

Abstract
Hominins with morphology similar to present-day humans appear in the fossil record across Eurasia between 40,000 and 50,000 y ago. The genetic relationships between these early modern humans and present-day human populations have not been established. We have extracted DNA from a 40,000-y-old anatomically modern human from Tianyuan Cave outside Beijing, China. Using a highly scalable hybridization enrichment strategy, we determined the DNA sequences of the mitochondrial genome, the entire nonrepetitive portion of chromosome 21 (∼30 Mbp), and over 3,000 polymorphic sites across the nuclear genome of this individual. The nuclear DNA sequences determined from this early modern human reveal that the Tianyuan individual derived from a population that was ancestral to many present-day Asians and Native Americans but postdated the divergence of Asians from Europeans. They also show that this individual carried proportions of DNA variants derived from archaic humans similar to present-day people in mainland Asia.
Keywords: ancient DNA, human evolution, nuclear capture strategy, paleogenetics
The term “early modern humans” generally refers to humans who fall within the morphological variation of present-day humans and date to the Middle or Early Upper Paleolithic. The earliest modern humans appear in the Eurasian fossil record about 45,000 y ago, whereas the last remains that tend to be classified as early modern humans are about 25,000 y old (1). Early modern humans may exhibit some archaic features shared with other earlier forms of humans such as Neandertals. Although early modern humans are thus only vaguely defined as a group, their genetic relationship to present-day humans is unclear. Similarly, their relationship to archaic humans is of interest, given that they may have interacted directly with them.
To begin to explore the genetic relationships of early modern humans with present-day humans, we have analyzed a partial human skeleton that was unearthed in 2003, along with abundant late Pleistocene faunal remains, in the Tianyuan Cave near the Zhoukoudian site in northern China, about 50 km southwest of Beijing. The skeleton was radiocarbon-dated to 34,430 ± 510 y before present (BP) (uncalibrated), which corresponds to ∼40,000 calendar years BP (2). A morphological analysis of the skeleton (3) confirms initial assessments (4) that this individual is a modern human, but suggests that it carries some archaic traits that could indicate gene flow from earlier hominin forms. The Tianyuan skeleton is thus one of a small number of early modern humans more than 30,000 y old discovered across Eurasia (2) and an even smaller number known from East Asia (5).
Results and Discussion
DNA Extraction.
To evaluate DNA preservation and the degree of modern human contamination in the Tianyuan skeleton, we prepared two DNA extracts from the left femur (TY1301) and two from the right tibia (TY1305) of the human skeleton excavated in Tianyuan Cave (4) using less than 100 mg of bone material per extraction (Table S1). Sequencing of random DNA fragments from DNA libraries constructed from these four extracts revealed that about 0.01–0.03% of the DNA in the libraries was of human origin (SI Text, section 1 and Table S1). This low percentage of endogenous DNA precludes sequencing of the entire genome of this individual. Thus, we used DNA capture approaches to retrieve the mitochondrial (mt)DNA (6) and nuclear DNA sequences from the Tianyuan individual.
mtDNA Capture and Sequencing.
We used a protocol for targeted DNA sequence retrieval that is particularly suited for mtDNA (6) to isolate mtDNA fragments. In total, we sequenced 4,423,607 unique DNA fragments from both ends on the Illumina GAII platform from the four libraries. Of these, 0.1–7.7% (Table S1) could be aligned to a human mtDNA reference sequence (7). All four libraries yielded consensus mtDNA sequences that agreed with each other. This is in agreement with the observation of paleontologists that the two bones come from a single individual (2). For estimation of mtDNA contamination and phylogenetic analyses, we used DNA fragments isolated from the femur library that had the highest mtDNA content (Table S1) and yielded an average coverage of 35.6-fold of the mtDNA genome.
DNA Sequence Authenticity.
When studying modern humans, it is particularly difficult to exclude that DNA from present-day humans might have contaminated the samples or experiments. It has previously been argued (8) that the combination of two observations makes it likely that a DNA library contains a majority of endogenous ancient human DNA: First, that the patterns of DNA degradation, in particular nucleotide misincorporations resulting from deamination of cytosine residues at ends of DNA fragments, indicate that the mtDNA is ancient; and second, that deep sequencing of the mitochondrial genome indicates that the human mtDNA in the library comes from a single individual. We identified 78 distinct DNA fragments that cover three positions where the Tianyuan mtDNA consensus differs from at least 308 of 311 mtDNA genomes from around the world (9). All these fragments carry the consensus base at these positions, indicating that the vast majority [95% confidence interval (CI): 95.3–100%] of the endogenous mtDNA fragments come from a single source (SI Text, section 2). Alignments of the individual mtDNA fragments against the consensus sequence constructed from all fragments reveal C→T and G→A substitution frequencies of between 25 and 30% close to the ends of the DNA fragments (Fig. S1C), an extent of substitution indicative of cytosine deamination not seen in present-day human contamination (8, 10). We conclude that the human DNA extracted from the Tianyuan skeleton comes from a single individual and is likely to be endogenous to the skeleton.
mtDNA Analyses.
We estimated a phylogenetic tree for 311 modern human mtDNAs, the Tianyuan mtDNA, and a complete Neandertal mtDNA (Fig. S2). The Tianyuan mtDNA falls within the variation of present-day human mtDNA. It carries all substitutions that have been used to define a group of related mtDNA sequences—“haplogroup R”—and in addition a deletion of a 9-bp motif (5′-CCCCCTCTA-3′, revised Cambridge reference sequence positions 8,281–8,289) as well as a substitution at position 16,189 (Fig. S3A), which together have been used to define a group of related mtDNA sequences, “haplogroup B” (11–13), within haplogroup R. In addition, it carries four substitutions (5,348, 5,836, 11,257, 16,293) that are not defining subgroups of haplogroup B (14–17). Thus, it is related to the mtDNA that was ancestral to present-day haplogroup B (Fig. 1), which has been estimated to be around 50,000 y old (18) (50. 7 ka BP; 95% CI: 38.1–68.3 ka BP). We note that the age of the Tianyuan individual is compatible with this date.
Fig. 1.
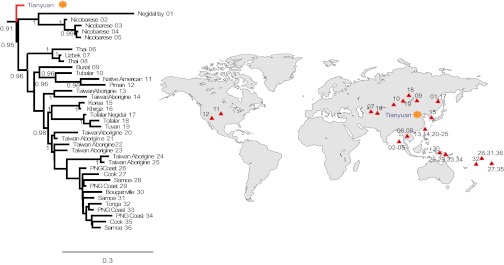
Tree of the Tianyuan and 36 present-day mtDNAs belonging to haplogroup B. The bar represents 0.3 substitutions per nucleotide site. Numbers indicate individuals in the tree and the map.
Today, mtDNA of haplogroup B occurs in Native Americans, populations of the Russian Far East, Central Asia, Korea, Taiwan, Melanesia, and Polynesia. It is thus widespread in Asia and America. The fact that an individual who lived in the Beijing area 40,000 y ago carried a mitochondrial genome that is potentially ancestral to mtDNAs in all these areas suggests that there is at least some population continuity from the earliest modern humans in East Asia to present-day populations in these areas. However, although genetic reconstructions of human population histories in Eurasia have relied heavily on mtDNA variation (19–21), the extent to which this accurately reflects older population histories in the region is unknown. We therefore proceeded to analyze nuclear DNA sequences of this individual who represents a member of an early population in Asia.
Chromosome 21 Capture and Sequencing.
The ability to sequence nuclear DNA sequences from the Tianyuan individual is limited by the fact that no more than 0.03% of the DNA extracted is endogenous to the bone. Previous studies (22–24) have shown that hybridization enrichment can be used to obtain nuclear DNA fragments from ancient samples. However, the commercially available hybridization systems used in these studies provide only a limited number of capture probes in each hybridization reaction. Because the enrichment of highly fragmented DNA requires large overlaps between probes, this limits the total size of genomic regions that can be targeted to a few megabases at best. To overcome this limitation, we modified a strategy previously described by Gnirke et al. (25) that uses oligonucleotides synthesized on arrays to construct probe libraries that are then amplified and converted into biotinylated RNA capture probes through in vitro transcription. Our approach differs in that first we use oligonucleotides from arrays with higher probe density (1 million probes per array); second, we exclude most of the linker sequence from oligonucleotide synthesis; third, we combine probe libraries from several arrays into a single “superprobe library”; and fourth, we use biotinylated DNA instead of RNA probes. This way, we produced a library consisting of 8.7 million different probes tiled at 3-bp intervals across the 29.8 Mbp of all nonrepetitive sequence in chromosome 21 (Fig. S4).
We produced additional DNA extracts and libraries from femur TY1301, this time using uracil DNA glycosylase and endonuclease VIII during library preparation to avoid nucleotide misincorporations induced by deaminated cytosine residues (26). From these libraries as well as the two initial libraries from the femur, we performed two successive enrichments for chromosome 21 fragments (Fig. S5). A total of 789,925 unique DNA fragments was identified that represents 1.75-fold coverage of the targeted regions. Separately, we also enriched for mtDNA by the same approach. This resulted in estimates of the average human mtDNA contamination in these libraries between 0.1% and 3.1% (SI Text, section 3.4). A comparison with shotgun sequencing results for two of the libraries used for capture showed that more than 70% of the total number of target DNA molecules present in the libraries was isolated by the capture procedure (SI Text, section 3.3). Thus, this procedure efficiently captures short molecules in very complex mixtures of DNA.
Chromosome 21 Analyses.
To investigate the relationship of the Tianyuan individual to present-day populations, we compared it to chromosome 21 sequences from 11 present-day humans from different parts of the world (a San, a Mbuti, a Yoruba, a Mandenka, and a Dinka from Africa, a French and a Sardinian from Europe, a Papuan, a Dai, and a Han from Asia, and a Karitiana from South America) and a Denisovan individual, each sequenced to 24- to 33-fold genomic coverage (27). Denisovans are an extinct group of Asian hominins related to Neandertals (28). In the combined dataset, 86,525 positions variable in at least one individual are of high quality in all 13 individuals. Table 1 shows that the Tianyuan individual differs by 21,944–23,756 substitutions from the Eurasian individuals, by 30,297–35,938 substitutions from the African individuals, and by 43,893 substitutions from the Denisovan individual. Thus, the Tianyuan individual was clearly more similar to present-day humans than to Denisovans, and more similar to present-day Eurasians than to present-day Africans.
Table 1.
Pairwise nucleotide differences among chromosome 21 sequences analyzed
| San | Mbuti | Yoruba | Mandenka | Dinka | French | Sardinian | Papuan | Karitiana | Han | Dai | Tianyuan | |
| Mbuti | 33,860 | |||||||||||
| Yoruba | 33,253 | 30,683 | ||||||||||
| Mandenka | 33,783 | 31,489 | 26,684 | |||||||||
| Dinka | 33,207 | 31,099 | 27,624 | 27,906 | ||||||||
| French | 34,076 | 31,720 | 27,559 | 28,063 | 27,135 | |||||||
| Sardinian | 34,138 | 31,936 | 27,901 | 27,331 | 27,245 | 18,326 | ||||||
| Papuan | 34,672 | 32,754 | 29,023 | 28,871 | 28,931 | 22,630 | 21,968 | |||||
| Karitiana | 34,622 | 32,644 | 28,917 | 29,077 | 28,187 | 20,336 | 20,470 | 22,210 | ||||
| Han | 33,984 | 32,054 | 28,633 | 27,981 | 27,923 | 20,484 | 21,278 | 21,606 | 18,768 | |||
| Dai | 34,173 | 32,209 | 28,092 | 28,332 | 27,704 | 20,211 | 21,683 | 21,297 | 20,131 | 18,525 | ||
| Tianyuan | 35,938 | 33,390 | 31,059 | 30,333 | 30,297 | 23,168 | 22,906 | 23,756 | 21,944 | 22,802 | 23,339 | |
| Denisovan | 47,535 | 47,169 | 45,876 | 45,850 | 46,208 | 45,633 | 45,909 | 43,935 | 45,765 | 45,507 | 45,160 | 43,893 |
To more accurately gauge how the population from which the Tianyuan individual is derived was related to Eurasian populations, while taking gene flow between populations into account, we used a recent approach (29) that estimates a maximum-likelihood tree of populations and then identifies relationships between populations that are a poor fit to the tree model and that may be due to gene flow. As suggested by the nucleotide differences (Table 1), the maximum-likelihood tree (Fig. S6A) shows that the branch leading to the Tianyuan individual is long, due to its lower sequence quality. However, among Eurasian populations, Tianyuan clearly falls with Asian rather than European populations (bootstrap support 100%). The strongest signal not compatible with a bifurcating tree (Fig. S6B) is an inferred gene-flow event that suggests that 6.7% of chromosome 21 in the Papuan individual is derived from Denisovans, in agreement with previous findings (28, 30). When this is taken into account, the Tianyuan individual appears ancestral to all Asian individuals studied (Fig. 2). We note, however, that the relationship of the Tianyuan and Papuan individuals is not resolved (bootstrap support 31%). Further work is necessary to clarify whether this reflects the age of the Tianyuan individual relative to the divergence between modern human populations.
Fig. 2.

Maximum-likelihood tree relating the chromosome 21 sequences of the Tianyuan individual, 11 present-day humans, and the Denisovan genome. The most strongly supported gene-flow event is shown in yellow. Bootstrap support for all internal edges is 100% except for the edge putting Tianyuan outside the four Asians, which is 31%. The scale bar shows 10 times the average standard error of the entries in the covariance matrix.
Archaic Admixture.
It has been shown that a population related to the Denisovan individual contributed genetic material to the ancestors of present-day Melanesians (28, 30), and this has also been suggested to be the case for some mainland Asian populations (31) (but see also ref. 27). It is therefore of interest to analyze whether the Tianyuan individual shows evidence of any Denisovan genetic contribution. For chromosome 21, we find that any putative admixture from Denisovans must be smaller than that in present-day Papuans and not larger than in other present-day mainland Asians analyzed (SI Text, section 3.7 and Fig. S6B). However, because archaic admixture may show systematic differences among chromosomes (27), we decided to analyze additional parts of the Tianyuan genome for traces of archaic admixture. To do this, we identified 1,666 single-nucleotide polymorphisms (SNPs) where both the Neandertal (32) and Denisovan (27) genomes differ from the genomes of individuals from seven African populations in the Foundation Jean Dausset-Centre d'Etude du Polymorphisme Humain (CEPH)-Human Genome Diversity Panel (HGDP-CEPH) (33), and 1,800 SNPs where the Denisovan genome differs from the Neandertal as well as the seven Africans (SI Text, section 3.7). We synthesized capture probes for these 3,466 sites and generated additional DNA libraries from the Tianyuan individual. After enrichment and sequencing, we identified 834 and 843 sites, respectively, for which data were available for individuals from the HGDP-CEPH and the Tianyuan individual. Fig. 3 shows that all non-African populations share more alleles with the two archaic individuals. Over and above the alleles shared with the Neandertal, Melanesians share additional alleles with the Denisovan, as previously described (27, 28, 30), whereas the Tianyuan individual falls within the range of present-day Eurasian mainland populations. This indicates that the Tianyuan individual is most similar to the latter populations in carrying a genomic component related to the Neandertal genome, but no Denisovan component is discernable with these analyses (see also Fig. S7).
Fig. 3.

Proportion of alleles shared with the Neandertal and Denisovan genomes for the Tianyuan and present-day individuals. The x axis shows the percent of alleles that match the Neandertal and Denisovan genomes at sites where both of these differ from seven Africans, and the y axis indicates the percent of alleles that match the Denisovan genome where this differs from the Neandertal as well as from the seven Africans.
Conclusion
The DNA hybridization capture strategy described here allows sequencing of large sections (>>1 Mbp) of the nuclear genome from mammalian samples even in the presence of a large excess of microbial DNA, a situation typical of almost all ancient samples outside permafrost regions. This opens the possibility of generating DNA sequences from previously inaccessible ancient samples. We use this capture strategy to analyze an early modern human, the Tianyuan individual, who contains less than 0.03% endogenous DNA.
The results show that early modern humans present in the Beijing area 40,000 y ago were related to the ancestors of many present-day Asians as well as Native Americans. However, they had already diverged from the ancestors of present-day Europeans.
That Europeans and East Asians had diverged by 40,000 y ago is consistent with dates for the first archaeological appearance of modern humans in Europe and also with the upper end of an estimate [23 ka BP (95% CI: 17–43 ka BP)] for the divergence of East Asian and European populations from nuclear DNA variation in present-day populations (34). The results also show that the Tianyuan individual did not carry any larger proportion of Neandertal or Denisovan DNA sequences in its genome than present-day people in the region. More analyses of additional early modern humans across Eurasia will further refine our understanding of when and how modern humans spread across Eurasia.
Materials and Methods
DNA Library Preparation.
The libraries used for mitochondrial capture and shotgun sequencing were produced from 20 μL of each extract as described (35) without enzymatic removal of deaminated cytosines (uracils) (Table S1). Both library adapters (P5 and P7) carried a 4-bp “clean-room key” sequence (5′-GTCT-3′) that is used exclusively for ancient DNA (36). In addition, sample-specific barcodes were introduced into both library adaptors during amplification (35), which was performed using AmpliTaq Gold DNA polymerase (Applied Biosystems).
For the chromosome 21 and SNP capture, additional extracts prepared from TY1301 were converted into sequencing libraries carrying the clean-room keys as described above. Miscoding DNA damage was removed during library preparation by treatment with uracil-DNA-glycosylase and endonuclease (EndoVIII) as described (26) (SI Text, section 3.1). Libraries were amplified with Herculase II Fusion DNA polymerase (Agilent) as described (37).
mtDNA Capture.
mtDNA was enriched from the libraries separately using sheared human mtDNA from a European individual as bait as described (6).The enriched libraries were amplified using the primers IS5 and IS6 (38). After purification with Qiagen’s MinElute PCR Purification Kit, library concentrations were determined on an Agilent 2100 Bioanalyzer DNA 1000 chip, pooled in equimolar amounts, and sequenced on a quarter of one lane of an Illumina Genome Analyzer IIx. The GenBank accession number of complete mtDNA genome is KC417443.
Nuclear DNA Capture.
Nonrepetitive regions of chromosome 21, as well as polymorphic positions across the genome selected to detect archaic human admixture, were captured using single-stranded biotinylated capture probes prepared from a commercial array (SI Text, section 3.2). Libraries were reamplified and captures were performed twice (SI Text, section3.1). The Sequence Read Archive accession number of chromosome 21 as well as polymorphic position sequences is ERP002037.
mtDNA Phylogenetic Reconstruction.
A Bayesian tree (Fig. S2) was estimated using MrBayes (39) with 50,000,000 Markov chain Monte Carlo iterations (5,000,000 burn-ins) using the Tianyuan consensus sequence, 311 human mtDNAs, and a Neandertal mtDNA (Vindija 33.25) (40). The general time-reversible sequence evolution model was applied with a fraction of invariable sites determined by the best-fit model approach of MODELTEST in conjunction with PAUP* (41). The haplogroup for each mtDNA was determined using Phylotree (Phylotree.org-mtDNA, build 15).
Chromosome 21 Sequence Determination.
The Unified Genotyper from the Genome Analysis Toolkit was used to produce genotype calls, and a variant call format file combining the information with 13 other individuals was produced as described (27). We required the difference in Phred-scaled likelihoods between the two most likely genotypes to be at least 50 (corresponding to an error rate of no more than 10−5). When this did not result in a genotype call, we considered the two most likely homozygous genotypes and called the most likely one if their difference in Phred score was at least 50. The sites that are variable in 13 individuals were converted to TreeMix (29) input (SI Text, section 3.5). To compute pairwise distances, we restricted the analysis to 86,525 sites where a haploid or diploid genotype was called in all individuals, and not more than one nonreference allele was called across all individuals. The distance between two individuals at a site was defined as the difference in the number of reference alleles (Table S2), and distances were summed over all sites (Table 1).
Supplementary Material
Acknowledgments
We thank Wu Xinzhi and Tong Haowen for their continual support, which made our work possible; Emily M. Leproust, Götz Frommer, and Leonardo Brizuela from Agilent Technologies for kindly providing special oligonucleotide arrays and technical advice; Martin Kircher and Birgit Nickel for invaluable technical help; Johannes Krause, Michael Lachmann, Daniel Lawson, Nick Patterson, Joseph Pickrell, David Reich, and Mark Stoneking for comments on the manuscript; and The Max Planck Society and its Presidential Innovation Fund, the Chinese Academy of Sciences Strategic Priority Research Program (Grant XDA05130202), and the Basic Research Data Projects (Grant 2007FY110200) of the Ministry of Science and Technology of China for financial support.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession no. KC417443) and Sequence Read Archive (accession no. ERP002037).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221359110/-/DCSupplemental.
References
- 1.Trinkaus E. European early modern humans and the fate of the Neandertals. Proc Natl Acad Sci USA. 2007;104(18):7367–7372. doi: 10.1073/pnas.0702214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shang H, Tong H, Zhang S, Chen F, Trinkaus E. An early modern human from Tianyuan Cave, Zhoukoudian, China. Proc Natl Acad Sci USA. 2007;104(16):6573–6578. doi: 10.1073/pnas.0702169104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shang H, Trinkaus E. The Early Modern Human from Tianyuan Cave. College Station, TX: Texas A&M Univ Press; 2010. [Google Scholar]
- 4.Tong HW, Shang H, Zhang SQ, Chen FY. A preliminary report on the newly found Tianyuan Cave, a Late Pleistocene human fossil site near Zhoukoudian. Chin Sci Bull. 2004;49(8):853–857. [Google Scholar]
- 5.Demeter F, et al. Anatomically modern human in Southeast Asia (Laos) by 46 ka. Proc Natl Acad Sci USA. 2012;109(36):14375–14380. doi: 10.1073/pnas.1208104109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One. 2010;5(11):e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrews RM, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23(2):147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 8.Krause J, et al. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol. 2010;20(3):231–236. doi: 10.1016/j.cub.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 9.Green RE, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134(3):416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One. 2012;7(3):e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melton T, et al. Polynesian genetic affinities with Southeast Asian populations as identified by mtDNA analysis. Am J Hum Genet. 1995;57(2):403–414. [PMC free article] [PubMed] [Google Scholar]
- 12.Redd AJ, et al. Evolutionary history of the COII/tRNALys intergenic 9 base pair deletion in human mitochondrial DNAs from the Pacific. Mol Biol Evol. 1995;12(4):604–615. doi: 10.1093/oxfordjournals.molbev.a040240. [DOI] [PubMed] [Google Scholar]
- 13.Hagelberg E, Cox M, Schiefenhövel W, Frame I. Past Human Migrations in East Asia: Matching Archaeology, Linguistics and Genetics. 2008. A genetic perspective on the origins and dispersal of the Austronesians. Routledge Studies in the Early History of Asia, eds Sanchez-Mazas A, Blench R, Ross MD, Peiros I, Lin M (Routledge, New York), pp 356–375. [Google Scholar]
- 14.Kong QP, et al. Updating the East Asian mtDNA phylogeny: A prerequisite for the identification of pathogenic mutations. Hum Mol Genet. 2006;15(13):2076–2086. doi: 10.1093/hmg/ddl130. [DOI] [PubMed] [Google Scholar]
- 15.Kivisild T, et al. The emerging limbs and twigs of the East Asian mtDNA tree. Mol Biol Evol. 2002;19(10):1737–1751. doi: 10.1093/oxfordjournals.molbev.a003996. [DOI] [PubMed] [Google Scholar]
- 16.Yao YG, Kong QP, Bandelt HJ, Kivisild T, Zhang YP. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet. 2002;70(3):635–651. doi: 10.1086/338999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, et al. Large scale mitochondrial sequencing in Mexican Americans suggests a reappraisal of Native American origins. BMC Evol Biol. 2011;11:293. doi: 10.1186/1471-2148-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soares P, et al. Correcting for purifying selection: An improved human mitochondrial molecular clock. Am J Hum Genet. 2009;84(6):740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoneking M, Delfin F. The human genetic history of East Asia: Weaving a complex tapestry. Curr Biol. 2010;20(4):R188–R193. doi: 10.1016/j.cub.2009.11.052. [DOI] [PubMed] [Google Scholar]
- 20.Soares P, et al. The archaeogenetics of Europe. Curr Biol. 2010;20(4):R174–R183. doi: 10.1016/j.cub.2009.11.054. [DOI] [PubMed] [Google Scholar]
- 21.O’Rourke DH, Raff JA. The human genetic history of the Americas: The final frontier. Curr Biol. 2010;20(4):R202–R207. doi: 10.1016/j.cub.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 22.Burbano HA, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328(5979):723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avila-Arcos MC, et al. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Sci Rep. 2011;1 doi: 10.1038/srep00074. Article 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burbano HA, et al. Analysis of human accelerated DNA regions using archaic hominin genomes. PLoS One. 2012;7(3):e32877. doi: 10.1371/journal.pone.0032877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27(2):182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Briggs AW, et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38(6):e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468(7327):1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89(4):516–528. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skoglund P, Jakobsson M. Archaic human ancestry in East Asia. Proc Natl Acad Sci USA. 2011;108(45):18301–18306. doi: 10.1073/pnas.1108181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patterson N, et al. Ancient admixture in human history. Genetics. 2012;192(3):1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5(10):e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40(1):e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104(37):14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dabney J, Meyer M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques. 2012;52(2):87–94. doi: 10.2144/000113809. [DOI] [PubMed] [Google Scholar]
- 38.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 40.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325(5938):318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 41.Posada D, Crandall KA. MODELTEST: Testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.