mutants indicated that the morphology defect seen in Δnum1 cells was distinct from both the characteristic mitochondrial division-defective nets observed in Δdnm1 cells and the mitochondrial fusion-defective fragments observed in fzo1-1 cells at the nonpermissive temperature (Fig. 1C). In addition, the morphology of mitochondria observed in Δdnm1 Δnum1 and fzo1-1 Δnum1 double-mutant cells was distinct from that of each single mutant. Specifically, and in contrast to Δdnm1 and fzo1-1 cells, mitochondria were not localized at the cell cortex of Δdnm1 Δnum1 and fzo1-1 Δnum1 double-mutant cells (Fig. 1C). These results are consistent with the idea that mitochondrial dynamics and Num1 function in parallel pathways and suggest that Num1 is important for the association of mitochondria with the cell cortex.
In fzo1-1 Δdnm1 Δnum1 triple-mutant cells, we uniquely observed, in addition to a mitochondrial morphological defect, a population of cells devoid of mitochondria. Thus, we analyzed mitochondrial distribution further in the conditional triple and double mutants by staining the cells with the cell-wall dye calcofluor to distinguish mother and daughter cells (Fig. 1D). Our analysis revealed that fzo1-1 Δdnm1 Δnum1 triple-mutant cells had a significant defect in the retention of mitochondria in mother cells, as evidenced by the localization of the entire mitochondrial network in daughter buds (16 ± 4% and 32 ± 3% of cells at permissive and nonpermissive temperatures, respectively) (Fig. 1D). In fzo1-1 Δnum1 double-mutant cells, we observed a relatively minor defect in mother cell mitochondrial retention under nonpermissive conditions; this defect correlated with the relatively subtle growth defect of this strain at the nonpermissive temperature (Fig. 1 B and D). In contrast, no significant defects were observed in mother cell mitochondrial retention or growth in Δdnm1 Δnum1 and fzo1-1 Δdnm1 cells (Fig. 1 B and D). Thus, our analysis indicates that the severe growth defect observed in the fzo1-1 Δdnm1 Δnum1 triple mutant is a consequence of a defect in mitochondrial distribution, specifically in retention of mitochondria in the mother cell.
Num1-Mediated Tethering of Mitochondria to the Cell Cortex Is Essential in Δfzo1 Δdnm1 Cells.
To gain insight into how Num1 functions to control mitochondrial distribution, we analyzed the behavior of mitochondria and Num1 over time in wild-type cells expressing a chromosomal functional Num1-GFP fusion and mito-dsRED. Consistent with previously published observations, multiple Num1 molecules were localized to clusters positioned at the cortex (Fig. 2A) (13, 16). In every case, Num1 clusters were observed adjacent to regions of the mitochondrial network that were localized persistently at the cell cortex (Fig. 2A). Mitochondrial regions associated with cortical Num1 clusters were relatively static and restricted in comparison with the movements and changes in shape of distal but contiguous regions of the organelle (Movie S1). Num1 foci also always were observed adjacent to mitochondria in Δfzo1 Δdnm1 cells as well as in Δdyn1 cells in which dynein-mediated nuclear migration is disrupted (Fig. 2B and Fig. S1A). These data, which are consistent with previous studies (17), indicate that the Num1–mitochondria interaction is independent of mitochondrial division and fusion and of Num1’s role in nuclear migration, respectively. Together the localization of Num1 clusters, their association with mitochondria, and the behavior of mitochondria in their proximity suggest that Num1 functions to tether mitochondria to the cortex physically to ensure accurate distribution of the organelle between mother and daughter cells. Significantly, Num1 clusters are observed in the mother cell and larger buds but are absent in small buds (13, 16). Thus, early in the cell cycle the mitochondrial tethering activity of Num1 is predominantly mother-cell specific.
Fig. 2.

Num1 physically tethers mitochondria to the cell cortex. (A) Time-lapse images of calcofluor-stained wild-type yeast cells expressing Num1-GFP and mito-dsRED. A single focal plane is shown. Time is shown in seconds. (B) Num1-mediated tethering is independent of mitochondrial fusion and division and is independent of Num1’s role in dynein-mediated nuclear migration. Images of calcofluor-stained wild-type, Δdyn1, and Δfzo1 Δdnm1 cells expressing Num1-GFP and mito-dsRED are shown. (C) Images of Δnum1 cells (Upper) and fzo1-1 Δdnm1 Δnum1 cells (Lower) expressing MITO-GFP, GFP-PH, or MITO-GFP-PH as indicated. Δnum1 cells were grown at 30 °C before imaging, and fzo1-1 and fzo1-1 Δnum1 cells were imaged after the cells were shifted to the nonpermissive temperature (37 °C) for 5 h. The cell cortex was visualized using bright-field (BF) imaging (Upper) or by staining the cells with calcofluor before imaging (Lower). Single focal planes are shown. (D) Expression of a synthetic mitochondria–cortex tether rescues the growth of fzo1-1 Δdnm1 Δnum1 cells. Serial dilutions of fzo1-1 Δdnm1 Δnum1 cells expressing control constructs, MITO-GFP and GFP-PH, or the synthetic mitochondria–cortex tether, MITO-GFP-PH, were plated on SC−URA+Dex medium and grown at 23 °C or 37 °C, as indicated. EV, empty vector. (Scale bars, 2 μm.)
To test whether the loss of Num1-dependent mitochondrial tethering is the basis for the lethality observed in Δfzo1 Δdnm1 Δnum1 cells, we investigated whether expression of a synthetic mitochondria–cortex tether could rescue the growth defect of the conditional triple-mutant strain. We created a mitochondria–cortex tether by fusing the mitochondrial targeting sequence and transmembrane domain of the mitochondrial outer membrane protein Tom70 and the pleckstrin homology (PH) domain of Num1, which previously was shown to be both necessary and sufficient for cortical targeting, to the N and C termini, respectively, of GFP (MITO-GFP-PH) (16, 18). We expressed MITO-GFP-PH or constructs lacking either the mitochondria-targeting (GFP-PH) or cortex-targeting (MITO-GFP) portions in Δnum1 and fzo1-1 Δdnm1 Δnum1 cells. In contrast to the GFP-PH or MITO-GFP constructs, expression of MITO-GFP-PH restored the cortical localization of mitochondria in Δnum1 and fzo1-1 Δdnm1 Δnum1 cells and uniquely suppressed the growth defect of fzo1-1 Δdnm1 Δnum1 cells at the nonpermissive temperature (Fig. 2 C and D). Consistently, expression of MITO-GFP-PH also alleviated the mitochondrial-inheritance defect of fzo1-1 Δdnm1 Δnum1 cells at the nonpermissive temperature; mitochondrial-inheritance defects were observed in 8% of fzo1-1 Δdnm1 Δnum1 cells expressing MITO-GFP-PH and in 26% and 27% of fzo1-1 Δdnm1 Δnum1 cells expressing MITO-GFP and GFP-PH, respectively (n = 100 cells). Although it restored the cortical association of mitochondria, expression of MITO-GFP-PH did not restore a wild-type or static tubular mitochondrial morphology in Δnum1 and fzo1-1 Δdnm1 Δnum1 cells, respectively, suggesting that the native tether possesses additional functionality (Fig. 2C). Consistent with Num1 having separate roles in nuclear migration and mitochondrial distribution, the expression of MITO-GFP-PH did not rescue the nuclear-segregation defect of Δnum1 cells (Fig. S1B). Together, our data indicate that the tethering of mitochondria to the cell cortex via Num1 is essential for accurate distribution of a static mitochondrial network.
Coiled-Coil and PH Regions of Num1 Are Required for the Cortical Tethering of Mitochondria.
Num1 is a 313-kDa protein that contains an N-terminal coiled-coil (CC) domain, an EF hand-like motif, a region of twelve 64-aa repeats, and a C-terminal PH domain, which binds with high specificity to phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] (Fig. 3A) (12, 19). To gain insight into the molecular role of Num1 in mitochondria–cortex tethering, we performed structure–function analysis to test the roles of the repeat, CC, and PH regions. The localization pattern and tethering functions of GFP-Num1 constructs lacking the repeat region expressed in Δnum1 cells were indistinguishable from wild-type Num1, indicating that this region is dispensable for the normal distribution and mitochondrial tethering function of Num1. However, consistent with published data, in Δnum1 cells expressing GFP-Num1 constructs lacking the CC (Num1ΔCC) or PH domain (Num1ΔPH), the mitochondrial distribution, as assessed by steady-state mitochondrial morphology, was defective in a manner and extent similar to that observed in Δnum1 cells (Fig. 3B) (17, 18). In addition, as is consistent with published observations (17, 18), the localization of both Num1ΔCC and Num1ΔPH was altered in Δnum1 cells as compared with cells expressing the full-length Num1 construct (Fig. 3B). Specifically, although Num1ΔCC was localized to the cell cortex, it was distributed into smaller clusters that circumscribed the cortex more uniformly, and these clusters were present in areas not associated with mitochondria. This localization pattern indicates that the CC region is important for both Num1 cluster formation and for the association of Num1 with mitochondria. In contrast, Num1ΔPH localized into mitochondria-associated clusters that were similar in size and intensity to those in wild-type Num1. However, these clusters were not stably associated with the cortex. In addition, we observed a pool of Num1ΔPH that was diffusely mislocalized to the cytosol (Fig. 3B). These observations suggest that the PH domain has an important role in targeting Num1 to the cortex and are consistent with its known ability to bind plasma membrane PI(4,5)P2 with high specificity (18, 19). In addition, only Num1 constructs that retained the CC and PH regions were able to rescue the growth of the conditional fzo1-1 Δdnm1 Δnum1 mutant at the nonpermissive temperature (Fig. 3C). It should be noted, however, that the CC and PH regions of Num1 also have been shown to be essential for Num1’s role in nuclear migration (Fig. S1A) (16–18). Thus, although our data indicate that Num1-dependent mitochondrial tethering and nuclear migration pathways do not share downstream components, such as dynein, the cortical localization of Num1 and its organization into larger foci are features required for the function of both pathways.
Fig. 3.

CC and PH domains of Num1 are required for mitochondria–cortex tethering. (A) Schematic of Num1. CC, coiled-coil; EF, EF-like hand motif; PH, pleckstrin homology domain. (B) Δnum1 cells expressing mito-dsRED and the indicated Num1 construct were analyzed by fluorescence microscopy. Cells were stained with calcofluor immediately before imaging. A single focal plane is shown. (Scale bar, 2 μm.) (C) Serial dilutions of fzo1-1 Δdnm1 Δnum1 cells expressing the indicated Num1 construct were plated on YPD medium and grown at 23 °C or 37 °C, as indicated. E.V., empty vector.
Sites of Num1-Mediated Mitochondrial Tethering Are Distinct from Sites of Mitochondrial Division.
Num1 has been reported to colocalize with the mitochondrial division DRP Dnm1 and has been proposed to function directly in mitochondrial division (14). To examine the spatial relationship between sites of Num1-mediated mitochondrial tethering and Dnm1-dependent mitochondrial division, mitochondrial-targeted blue fluorescent protein (mito-BFP), Dnm1-mCherry, and Num1-GFP were expressed simultaneously and imaged in wild-type cells over time. In 40 of the 41 division events observed, the sites of Dnm1-dependent mitochondrial division were not marked or associated with Num1 clusters (Fig. 4A, arrows). In addition, we observed Dnm1-dependent mitochondrial division events in Δnum1 cells (Fig. 4B, arrows). The observed rate of mitochondrial division in Δnum1 cells was reduced as compared with wild-type cells (Fig. 4C). Although this observation may suggest that division is slightly attenuated in the absence of Num1, the disorganized and rapidly moving mitochondrial network of Δnum1 cells made it more difficult to visualize and therefore detect division events accurately in these cells than in wild-type cells. Thus, the actual number of division events in Δnum1 cells is likely greater than determined. We also observed that in every case Num1 clusters were adjacent to cortical regions of the mitochondrial network in Δdnm1 cells (Fig. 4D). These data are consistent with Dnm1 and Num1 having separate mitochondrial functions, a notion that is supported further by the distinct defects in mitochondrial morphology observed in Δnum1, Δdnm1, and Δnum1 Δdnm1 cells (Fig. 1C and Fig. S2). Thus, our data are consistent with a model in which Num1-mediated tethering and Dnm1-mediated division function in parallel to control the distribution of mitochondria in cells.
Fig. 4.

Num1 does not play a significant role in mitochondrial division. (A) Time-lapse images of wild-type cells expressing Num1-GFP, Dnm1-mCherry, and mito-BFP. Arrows indicate sites of mitochondrial division, and arrowheads indicate sites of Num1 and Dnm1 colocalization. A single focal plane is shown. (B) Time-lapse images of Δnum1 cells expressing Dnm1-GFP and mito-dsRED. Arrows indicate sites of mitochondrial division. Whole-cell projections are shown. (C) Quantification of the average number of mitochondrial division events observed per minute in wild-type and Δnum1 cells expressing mito-dsRED. The mitochondrial division rate of 25 cells was determined for each strain, and data are shown as the mean ± SE. (D) Time-lapse images of Δdnm1 cells expressing Num1-GFP and mito-dsRED. A single focal plane is shown. (E) Wild-type and Δcaf4 cells expressing Num1-GFP and Dnm1-mCherry. A single focal plan is shown. Arrows indicated colocalization of Dnm1 and Num1. (F) Quantification of the number of Dnm1 foci persistently localized to the cell cortex in the indicated cells. To be considered persistently localized, foci had to remain stationary at the cortex for ≥1 min. Data are shown as the mean ± SE. For each cell type, Dnm1 foci in 20 cells were counted. For all time-lapse series, time is shown in seconds. (Scale bars, 2 μm.)
Although the division and tethering pathways are independent, as is consistent with previous results, we observed colocalization of a subset of Dnm1 puncta with a subset of Num1 clusters (Fig. 4A, arrowheads) (14). Previous work has demonstrated that a distinct class of Dnm1 puncta is localized to the cortical side of mitochondrial tubules (20). This cortically polarized class of Dnm1 puncta has been shown to be dependent on Caf4, a paralog of the mitochondrial-division effector Mdv1 that plays, at most, a minor role in mitochondrial division (20, 21). Given the cortical localization of Num1, we examined the nature of Dnm1 puncta associated with Num1 clusters. As shown in Fig. 4A, Dnm1 puncta that colocalized with Num1 clusters were exclusively cortically polarized on mitochondrial tubules and were persistently localized at Num1-marked cortical sites over time. In contrast, Dnm1 puncta at sites of mitochondrial division were not polarized on mitochondrial tubules and were dynamic in nature (Fig. 4A, compare Dnm1 marked by arrowheads versus Dnm1 marked by arrows). Consistently, we observed that the colocalization of Dnm1 with Num1 clusters at the cell cortex was Caf4-dependent and that the number of Dnm1 puncta persistently localized to the cell cortex was reduced significantly in both Δnum1 and Δcaf4 cells (Fig. 4 E and F). Δcaf4 cells exhibit normal mitochondrial morphology and division rates (21). Thus, the localization of Dnm1 to Num1 clusters is not required for mitochondrial division, and this result is consistent with our data indicating that Dnm1-dependent division and Num1-dependent tethering are independent pathways (Fig. 4 B and D). Although the functional significance of Dnm1 at Num1 clusters is not clear, its dependence on Caf4 suggests that Dnm1 activity at Num1-tethering sites is different from its activity at mitochondrial-division sites.
Mdm36 Is a Component of the Num1 Tether Complex.
To identify additional components of the Num1 mitochondria–cortex tether complex, we purified Num1-GFP from cross-linked, detergent-solubilized, whole-cell extracts using anti-GFP antibodies and identified interacting proteins using LC-MS/MS (Fig. 5A). As a control, we performed the identical analysis of a wild-type untagged strain. The most robust interacting protein was Mdm36, which previously had been proposed to function with Num1 in mitochondrial distribution and division (22). In contrast, in our proteomic analysis we did not identify proteins that function with Num1 in the dynein-mediated nuclear migration pathway. Both genetic and cytological evidence suggests that Num1 interacts physically with components in this pathway. Thus, the absence of dynein/dynactin proteins in our analyses may indicate that the interactions of Num1 with dynein pathway components are dynamic or that the purification method disrupts these interactions. The Num1–Mdm36 interaction was verified in a reciprocal anti-FLAG purification from extracts of cells expressing a functional FLAG-tagged version of Mdm36 (Fig. 5A). In addition, using a yeast two-hybrid assay, we observed a robust Mdm36–Num1 interaction (Fig. 5D). Thus, our evidence indicates that Mdm36 and Num1 interact physically in a complex.
Fig. 5.
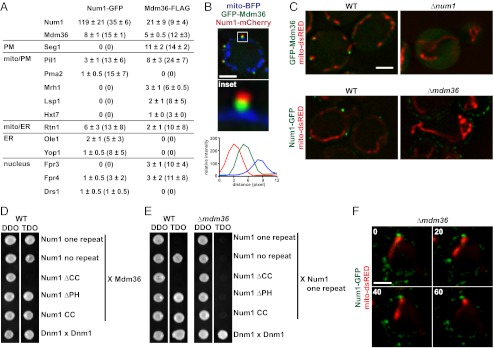
Mdm36 is a component of the Num1 mitochondria–cell cortex tether. (A) Proteomic analysis of Num1-GFP and Mdm36-FLAG immunoprecipitations as described in Methods. For each on-bead digest of the indicated purification, the number of peptides (and percent coverage) is shown for each identified protein. Data are shown as the mean ± SE of three independent experiments. (B) Cells expressing mito-BFP, GFP-Mdm36, and Num1-mCherry were analyzed by fluorescence microscopy. A single focal plane is shown. The boxed area is magnified fivefold in the Inset . The graph shows the relative fluorescence intensities of Num1 (red), Mdm36 (green), and mitochondria (blue) along a line drawn through the center of the Num1 and Mdm36 foci. (C) Images of wild-type and Δnum1 cells expressing GFP-Mdm36 and mito-dsRED (Upper) and wild-type and Δmdm36 cells expressing Num1-GFP and mito-dsRED (Lower). A single focal plane is shown. (D) Yeast two-hybrid analysis to assess the interaction of a Gal4BD-Mdm36 fusion with the indicated Gal4AD-Num1 fusions. Protein–protein interactions were assessed by growth on triple-dropout (TDO) medium. Dnm1 self-interaction was used as a positive control. TDO, SC−Leu−Trp−Ade; double-dropout medium (DDO), SC−Leu−Trp. (E) Yeast two-hybrid analysis to assess the interaction of a Gal4AD-Num1 one repeat fusion with the indicated Gal4BD-Num1 fusions in the presence (WT) or absence (Δmdm36) of Mdm36. Protein–protein interactions were assessed as in D. (F) Time-lapse images of Δmdm36 cells expressing Num1-GFP and mito-dsRED. A single focal plane is shown. Time is shown in seconds. (Scale bars, 2 μm.)
To test further the significance of the Num1–Mdm36 interaction in vivo, we examined the subcellular distribution of a functional GFP-Mdm36 fusion in cells expressing Num1-mCherry and mito-BFP. We observed that Mdm36 formed mitochondrial-associated foci on the cortical side of mitochondrial tubules that were adjacent to but only partially colocalized with Num1 clusters (Fig. 5B and Fig. S3). Consistent with a physical and functional Num1–Mdm36 interaction, the localization patterns of Mdm36 and Num1 were interdependent. In the absence of Num1, Mdm36 was distributed diffusely in the cytosol (Fig. 5C). In absence of Mdm36, Num1 was distributed in smaller clusters that uniformly circumscribed the cortex and were present in regions not associated with mitochondria (Fig. 5C). This pattern of Num1 localization is similar to that of Num1ΔCC in Δnum1 cells, suggesting that the Num1ΔCC distribution defect is a consequence of its inability to interact with Mdm36 (Figs. 3B and 5C). Consistent with this interpretation, in the yeast two-hybrid assay we observed that the CC region of Num1 was both necessary and sufficient for the Mdm36 interaction (Fig. 5D). Thus, together our data indicate that Num1 and Mdm36 interact in a manner dependent on the CC region of Num1 and that this interaction is important for the correct localization and function of Num1 in cells.
To gain insight into the mechanistic role of Mdm36, we explored the idea that Mdm36 facilitates the assembly of Num1, because Num1 clusters were smaller and more diffusely distributed in Δmdm36 cells. In the yeast two-hybrid assay, we observed robust Num1–Num1 and Mdm36–Mdm36 interactions. Thus, we tested whether the Num1–Num1 interaction was dependent on the presence of Mdm36 by examining the two-hybrid interaction in Δmdm36 cells. We observed that the Num1–Num1 interaction indeed was dependent both on Mdm36 expression and on the presence of the Num1 CC (Fig. 5E). In contrast, the Mdm36–Mdm36 two-hybrid interaction was independent of Num1 expression (Fig. S4A). These data are consistent with a model in which Mdm36, via its interaction with the Num1 CC, functions as a molecular bridge to facilitate Num1 self-assembly into larger mitochondrial-associated clusters.
We investigated the in vivo role of Mdm36 in mitochondrial tethering. Deletion of MDM36 caused a significant growth defect in Δfzo1 Δdnm1 cells, but this growth defect was not as severe as that observed in Δfzo1 Δdnm1 Δnum1 cells (Fig. S4 B and C). In addition, Δfzo1 Δdnm1 Δmdm36 cells did not exhibit a significant defect in mitochondrial inheritance (Fig. S4D). Consistently, in Δmdm36 cells we observed regions of mitochondria adjacent to a subset of Num1 clusters at the cell cortex. Num1 clusters associated with mitochondria in Δmdm36 cells were larger than non–mitochondrial-associated clusters in the same cell and were fewer than those observed in wild-type cells (Fig. 5F and Fig. S4E). However, as in wild-type cells, mitochondrial regions associated with Num1 clusters in Δmdm36 cells were less mobile, indicating that this subset of Num1 clusters possessed tethering activity (Fig. 5F). Together our data suggest that Mdm36 bridges Num1 interactions to facilitate the formation of an adequate number of large Num1 clusters that are competent for mitochondrial tethering. These data also indicate that Mdm36, although important, is not essential for the biogenesis of functional Num1 tethers and suggest that other factors, in addition to Mdm36, are involved.
ER Is a Component of the Num1 Mitochondrial Tether.
To gain insight into other factors associated with the Num1 tether, we considered the nature of the proteins that copurified with Num1 or Mdm36 (Fig. 5A). In addition to proteins localized to the PM, we identified a significant fraction of ER-localized proteins. We also identified a significant fraction of ER proteins copurifying with Num1 when the protein was isolated from Δmdm36 cells, in which Num1 is more diffusely distributed at the cortex (Fig. S5). Yeast possesses cortical ER, which is estimated to cover 20–45% of the PM (23). Thus, the proteomic analysis suggests proximity between the Num1 tether complex and the cortical ER.
We examined the spatial relationship between the Num1 mitochondria–cortex tether and the ER by monitoring the localization of Num1-GFP, mito-BFP, and ER-dsRED simultaneously in live cells. We observed that in every case the ER was present at sites of Num1-mediated mitochondria–cell cortex tethering (Fig. 6A). We quantified the relationship between Num1, mitochondria, and ER by performing line-scan analyses. Consistent with a Num1–ER association, line-scan analyses through the center of Num1 clusters indicated that the profiles of fluorescence intensity versus distance were similar for Num1 and ER (Fig. 6A and Fig. S6A). In contrast, the peak intensities of the Num1 clusters and associated mitochondria were spatially resolved. To gain further insight into the architecture of Num1-tethering sites, we extended our line-scan analysis to include Mdm36 foci, mitochondria, and ER (Fig. 6B and Fig. S6B). In contrast to Num1, the peak intensity of Mdm36 was consistently between the peak intensities of the ER and mitochondria, as is consistent with the partial colocalization of Mdm36 with Num1. These data are consistent with a model in which interactions between Num1 and Mdm36 create a structure that links the ER and mitochondria at the cortex.
Fig. 6.

The ER is a component of the Num1 mitochondria–cell cortex tether. (A) Wild-type cells expressing Num1-GFP, mito-BFP, and ER-dsRED were analyzed by fluorescence microscopy. The boxed area is magnified fivefold in the Inset. The graphs show the relative fluorescence intensities of the ER (red), Num1 (green), and mitochondria (blue) along a line drawn through the center of the Num1 cluster. (B) Wild-type cells expressing GFP-Mdm36, mito-BFP, and ER-dsRED were analyzed by fluorescence microscopy. The boxed area is magnified fivefold in the Inset. The graph shows the relative fluorescence intensities of the ER (red), Mdm36 (green), and mitochondria (blue) along a line drawn through the center of the Mdm36 foci. (C) Δrtn1/2 Δyop1 cells expressing Num1-GFP, mito-BFP, and ER-dsRED were analyzed as described in A. (D) Wild-type and Δrtn1/2 Δyop1 cells expressing ER-dsRED and GFP-Num1 or GFP-Num1ΔCCΔEF, as indicated, were analyzed by fluorescence microscopy. In A–D, a single focal plane is shown. (Scale bars, 2 μm.) (E) Model of MECA-mediated mitochondria–cortex tethering. The MECA is comprised of two interacting proteins, Num1 and Mdm36, and three organelles, mitochondria (mito), the ER, and the PM. Num1 is a core component of the MECA and possesses at least three regions of unique functionality: The CC domain mediates Num1 self-assembly in a manner regulated by Mdm36-dependent bridging/stabilizing interaction; the PH domain targets the MECA to the cell cortex via interaction with PM-localized PI(4,5)P2; and a region of Num1 outside the CC and PH domains interacts with the ER directly or indirectly via additional, not-yet-identified MECA components (indicated by the black question mark). Factors independent of Mdm36 and the CC region of Num1 contribute to Num1 self-assembly (indicated by the orange question mark) and to the association of the MECA with mitochondria (light blue oval).
To test further that the ER is a component of the Num1 tether, we disrupted cortical ER distribution by deleting genes encoding the ER tubule-shaping proteins Rtn1/2 and Yop1 and examined Num1 and ER colocalization (24). In these cells, Num1 clusters were observed to localize only to regions of the cortex where the ER was present and were strictly absent from large ER-depleted cortical regions (Fig. 6C, Upper and Fig. S6C). In addition, the more diffusely distributed N-terminal Num1 deletion Num1ΔEFΔCC was enriched on regions of the cell cortex occupied by cortical ER in both wild-type and Δrtn1/2 Δyop1 cells (Fig. 6D), providing further evidence for a Num1–ER association. The cellular distribution of Num1 clusters in Δrtn1/2 Δyop1 cells was altered also. Specifically, a small fraction of Δrtn1/2 Δyop1 cells (∼5% of cells) contained Num1 clusters that were not localized to the cortex; significantly, these clusters remained colocalized with the ER and mitochondria (Fig. 6C, Lower and Fig. S6D). Based on the behavior of mitochondria localized proximal to the noncortical Num1 clusters, the ability of these noncortical clusters to tether mitochondria was maintained, suggesting that an interaction with the PM is not required for Num1 mitochondrial tethering and that the ER plays a primary role in Num1 localization. Thus, in total, our data indicate that Num1 is a component of a multisubunit tether that bridges the mitochondria and ER at the cell cortex. Based on our data, we term this complex “MECA” (for “mitochondria–ER–cortex anchor”) and suggest that the ER is a critical structural and functional component of the MECA.
Discussion
Our analysis indicates that mitochondrial dynamics and mitochondrial tethering are parallel pathways that work in concert to distribute mitochondria accurately in cells. In yeast, to date, two cortical mitochondrial tethers have been identified: the MECA, which we demonstrate functions in the distribution and mother-specific retention of mitochondria, and Mmr1, which functions in the inheritance of mitochondria by buds early in the cell cycle as a receptor for Myo2 (25, 26). In contrast to Mmr1, which is restricted to buds, the MECA is absent from small buds and is localized to larger buds and mother cells (13, 16, 25). Thus, these two cortical tethers work to ensure the proper distribution of mitochondria by generating opposing forces at spatially distinct and exclusive locations, a model supported by the strong positive genetic interactions observed between NUM1 and MMR1 (27).
Our data indicate that the MECA is an extended, multisubunit structure comprising at least two proteins, Num1 and Mdm36, and three organelles, mitochondria, the ER, and the PM (Fig. 6E). Num1 functions in a modular manner as an essential MECA core component by mediating interactions with the PM, the ER, and Mdm36. Mdm36, like Num1, but in contrast to another MECA-associated component, Dnm1, possesses a steady-state MECA localization and has been reported to interact with the mitochondrial outer membrane (22). This observation, in combination with our data demonstrating that the localization of Mdm36 within the MECA is more proximal to mitochondria, as opposed to the more cortical localization of Num1, raises the possibility that Mdm36 functions as the MECA mitochondrial receptor. However, our analysis indicates either that this function is unlikely or that Mdm36 does not act alone, because Mdm36 is not essential for MECA-mediated mitochondrial tethering. In cells lacking Mdm36, the distribution of Num1 cluster size/intensity is shifted toward smaller clusters, and these smaller clusters do not associate with mitochondria. Thus, our data are more consistent with a role for Mdm36 in MECA biogenesis/maintenance and suggest that Mdm36 facilitates and/or stabilizes Num1 self-assembly, which in turn promotes the formation of Num1 clusters that possess the necessary valency to anchor mitochondria robustly to the cell cortex.
Although Mdm36 plays a role, the mechanistic basis for the self-assembly of Num1—a critical feature of the MECA—is not understood completely. Our data demonstrate that Mdm36 interacts with Num1 via its N-terminal CC, which corresponds to a region suggested in a recent study to form a dimeric BAR domain (17). BAR domains possess the ability to self-assemble on membranes, suggesting that Mdm36 may function in MECA biogenesis and maintenance by peripherally binding to and stabilizing the putative Num1 BAR domain assembly interface(s). However, Num1 lacking the CC region also retains the ability to assemble into small clusters that do not associate with mitochondria, similar to those observed in the absence of Mdm36. Thus, it is likely that, in addition to Mdm36 and the Num1 CC region, other factors contribute to Num1 self-assembly and Num1’s association with mitochondria. Indeed, exactly how the MECA complex associates with mitochondria is an outstanding question.
We are only beginning to understand the exact and relative contributions of the cortical ER and PM to MECA function. Noncortical MECA complexes retain their ability to tether mitochondria and ER, indicating that PM association is not required for MECA-mediated tethering of the two organelles. However, the localization of the MECA to the cortex is critical for its role in mitochondrial distribution and is likely mediated by an interaction between the Num1 PH domain and the predominantly PM-localized lipid PI(4,5)P2 (19). Our analysis indicates that the ER, like Num1 but in contrast to the PM, is a core MECA component that plays a central role in the MECA’s tethering function. The molecular basis underlying the interaction between these two core components is not known, but our structure–function analysis indicates the interaction involves a region of Num1 outside the CC and PH domains and likely other not-yet-identified protein components.
The central role of the ER in mitochondrial tethering in yeast cells is underscored by the recent observation that, like the MECA, the bud-specific Mmr1 tether is associated with the ER (28). These observations raise the intriguing possibility that the ER is a conserved component of mitochondrial tethers in other cell types, such as neurons and activated immune cells, where mitochondria are tethered in cellular regions with high energy and calcium-buffering needs (5–7, 29). Contacts between mitochondria and the ER also play a conserved role in mitochondrial division (30). Thus, contacts between the ER and mitochondria participate actively in multiple pathways that position and shape the mitochondrial network.
The juxtaposition of three membrane systems within the MECA raises the possibility that the MECA, in addition to its tethering function, also serves to create specialized microdomains that facilitate the exchange of lipid, calcium, and/or other small molecules between membrane compartments (31). Indeed, ER-mitochondria encounter structure-mediated ER–mitochondria contacts have been implicated in lipid exchange in yeast (32). In this context, we speculate that the modular multidomain nature of Num1 allows it to serve as a master integrator of both temporal and spatial cues to regulate MECA functions at the cell cortex.
Methods
Strains and Plasmids.
Strains W303 (ade2–1; leu2–3; his3–11, 15; trp1–1; ura3–1; can1–100), W303 fzo1-1, and NDY257 (BY4741 his3Δ1 leu2Δ met15Δ ura3Δ rtn1::kanMX4 rtn2::kanMX4 yop1::kanMX4) were described previously (24, 33) The yeast two-hybrid strains Y187 and Y2HGold were purchased from Clontech.
The plasmids pVT100U-mito-dsRED (mito-dsREDa) (33), pXY142-mito-dsRED (mito-dsREDb) (30), pYES-mtBFP (mito-BFP) (34), pHS20 (Dnm1-GFP, a kind gift from R. Jensen Johns Hopkins University, Baltimore) (35), pHS20-mCherry (Dnm1-mCherry) (36), and pKW1803 (dsRED-HDEL, referred to as “ER-dsRED” in the text) (37) were described previously.
The following W303 gene-deletion strains were obtained by replacing the complete ORF of the genes by the indicated cassette using PCR-based targeted homologous recombination: Δcaf4::HIS3MX6, Δdnm1::KanMX6, Δdyn1::HIS3MX6, Δfzo1::NATMX6, Δmdm36::NATMX6, Δmdv1::KanMX6, and Δnum1::HIS3MX6 (38). The functional C-terminally tagged strains, Num1-yEGFP (Num1-GFP), Num1-yEmCherry (Num1-mCherry), and Mdm36-FLAG, were constructed by PCR-based targeted homologous recombination using yEGFP-SpHis5MX6 (39), yEmCherry-SpHis5MX6 (described below), and 3XFLAG-His3MX6 (described below) cassettes, respectively. Haploid double- or triple-mutant/tagged strains were generated by crossing followed by sporulation or by PCR-based targeted homologous recombination. W303 dsRED-HDEL and Δrtn1/2 Δyop1 dsRED-HDEL were constructed by transforming the indicated strain with EcoRV-linearized pKW1803.
To construct the yEmCherry integration cassette, the yEmCherry coding sequence from pMG2254 (a kind gift from J. Berman University of Minnesota, Minneapolis) was amplified by PCR and used to replace the yEGFP coding sequence in pKT128 using PacI/AscI (39, 40). To construct the 3XFLAG-His3MX6 integration cassette, the 3XFLAG sequence was amplified by PCR and used to replace the GFP(S56T) coding sequence from pFA6a-GFP(S65T)-HISMX6 using PacI/AscI (38).
To construct the synthetic mitochondria–cortex tether MITO-GFP-PH, the C-terminal ER tail-anchor of Ubc6 in p416GPD::ChiMERA [a kind gift from B. Kornmann and P. Walter, University of California, San Francisco (32)] was replaced with the PH domain (amino acids 2563–2692) of Num1. The expression level of the MITO-GFP-PH from this plasmid was determined by Western analysis to be ∼20-fold greater than that of Num1-GFP. p416GPD::MITO-GFP-PH then was used as a template to PCR amplify MITO-GFP and GFP-PH, which then were used to replace MITO-GFP-PH in p416GPD::MITO-GFP-PH using BamHI/XhoI and resulting in p416GPD::MITO-GFP (MITO-GFP) and p416GPD::GFP-PH (GFP-PH), respectively.
To construct p414MET25::yEGFP and p416MET25::yEGFP, yEGFP was amplified from pKT128 by PCR, digested with SpeI/BamHI, and inserted into similarly digested p414MET25 and p416MET25 (39, 41). To construct p414MET25::yEGFP-Num1 and p414MET25::yEGFP-Num1 (GFP-Num1), Num1 was amplified by PCR and inserted into the vectors using BamHI/XhoI sites. p414MET25::yEGFP-Num1 one repeat was constructed in multiple steps. Num1(1384–2784) was PCR amplified and inserted into pRS316 using XmaI/XhoI sites. Num1(1–656) then was PCR amplified and inserted into pRS316 Num1(1384–2784) using BamHI/XmaI sites to construct pRS316 Num1 one repeat. Num1 one repeat then was subcloned from pRS316 into p414MET25::yEGFP using BamHI/XhoI sites. The N-terminal and C-terminal deletion constructs were PCR amplified using p414MET25::yEGFP-Num1 one repeat as a template and were inserted into p414MET25::yEGFP using BamHI/XhoI sites. To construct p414MET25::yEGFP-Num1 no repeat, Num1(1–592) first was PCR amplified and inserted into pRS316 Num1(1384–2784) to construct pRS316 Num1 no repeat. Num1 no repeat then was subcloned from pRS316 into p414MET25::yEGFP using BamHI/XhoI sites. To construct the p416MET25::yEGFP-Num1 deletion plasmids, the indicated Num1 deletions from the p414MET25::yEGFP-Num1 deletion series were subcloned into p416MET25::yEGFP using BamHI/XhoI sites. To construct p414MET25::yEGFP-Mdm36 (GFP-Mdm36), Mdm36 was amplified by PCR, digested with BglII/XhoI, and inserted into p414MET25::yEGFP using the BamHI/XhoI sites.
To construct pGADT7-BamHI and pGBKT7-BamHI, the multiple cloning sites of pGADT7 and pGBKT7 (Clontech) from the NdeI–XhoI or SalI sites, respectively, were replaced with CATATGGGATCCACTAGTCTCGAG and CATATGGGATCCACTAGTGTCGAC, respectively. This substitution places a BamHI site in frame with the upstream Gal4AD and Gal4BD sequence. Num1 deletion constructs were amplified by PCR and inserted into pGADT7-BamHI and pGBKT7-BamHI using BamHI/XhoI and BamHI/SalI, respectively. Mdm36 was amplified by PCR, and the product was digested with BglII/XhoI or BglII/SalI and inserted into pGADT7-BamHI and pGBKT7-BamHI using BamHI/XhoI and BamHI/SalI, respectively.
Growth Assays.
For analysis of growth by serial dilution, cells were grown overnight in yeast extract/peptone/dextrose (YPD) medium or in synthetic complete medium-uracil (SC−Ura) + 2% (wt/vol) dextrose (Dex), pelleted, and resuspended in water at a concentration of 0.5 OD600/mL, and fivefold serial dilutions were performed. Cells were spotted onto YPD or SC−Ura+Dex, as indicated, and were grown at the indicated temperature.
Yeast Two-Hybrid Analysis.
Y187 and Y2HGold were transformed with the indicated Gal4AD and Gal4BD fusions, respectively. Y187 cells harboring the indicated Gal4AD fusions then were mated with Y2HGold cells harboring the indicated Gal4BD fusions. Diploids were selected by plating on SC–Leu–Trp+Dex medium, and protein–protein interactions were assessed by growth on SC–Leu–Trp–Ade+Dex medium. The expression of all Gal4AD and Gal4BD fusion proteins was confirmed by Western analysis.
Cytological Analysis.
To visualize mitochondrial morphology and inheritance, the indicated strains were transformed with pVT100U-dsRED. Before imaging, the strains were grown in SC−Ura+Dex, and calcofluor was added to the medium at a final concentration of 100 μg/mL for 5 min before imaging. To visualize Num1-mediated mitochondrial tethering, W303 Num1-yEFGP, W303 Num1-yEFGP Δfzo1 Δdnm1, W303 Num1-yEFGP Δdnm1, W303 Num1-yEFGP Δmdm36, and W303 Num1-yEFGP Δdyn1 cells harboring pYX142-mitodsRED were grown in SC−Leu+Dex medium and, where indicated, were stained with calcofluor before imaging. To visualize Dnm1-mediated mitochondrial division in the absence of Num1, Δnum1 cells harboring pVT100U-dsRED and pHS20 were grown in SC−Ura−Leu+Dex medium. To visualize the ER and Num1 in wild-type and Δrtn1/2 Δyop1 cells, wild-type dsRED-HDEL and Δrtn1/2 Δyop1 dsRED-HDEL cells harboring the indicated p416MET25::yEGFP-Num1 deletion were grown in SC−Ura+Dex medium. In all cases, cells were grown to log phase in the medium indicated above, concentrated by centrifugation, and resuspended in 1/100th volume of fresh medium. Cell monolayers then were created by mounting cells on a 4% (wt/vol) agarose pad or by flowing cells into the imaging chamber of a CellAsic Y04C plate using an ONIX Microfluidic Perfusion Platform (CellAsic). Z-series of cells were imaged over time using the spinning-disk module of a Marianas SDC Real Time 3D Confocal-TIRF microscope (Intelligent Imaging Innovations) fitted with a 100×, 1.46 NA objective and an EMCCD camera (Photometrics). A step size of 0.4 μm was used. Image capture and postcapture processing were done using SlideBook5 software (Intelligent Imaging Innovations), and Photoshop (Adobe) was used to make linear adjustments to brightness and contrast. Interpolated images are shown.
To visualize Num1, Dnm1, and mitochondria, W303 Num1-yEGFP harboring pHS20-mCherry and pYES-mitoBFP were grown in SC−Leu−Ura + 2% (wt/vol) galactose (Gal) medium. To visualize Num1, Mdm36, and mitochondria, W303 Num1-yEmCherry cells harboring p414-Met25::yEGFP-Mdm36 and pYES-mitoBFP were grown in SC−Trp−Ura+Gal medium. For the Num1–Mdm36 interdependence analysis, Δnum1 cells harboring p414-Met25::yEGFP-Mdm36 and pYX142-mitodsRED and Δmdm36 Num1-yEGFP cells harboring pYX142-mitodsRED were grown in SC−Leu−Trp+Dex and SC−Leu+Dex media, respectively. To visualize Num1, mitochondria, and the ER, wild-type Num1-yEGFP dsRED-HDEL cells and Δrtn1/2 Δyop1 Num1-yEGFP dsRED-HDEL cells harboring pYES-mitoBFP were grown in SC−Ura+Gal medium. To visualize Mdm36, mitochondria, and the ER, wild-type dsRED-HDEL cells harboring p414-Met25::yEGFP-Mdm36 and pYX142-mitodsRED cells were grown in SC−Leu−Trp+Dex. In all cases, cells were grown to log phase in the indicated medium, were concentrated by centrifugation, and were resuspended in a 1/100th volume of fresh medium. Cell monolayers then were created by mounting cells on a 4% agarose pad. Either single focal planes over time or whole-cell z-series at a single time point were captured using a DeltaVision-Real Time microscope fitted with a 60×, 1.4 NA objective and CoolSnap HQ camera. For z-series, a step size of 0.3 μm was used. Image capture and postcapture processing were done using softWoRx software (Applied Precision), and Photoshop (Adobe) was used to make linear adjustments to brightness and contrast. Deconvolved images are shown.
For the nuclear segregation assay, the indicated strains were grown at 30 °C to early-log phase and then were shifted to 12 °C for 16 h. Cells were fixed with 70% ethanol and stained with DAPI at a final concentration of 100 ng/mL to visualize nuclei.
Proteomic Analysis.
W303, W303 Num1-yEGFP, W303 Mdm36-3xFLAG, and W303 Δmdm36 Num1-yEGFP strains were grown at 30 °C in YPD to an OD600 of 1.0. Cells were pelleted, washed in H2O, and resuspended in one pellet-volume of immunoprecipitation lysis buffer [IPLB: 20 mM Hepes KOH (pH 7.4), 150 mM KOAc, 2 mM Mg(Ac)2, 1 mM EGTA, and 0.6 M sorbitol] + 1× Protease Inhibitor Mixture I (PIC; Calbiochem). The cell–buffer suspension was added dropwise to liquid N2 to create yeast cell–buffer pellets, and the pellets were stored at −80 °C. The frozen cells were lysed using a Freezer/Mill (SPEX), and the resulting cell lysate powder was thawed in an H2O bath at room temperature. Then 1× PIC was added to the lysate. Large cellular debris was cleared with a low-speed spin, 500 × g for 10 min at 4 °C. Dithiobis(succinimidyl propionate), to a final concentration of 1 mM, was added to the supernatant, and the samples were placed on ice for 30 min. Tris (pH 7.5) was added to a final concentration of 100 mM to quench the cross-linking reaction, and the lysates were placed on ice for 15 min. To solubilize membranes, digitonin was added to a final concentration of 1%, and the samples were incubated on ice for 30 min. The lysates then were cleared with a 12,000 × g spin for 10 min at 4 °C. The resulting supernatants were incubated on ice for 30 min with 50 μL μMACS Anti-GFP MicroBeads (Miltenyi Biotec) or 3 μg anti-FLAG antibody (Sigma-Aldrich) and 50 μL μMACS protein G beads (Miltenyi Biotec). For each immunoprecipitation, cells at OD500 were used. The beads were isolated using Miltenyi μ columns and a μMACS separator (Miltenyi Biotec), washed three times with 800 μL IPLB + 0.1% digitonin + PIC, and washed twice with 500 μL IPLB. On-bead trypsin digestion was performed essentially as described previously (42). In brief, beads were incubated for 30 min at room temperature in 25 μL of elution buffer I [2 M urea, 50 mM Tris (pH 7.5), 1 mM DTT, and 5 μg/mL trypsin]. This incubation was followed by two 50-μL applications of elution buffer II [2 M urea, 50 mM Tris (pH 7.5), and 5 mM chloroacetamide]. Elutions were collected, and digestion was allowed to continue at room temperature overnight. Reactions were stopped with 1 μL trifluoroacetic acid. The peptides were submitted to the Genome Center Proteomics Core at the University of California, Davis, for LC-MS/MS–based protein identification. Sample processing and analysis were performed as described previously (27).
Supplementary Material
Acknowledgments
We thank Megan Wemmer and other members of the J.N. laboratory for critical discussions and comments on the manuscript. We thank Angelica Kowalchuk for help with strain construction, Dr. Christof Osman for technical advice on the synthetic lethality screen, and Dr. Thomas Langer for providing the reagents and equipment necessary to conduct the screen. All imaging was conducted in the University of California, Davis (UC Davis) Molecular and Cellular Biology Light Microscopy Imaging Facility with technical advice from Dr. Michael Paddy. All proteomic analyses were conducted by the UC Davis Proteomics Core Facility with technical advice and assistance from Dr. Brett Phinney, Diana Tran, and Darren Weber. A.M. is supported by National Institutes of Health (NIH) Training Grant 5T32GM007377-34. J.N. is supported by the NIH Grant R01GM097432.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 1986 (volume 110, number 6).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215232110/-/DCSupplemental.
References
- 1.Nunnari J, Suomalainen A. Mitochondria: In sickness and in health. Cell. 2012;148(6):1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- 3.Simon VR, Karmon SL, Pon LA. Mitochondrial inheritance: Cell cycle and actin cable dependence of polarized mitochondrial movements in Saccharomyces cerevisiae. Cell Motil Cytoskeleton. 1997;37(3):199–210. doi: 10.1002/(SICI)1097-0169(1997)37:3<199::AID-CM2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Yang HC, Palazzo A, Swayne TC, Pon LA. A retention mechanism for distribution of mitochondria during cell division in budding yeast. Curr Biol. 1999;9(19):1111–1114. doi: 10.1016/s0960-9822(99)80480-1. [DOI] [PubMed] [Google Scholar]
- 5.Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118(Pt 23):5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Cai Q, Sheng ZH. Mitochondrial transport and docking in axons. Exp Neurol. 2009;218(2):257–267. doi: 10.1016/j.expneurol.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csordás G, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174(7):915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boncompagni S, et al. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009;20(3):1058–1067. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perkins GA, et al. Electron tomographic analysis of cytoskeletal cross-bridges in the paranodal region of the node of Ranvier in peripheral nerves. J Struct Biol. 2008;161(3):469–480. doi: 10.1016/j.jsb.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins GA, et al. The micro-architecture of mitochondria at active zones: Electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J Neurosci. 2010;30(3):1015–1026. doi: 10.1523/JNEUROSCI.1517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kormanec J, Schaaff-Gerstenschläger I, Zimmermann FK, Perecko D, Küntzel H. Nuclear migration in Saccharomyces cerevisiae is controlled by the highly repetitive 313 kDa NUM1 protein. Mol Gen Genet. 1991;230(1-2):277–287. doi: 10.1007/BF00290678. [DOI] [PubMed] [Google Scholar]
- 13.Heil-Chapdelaine RA, Oberle JR, Cooper JA. The cortical protein Num1p is essential for dynein-dependent interactions of microtubules with the cortex. J Cell Biol. 2000;151(6):1337–1344. doi: 10.1083/jcb.151.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerveny KL, Studer SL, Jensen RE, Sesaki H. Yeast mitochondrial division and distribution require the cortical num1 protein. Dev Cell. 2007;12(3):363–375. doi: 10.1016/j.devcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Hermann GJ, et al. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143(2):359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farkasovsky M, Küntzel H. Yeast Num1p associates with the mother cell cortex during S/G2 phase and affects microtubular functions. J Cell Biol. 1995;131(4):1003–1014. doi: 10.1083/jcb.131.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang X, Germain BS, Lee WL. A novel patch assembly domain in Num1 mediates dynein anchoring at the cortex during spindle positioning. J Cell Biol. 2012;196(6):743–756. doi: 10.1083/jcb.201112017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang X, Punch JJ, Lee WL. A CAAX motif can compensate for the PH domain of Num1 for cortical dynein attachment. Cell Cycle. 2009;8(19):3182–3190. doi: 10.4161/cc.8.19.9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu JW, et al. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell. 2004;13(5):677–688. doi: 10.1016/s1097-2765(04)00083-8. [DOI] [PubMed] [Google Scholar]
- 20.Schauss AC, Bewersdorf J, Jakobs S. Fis1p and Caf4p, but not Mdv1p, determine the polar localization of Dnm1p clusters on the mitochondrial surface. J Cell Sci. 2006;119(Pt 15):3098–3106. doi: 10.1242/jcs.03026. [DOI] [PubMed] [Google Scholar]
- 21.Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170(2):237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammermeister M, Schödel K, Westermann B. Mdm36 is a mitochondrial fission-promoting protein in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21(14):2443–2452. doi: 10.1091/mbc.E10-02-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011;193(2):333–346. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124(3):573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 25.Itoh T, Toh-E A, Matsui Y. Mmr1p is a mitochondrial factor for Myo2p-dependent inheritance of mitochondria in the budding yeast. EMBO J. 2004;23(13):2520–2530. doi: 10.1038/sj.emboj.7600271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frederick RL, Okamoto K, Shaw JM. Multiple pathways influence mitochondrial inheritance in budding yeast. Genetics. 2008;178(2):825–837. doi: 10.1534/genetics.107.083055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoppins S, et al. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195(2):323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swayne TC, et al. Role for cER and Mmr1p in anchorage of mitochondria at sites of polarized surface growth in budding yeast. Curr Biol. 2011;21(23):1994–1999. doi: 10.1016/j.cub.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quintana A, et al. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci USA. 2007;104(36):14418–14423. doi: 10.1073/pnas.0703126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman JR, et al. ER tubules mark sites of mitochondrial division. Science. 2011;334(6054):358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toulmay A, Prinz WA. Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Curr Opin Cell Biol. 2011;23(4):458–463. doi: 10.1016/j.ceb.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kornmann B, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325(5939):477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naylor K, et al. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J Biol Chem. 2006;281(4):2177–2183. doi: 10.1074/jbc.M507943200. [DOI] [PubMed] [Google Scholar]
- 34.Westermann B, Neupert W. Mitochondria-targeted green fluorescent proteins: Convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 2000;16(15):1421–1427. doi: 10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 35.Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147(4):699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lackner LL, Horner JS, Nunnari J. Mechanistic analysis of a dynamin effector. Science. 2009;325(5942):874–877. doi: 10.1126/science.1176921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madrid AS, Mancuso J, Cande WZ, Weis K. The role of the integral membrane nucleoporins Ndc1p and Pom152p in nuclear pore complex assembly and function. J Cell Biol. 2006;173(3):361–371. doi: 10.1083/jcb.200506199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 39.Sheff MA, Thorn KS. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21(8):661–670. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- 40.Gerami-Nejad M, Dulmage K, Berman J. Additional cassettes for epitope and fluorescent fusion proteins in Candida albicans. Yeast. 2009;26(7):399–406. doi: 10.1002/yea.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mumberg D, Müller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae: Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22(25):5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubner NC, et al. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol. 2010;189(4):739–754. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
