

Abstract
Many drugs and drug candidates are suboptimal because of short duration of action. For example, peptides and proteins often have serum half-lives of only minutes to hours. One solution to this problem involves conjugation to circulating carriers, such as PEG, that retard kidney filtration and hence increase plasma half-life of the attached drug. We recently reported an approach to half-life extension that uses sets of self-cleaving linkers to attach drugs to macromolecular carriers. The linkers undergo β-eliminative cleavage to release the native drug with predictable half-lives ranging from a few hours to over 1 y; however, half-life extension becomes limited by the renal elimination rate of the circulating carrier. An approach to overcoming this constraint is to use noncirculating, biodegradable s.c. implants as drug carriers that are stable throughout the duration of drug release. Here, we use β-eliminative linkers to both tether drugs to and cross-link PEG hydrogels, and demonstrate tunable drug release and hydrogel erosion rates over a very wide range. By using one β-eliminative linker to tether a drug to the hydrogel, and another β-eliminative linker with a longer half-life to control polymer degradation, the system can be coordinated to release the drug before the gel undergoes complete erosion. The practical utility is illustrated by a PEG hydrogel–exenatide conjugate that should allow once-a-month administration, and results indicate that the technology may serve as a generic platform for tunable ultralong half-life extension of potent therapeutics.
Keywords: click chemistry, regenerative medicine, tetra-PEG
Conjugation of drugs to macromolecular carriers is a proven strategy for improving pharmacokinetics. In one approach, the drug is covalently attached to a long-lived circulating macromolecule—such as PEG—through a linker that is slowly cleaved to release the native drug (1, 2). We have recently reported such conjugation linkers that self-cleave by a nonenzymatic β-elimination reaction in a highly predictable manner, and with half-lives of cleavage spanning from hours to over a year (3). In this approach, a macromolecular carrier is attached to a linker that is attached to a drug or prodrug via a carbamate group (1; Scheme 1); the β-carbon has an acidic carbon–hydrogen bond (C–H) and also contains an electron-withdrawing “modulator” (Mod) that controls the pKa of that C–H. Upon hydroxide ion-catalyzed proton removal to give 2, a rapid β-elimination occurs to cleave the linker-carbamate bond and release the free drug or prodrug and a substituted alkene 3. The rate of drug release is proportional to the acidity of the proton, and that is controlled by the chemical nature of the modulator; thus, the rate of drug release is controlled by the modulator. It was shown that both in vitro and in vivo cleavages were linearly correlated with electron-withdrawing effects of the modulators and, unlike ester bonds commonly used in releasable linkers (2), the β-elimination reaction was not catalyzed by general bases or serum enzymes. Although the PEG-Michael acceptor products are potentially reactive toward nucleophiles, their reactivities correlate with their rates of formation and are in general slow compared with renal elimination (3).
Scheme 1.
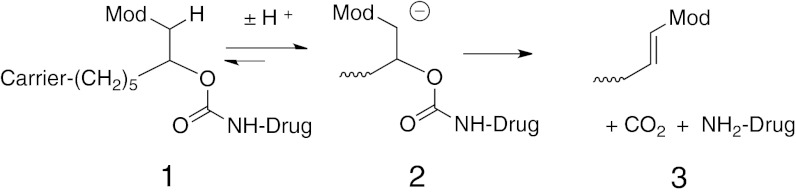
The in vivo elimination rate of a drug (kdrug) released from a circulating carrier is described by kdrug = k1 + k3, where k1 is the rate of linker cleavage and k3 is the rate of clearance of the conjugate (3). With very slowly cleaving linkers, the limitation to half-life extension is therefore not the cleavage rate (k1) of the linker, but rather the elimination rate (k3) of the circulating conjugate; for PEG conjugates, the elimination t1/2 is usually no longer than ∼7 d in humans (4, 5). To overcome this limitation, we used our linkers to conjugate the 39 amino acid peptide exenatide to a noncirculating, nondegradable polymeric hydrogel (3); here, k3 is negligible so the overall elimination rate of the drug, kdrug, approaches the rate of linker cleavage, k1. Indeed, when the hydrogel was implanted s.c. in rats, the half-life of the peptide in serum was increased 150-fold over that for a bolus injection. Although this demonstrated the utility of the β-eliminative linkers for in vivo s.c. implants, the gel used was nondegradable, and a biocompatible, biodegradable implant is essential for practical use.
Most biodegradable drug-delivery implants encapsulate a drug within gels of limiting pore size that retards diffusion, and release the drug concomitant with hydrolytic degradation—often of ester bonds—of the polymer (6, 7) (Fig. 1A). In contrast, for our drug-releasing linkers, we required a porous gel that readily allows diffusion, is substantially stable over the duration of drug release, and degrades subsequent to release of the drug (Fig. 1B). Although conventional ester-containing degradable gels could serve as a scaffold, ester linkages per se do not provide wide variation in cleavage rates, and it is difficult to achieve tunable, slow gel erosion rates without modifying gel network properties. It was our desire to develop a system in which we could keep gel properties largely constant, and achieve different degradation rates by modifying the rate of gel scaffold cleavage. In the present work, we tether drugs or drug surrogates to large-pore hydrogels prepared from two multi-arm PEG macromonomers using β-eliminative linkers that cleave at desired rates of drug release; similar β-eliminative linkers having slower cleavage rates are used to cross-link four arms of each macromonomer to form Tetra-PEG gels (8, 9). In this manner, either or both cleavage rates can be tuned to coordinate drug release and gel degradation without introducing major structural differences in the gels.
Fig. 1.

Drug release from polymeric gels. (A) Encapsulated drug released concomitant with gel degradation. (B) Release by linker cleavage of covalently tethered drug, followed by gel degradation.
Results
Design and Synthesis of Hydrogels.
PEG was chosen as a hydrogel scaffold because star polymers for step-growth polymerization are available, precise chemical modifications are facile, it is biocompatible, and is a Generally Recognized as Safe substance. The ∼7-nm (10 kDa PEG) distance between nodes of the gels used here provides a porous gel (mesh size >5 nm) that for our purposes would not significantly retard diffusion of proteins of up to at least 100 kDa (10, 11). The general approaches used for tethering drugs to hydrogels and for hydrogel syntheses are depicted in Scheme 2 and Fig. 2 and detailed in SI Text, Preparation of End-Group Modified Multivalent PEGs. 6-Azidohexylcarbamoyl-hydroxysuccinide or azido-linker succinimido carbonates (HSC) 4 possessing one of the modulators A–D were reacted with a four-arm amine-derivatized PEG20kDa to provide stable PEG20kDa(L-N3)4 and cleavable PEG20kDa(L-Mod-N3)4, 5. Likewise, 4, A–D, is reacted with an amine group of a drug to form carbamates 6, A–D. The azido-linker drug 6 is then reacted by click chemistry with four arms of an octa-armed cyclooctyne(CO)-derivatized PEG [dibenzocyclooctyne (DBCO) (12) or bicyclononyne (BCN) (13)] to give 7.
Scheme 2.

Fig. 2.

Idealized hydrogels formed from four-arm PEG azide and (A) four-arm PEG-CO (4 × 4 gel) or (B) eight-arm PEG-CO (4 × 8 gel) with four available cyclooctynes. Circles represent a drug or drug surrogate (D) tethered to the network by a releasable linker, or an erosion probe (EP) connected by a stable linker; L1 is a β-eliminative linker that releases the drug, and L2 is a β-eliminative cleavable cross-link of the gel.
A key reaction used in synthesis of the hydrogels described here is Cu-free azide-alkyne click chemistry, which has become a popular method for polymerizing hydrogels (14–18). In one format (Fig. 2A), “4 × 4” hydrogels were prepared by step-growth polymerization of 20-kDa four-armed azido-PEG 5 with a stoichiometrically balanced amount of 20-kDa four-arm CO-derivatized PEG [PEG20kDa(CO)4] to provide a gel with 10-kDa PEG chains between nodes. Nondegradable gels used the noncleavable 6-azidohexylcarbamoyl PEG [PEG20kDa(L-N3)4], and degradable gels contained β-eliminative carbamates to connect the azide and amino-PEG moieties [PEG20kDa(L2-Mod-N3)4]. To monitor gel dissolution, a trace amount of absorbing and/or fluorescent “erosion” probe was covalently attached by a stable linker to CO residues before hydrogel formation (19). Although we have not studied mechanical or structural properties of these gels, analogous Tetra-PEG gels have been intensively studied and shown to possess near-ideal homogeneous polymer network properties with minimal defects (8, 9, 20). In another format (Fig. 2B), a drug or drug surrogate was tethered to the eight-arm component of a 4 × 8 hydrogel by a releasable linker. Azides of the β-eliminative carbamoyl drug 6, a trace amount of erosion probe, and, when specified, an inert PEG cap were attached to an eight-arm CO-derivatized PEG [PEG40kDa(CO)8] to modify an average of four of the eight CO end-groups. Recognizing that, unlike the above 4 × 4 Tetra-PEG gels, the nonuniform modification of the number of CO end-groups and increased intramolecular coupling at low monomer concentrations may lead to gel imperfections (19–21), the remaining end-groups were polymerized with stoichiometric amounts of PEG20kDa(L2-Mod-N3)4 to give 4% wt/vol gels.
Diffusion of Molecules from Hydrogels.
We determined the rates of diffusion of noncovalently bound, encapsulated proteins (MW 17.7–66 kDa) from a nondegradable 4 × 4 PEG hydrogel having 10-kDa PEG chains between nodes (Fig. 2A). Gels were prepared by reaction of PEG20kDa(L-N3)4 with PEG20kDa(CO)4 in the presence of various proteins. Upon incubation in pH 7.4 buffer, 37 °C, the proteins diffused from the gel in the expected reverse-order of their molecular weights (SI Text, Protein Diffusion in Encapsulating Gels), with the largest protein (BSA, 66 kDa) diffusing much faster (t1/2 ∼2.5 h) than the anticipated cleavage of our β-eliminative linkers; thus, diffusion is not an obstacle to controlled release.
β-Eliminative Degradation of Hydrogels.
Next, we showed that hydrogels containing β-eliminative linkers within the gel network degraded in a predictable manner. PEG 4 × 4 gels were prepared that contained a trace of fluorescein erosion probe and various modulators controlling the cleavable cross-links [PEG20kDa(L2-Mod-N3)4]. Gels released PEG-fluorescein fragments slowly until the reverse gelation time (tRGEL), at which time the remainder of the gel rapidly and completely solubilized (Fig. 3A). As the erosion of a gel with Mod = PhSO2− progressed, samples of the soluble fraction were analyzed by size-exclusion chromatography (SEC). In accord with degradation studies of step-growth polymers (19), released gel fragments were primarily 20-kDa monomer components at early stages, but the proportion of dimers and larger fragments increased dramatically as the reverse-gelation point neared (SI Text, SEC-HPLC Analysis of Degrading Gel). In an idealized step-growth network, tRGEL should occur when an average of two cross-links remain attached to each node (19, 21). Thus, for the 4 × 4 gel erosions in Fig. 3A, tRGEL should occur when two of the four cross-links per node are cleaved, and correlate with the t1/2 of linker cleavage. As shown in Table 1, tRGEL varied over a range of ∼1 to ∼100 d depending on the modulator used, and were slightly lower than, but highly correlated (R2 = 0.990) with, the half-lives of β-eliminative cleavage of the same linkers in soluble PEG conjugates (3). As with cleavage rates of soluble PEG conjugates, tRGEL of a 4 × 4 gel containing the ClPhSO2− modulator was first-order with respect to hydroxide ion up to at least pH 9.0 (Fig. S1) with kOH = 5.1 × 104 M−1⋅h−1. These results show that hydrogels containing β-eliminative linkers in the cross-linking chains have predictable degradation rates that can be controlled over very long periods simply by varying the modulator used.
Fig. 3.
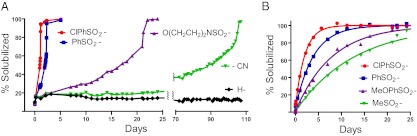
Degradation of and drug release from hydrogels. (A) Degradation of PEG hydrogels at pH 7.4, 37 °C, as measured by solubilized PEG-fluorescein fragments. Reverse-gelation times using different modulators are ClPhSO2− = 30 h, PhSO2− = 55 h, O(CH2CH2)2NSO2− = 22 d, CN = 105 d; the lines connecting the points are a visual guide in observing the trends of the data. (B) Release of a drug surrogate, aminoacetyl-fluorescein (AAF), using β-eliminative linkers with varying cleavage t1/2 values. The lines show the best fit of the data to a first-order rate equation, and SEs were <4.2% of the rate constants (SI Text, Drug Release from β-Eliminative Linkers); tRGEL was 630 ± 39 (SD) h (n = 8), and observed t1/2 values for total drug release using different modulators were ClPhSO2− = 33 h, PhSO2− = 65 h, MeOPhSO2− = 131 h, and MeSO2− = 212 h.
Table 1.
Summary of β-eliminative linker cleavage half-lives in PEG conjugates and hydrogels
Tunable Drug Release with Hydrogel Degradation.
To show that the technology provides predictable, tunable drug release, we studied 4 × 8 hydrogels that contained a surrogate drug tethered to hydrogels by β-eliminative linkers with varying cleavage rates, and a β-eliminative linker with a slower cleavage rate (Mod = MeSO2−) to cross-link the gel. An average of four of eight end-groups of PEG40kDa(DBCO)8 were tethered to fluorescein as a drug surrogate via β-eliminative linkers, a BODIPY erosion probe, and a small PEG cap; the approximately four remaining DBCO end groups were reacted with tetra-arm PEG20kDa(L2-MeSO2-N3)4 to give 4 × 8 gels depicted in Fig. 2B. Fig. 3B shows the fluorescein drug surrogate released into solution over time. Here, the solubilized fluorescein is the sum of that released from its tether by direct cleavage at L1 of the hydrogel, and that released as PEG-fluorescein fragments upon cleavages at L2 cross-links; the latter ultimately releases free fluorescein molecules in solution that have the same absorbance as the PEG-fluorescein fragments and cannot be distinguished. The rate of fluorescein release directly from the intact gel may be described by [1 – D(t)/D∞]/[1 – EP(t)/EP∞] = e−kL1t, where D(t)/D∞ and EP(t)/EP∞ are the fractions of drug and erosion probe solubilized at time t, respectively, and kL1 is the rate constant for drug release (Fig. S2; Eq. S6). Expectedly, the drug released directly from the gel is slower than the total drug released (Table 1), and becomes more difficult to estimate accurately as the cleavage rate at L1 approaches that for L2 (e.g., when both are MeSO2−). Rates of cleavage of the tethered fluorescein from the gel via L1 are slightly slower than but—as with gel erosion rates—highly correlated (R2 = 0.998) with those from corresponding soluble PEG–fluorescein conjugates (Table 1). Further, as with PEG conjugates, cleavage of fluorescein tethered to a 4 × 8 gel by a linker controlled by Mod = ClPhSO2− was first-order in hydroxide ion with kOH = 7.6 × 10−4 M−1⋅h−1 (Fig. S3). The tRGEL was 26 d, which is somewhat slower than the cleavage t1/2 of the corresponding PEG conjugate; this is likely due to a substantial fraction of free DBCO end-groups (i.e., >4) remaining on the randomly modified eight-arm PEG, which leads to higher cross-linking density and hence longer tRGEL for complete erosion (Table S1). These experiments clearly show that gels can be designed whereby a tethered drug is completely released at a predictable rate, followed by gel degradation.
Hydrogel–Exenatide Conjugate.
The potential utility of the hydrogel system for ultraslow release of a drug was demonstrated with exenatide, a 39-amino acid peptide that is a potent glucagon-like peptide-1 (GLP-1) agonist (22). Because exenatide has a t1/2 of ∼2.5 h in humans and requires twice-daily injections, there is considerable interest in developing long-acting GLP-1 agonists (23); currently, several such agonists—one marketed and several in clinical trials—require once-a-week administration. We attached a β-eliminative Nα-azido–linker exenatide (6, Mod = MeSO2−) (3) to ∼25% of the eight CO groups of PEG40kDa(BCN)8 (13) along with a trace BODIPY erosion probe; the remaining ∼6 BCN end-groups were reacted with stoichiometric amounts of PEG20kDa(L2-CN-N3). To decrease the time required to measure drug release and gel erosion, the kinetic study was conducted at pH 8.8, 37 °C (SI Text, Exenatide–PEG Hydrogel). Because the β-eliminative cleavages are first-order in hydroxide ion (above and ref. 3), we could estimate that exenatide is released from the gel with a t1/2 of 22 d—similar to the cleavage t1/2 of the same linker in a soluble PEG conjugate (Table 1)—and that the gel erodes with a tRGEL of 180 d at pH 7.4, 37 °C (Fig. 4). The longer tRGEL compared with that of the 4 × 4 gel in Fig. 3A (105 d) is ascribed to this gel having a higher cross-linking density of ∼4.7 vs. four cross-links per node, which can significantly increase tRGEL (Table S1). We also estimated (Eqs. S6 and S7) that ∼88% of the solubilized exenatide was directly released from the gel with a t1/2 of 25 d, and ∼12% was released as PEG–exenatide fragments from gel erosion—the activity of PEG–exenatide fragments should be minimal because the attachment on exenatide abolishes its binding to the GLP-1 receptor (3). If desired, the rate of erosion—and hence the fraction of released PEG–exenatide fragments—could be further decreased by simply using a β-eliminative linker in the gel cross-links with a slower cleavage rate (Fig. 3A). Studies with 2 mg of exenatide (Bydureon) once weekly have shown that the therapeutic steady-state concentration of exenatide in humans is ∼70 pM (24). To maintain exenatide concentrations over 70 pM for 1 mo in humans with a hydrogel–exenatide conjugate having a drug release t1/2 of ∼21 d, it would be necessary to attain a maximum serum concentration of ∼0.18 nM exenatide. Using an exenatide volume of distribution at steady state of ∼0.4 L/kg (25), we estimate this state could be achieved by an average of 10.4 μg·kg−1 exenatide/wk, which, assuming complete bioavailability, could be realized by a monthly s.c. hydrogel containing ∼3.2 mg exenatide, which compares favorably with 2-mg once-weekly exenatide. Thus, if the in vivo cleavage rates are similar to the in vitro rates, and if the hydrogel-bound exenatide survives the inflammatory process, an optimized form of this hydrogel should provide an exenatide delivery system that would require only once-a-month s.c. administration.
Fig. 4.

Exenatide release from and erosion of hydrogel. Data obtained at pH 8.8 [k = 0.75 ± 0.057 (SE) d−1] was estimated at pH 7.4 using tpH7.4 = tpH8.8 × 10pH8.8–7.4 based on observations that β-eliminative cleavages are first-order in hydroxide ion for drug release (3) and gel degradation (Fig. S1); (–●–) Total exenatide released from gel (free and PEG fragments); (–■–) gel erosion. The line for exenatide release is the best exponential fit of the data, and the lines connecting the gel degradation points are a visual guide in observing the trends of the data.
Discussion
We recently reported an approach to half-life extension of therapeutics that uses a set of self-cleaving linkers to attach drugs to circulating macromolecular carriers (3). The linkers undergo β-eliminative cleavage controlled by an electron-withdrawing modulator to release the native drug with half-lives adjustable from a few hours to over 1 y. These linkers were not susceptible to catalysis by serum enzymes, and showed an excellent correlation between in vitro and in vivo cleavage rates. However, with slowly cleaving linkers, the half-life of the released drug is limited by the elimination of the large conjugate, which is usually no longer than ∼1 wk in humans. We therefore sought to use our β-eliminative linkers to tether drugs to noncirculating s.c. implants, where the elimination half-life of a released molecule directly reflects the half-life of the linker used. However, implementation of this approach in vivo also required a carrier that had tunable, long degradation rates, so that the depleted carrier degrades to small, nontoxic components after the drug is released.
In the present work, we have used these β-eliminative linkers to tether drugs or drug surrogates to large-pore PEG hydrogels—analogous to the near-ideal Tetra-PEG gels (8, 9, 20)—and have shown they are released over predictable periods. Using similar β-elimination linkers as polymer cross-links, the hydrogels were also tuned to degrade at predictable times over a wide range of ∼1–100 d. As shown with Tetra-PEG gels containing ester cross-links, replacing a fraction of the cleavable linkers by stable ones should incrementally decrease the degradation rate of each gel up to at least threefold (26); in this manner, using the reported β-eliminative linkers (3), one could produce hydrogels with finely tunable degradation rates spanning ∼1 d to well over a year. Thus, by using one β-eliminative linker to tether a drug to the hydrogel, and another with a longer half-life to control polymer degradation, the cleavage rates can be optimally coordinated to release the drug before the gel undergoes complete degradation.
Cleavage rates of β-eliminative linkers attaching drugs to a gel or cross-linking the gel are highly correlated with their cleavage rates in soluble PEG conjugates, which in turn are tightly correlated with the electron-withdrawing ability of the modulator controlling linker cleavage. As for PEG conjugates, β-eliminative cleavage rates of the linkers are first-order in hydroxide ion. Hence, by raising the pH, rates of reactions that are very slow can be studied over reasonable periods of time; by lowering the pH, the gels or gel components can be stabilized for long periods required for storage. As described, the technology should be applicable to almost any drug—small molecule, peptide, or protein—and any gel sufficiently porous to allow unimpaired drug diffusion— dextran, hyaluronic acid, and others.
As an illustration of the potential practical utility of this technology platform, we prepared a PEG hydrogel–exenatide conjugate and showed that the peptide was slowly released with an estimated half-life of ∼3 wk at pH 7.4; subsequently, the gel degraded with complete dissolution occurring at ∼6 mo. If these in vitro cleavage rates translate in vivo, an optimized form of this hydrogel conjugate should provide an exenatide delivery system that would require only once-a-month s.c. administration, and there is no reason to expect this would not also apply to other potent drugs.
The delivery system described here overcomes certain limitations of conventional drug-encapsulating implants. First, covalently cross-linked encapsulating gels depend on limiting pore size for drug retention and gel degradation—usually by hydrolysis of ester linkages within the cross-links—for drug release. Because hydrolysis rates of esters commonly used in polymers are not easily tuned over a wide range, gel network chemistry and structure play the larger role in gel degradation rates, and individually optimized polymers are required for any given drug delivery system (19, 21). In the present work, we desired to avoid changes in polymer network properties that might affect cleavage rates of our β-eliminative linkers. Here, drug release and gel degradation are governed by the small modulators dictating linker cleavage rates, and gel network structure and chemistry are kept relatively constant; these systems do not require individually optimized polymer structures for each drug, and do not require that drug release be dependent on gel degradation rates. Drug-encapsulating systems can also show initial “burst release” or terminal “drug-dumping” effects at the beginning and end of gel degradation. Here, because the drug is covalently fixed to the hydrogel from the outset, such effects should not occur.
Second, drug encapsulation requires that gel polymerization be performed in the presence of the drug, usually concomitant with formation of injectable micro- or nanoparticles. The process often requires the use of organic solvents incompatible with some drugs—e.g., peptides and proteins—and commonly uses radical-induced polymerization that can lead to irreversible attachment of the drug to the polymer (27). In the present work, the tethering of molecules and gel polymerization reactions both use Cu-free [3 + 2] dipolar cycloaddition chemistry that is orthogonal to biological functional groups and can be performed in aqueous media; this expands the reach of the technology to molecules sensitive to harsh conditions (e.g., peptides and proteins) and should also avoid undesirable irreversible attachment of the drug to the gel (28). Other bioorthogonal coupling reactions, such as Michael addition of thiols (29) or tetrazine cycloadditions with dienophiles (30), should provide similar benefits. Also, because drugs are tethered to components of the hydrogel before cross-linking, it should be possible, but not necessary, to perform the polymerization in situ by mixing of filter-sterilized components shortly before injection (30).
Last, ester cross-linked polymers commonly used in encapsulating gels, such as those derived from glycolic or lactic acid (6), produce acid upon hydrolytic degradation that can cause undesirable modifications of peptidic drugs (31). With the present system, the carbamate bonds cleaved upon gel degradation do not produce acid and thus avoid such problems.
There are several relevant extensions of the technology platform. For example, more than one drug could be tethered to a hydrogel by one or more individually tuned linkers to provide a delivery system that releases the drug combination at the same or different rates. Also, absent the tethered drug, smaller mesh-size hydrogels cross-linked by β-eliminative linkers could be used for encapsulation of larger proteins and erosion of the polymer tuned to the desired rate of drug release. Moreover, in addition to s.c. drug delivery, the tunable hydrogels should find utility in other applications, such as regenerative medicine, orthopedic implants, single-shot vaccines, and others.
Materials and Methods
The source of specialized materials is provided, along with their use, in SI Text. Detailed synthetic procedures for hydrogel formation are described. In vitro kinetic procedures for drug release and hydrogel degradation methodologies are provided. Derivation of equations used to estimate the rate and extent of release of molecules directly from gel are presented in SI Text, Derivation and Use of Equations.
Supplementary Material
Acknowledgments
We thank Joe Dougherty, Linda Pullan, and Ron Zuckerman for their comments on this manuscript, and NOF America Corp. for generous gifts of modified polyethylene glycols.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215498110/-/DCSupplemental.
References
- 1.Greenwald RB, et al. Controlled release of proteins from their poly(ethylene glycol) conjugates: Drug delivery systems employing 1,6-elimination. Bioconjug Chem. 2003;14(2):395–403. doi: 10.1021/bc025652m. [DOI] [PubMed] [Google Scholar]
- 2.Filpula D, Zhao H. Releasable PEGylation of proteins with customized linkers. Adv Drug Deliv Rev. 2008;60(1):29–49. doi: 10.1016/j.addr.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Santi DV, Schneider EL, Reid R, Robinson L, Ashley GW. Predictable and tunable half-life extension of therapeutic agents by controlled chemical release from macromolecular conjugates. Proc Natl Acad Sci USA. 2012;109(16):6211–6216. doi: 10.1073/pnas.1117147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alconcel SNS, Baas AS, Maynard HD. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym Chem. 2011;2(7):1442–1448. [Google Scholar]
- 5.Fishburn CS. The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97(10):4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 6.Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—a review. Int J Pharm. 2011;415(1-2):34–52. doi: 10.1016/j.ijpharm.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 7.Censi R, Di Martino P, Vermonden T, Hennink WE. Hydrogels for protein delivery in tissue engineering. J Control Release. 2012;161(2):680–692. doi: 10.1016/j.jconrel.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Sakai T, et al. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules. 2008;41(14):5379–5384. [Google Scholar]
- 9.Matsunaga T, Sakai T, Akagi Y, Chung U, Shibayama M. SANS and SLS studies on tetra-arm PEG gels in as-prepared and swollen states. Macromolecules. 2009;42(4):6245–6252. [Google Scholar]
- 10.Mason MN, Metters AT, Bowman CN, Anseth KS. Predicting controlled-release behavior of degradable PLA-b-PEG-b-PLA hydrogels. Macromolecules. 2001;34(13):4630–4635. [Google Scholar]
- 11. Flory PJ (1953) Principles of Polymer Chemistry (Cornell Univ Press, Ithaca, NY)
- 12.Debets MF, et al. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem Commun (Camb) 2010;46(1):97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 13.Dommerholt J, et al. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew Chem Int Ed Engl. 2010;49(49):9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Dijk M, Rijkers DT, Liskamp RM, van Nostrum CF, Hennink WE. Synthesis and applications of biomedical and pharmaceutical polymers via click chemistry methodologies. Bioconjug Chem. 2009;20(11):2001–2016. doi: 10.1021/bc900087a. [DOI] [PubMed] [Google Scholar]
- 15.Deforest CA, Sims EA, Anseth KS. Peptide-functionalized click hydrogels with independently tunable mechanics and chemical functionality for 3D cell culture. Chem Mater. 2010;22(16):4783–4790. doi: 10.1021/cm101391y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson JA, Baskin JM, Bertozzi CR, Koberstein JT, Turro NJ. Copper-free click chemistry for the in situ crosslinking of photodegradable star polymers. Chem Commun (Camb) 2008;(26):3064–3066. doi: 10.1039/b803043j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Filion TM, Prifti F, Song J. Cytocompatible poly(ethylene glycol)-co-polycarbonate hydrogels cross-linked by copper-free, strain-promoted click chemistry. Chem Asian J. 2011;6(10):2730–2737. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nimmo CM, Shoichet MS. Regenerative biomaterials that “click”: Simple, aqueous-based protocols for hydrogel synthesis, surface immobilization, and 3D patterning. Bioconjug Chem. 2011;22(11):2199–2209. doi: 10.1021/bc200281k. [DOI] [PubMed] [Google Scholar]
- 19.DuBose JW, Cutshall C, Metters AT. Controlled release of tethered molecules via engineered hydrogel degradation: Model development and validation. J Biomed Mater Res A. 2005;74(1):104–116. doi: 10.1002/jbm.a.30307. [DOI] [PubMed] [Google Scholar]
- 20.Akagi Y, Matsunaga T, Shibayama M, Chung U, Sakai T. Evaluation of topological defects in tetra-PEG gels. Macromolecules. 2010;43(1):488–493. [Google Scholar]
- 21.Metters A, Hubbell J. Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules. 2005;6(1):290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 22.Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5(5):262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- 23.Madsbad S, et al. An overview of once-weekly glucagon-like peptide-1 receptor agonists—available efficacy and safety data and perspectives for the future. Diabetes Obes Metab. 2011;13(5):394–407. doi: 10.1111/j.1463-1326.2011.01357.x. [DOI] [PubMed] [Google Scholar]
- 24.Drucker DJ, et al. DURATION-1 Study Group Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet. 2008;372(9645):1240–1250. doi: 10.1016/S0140-6736(08)61206-4. [DOI] [PubMed] [Google Scholar]
- 25.Iwamoto K, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of exenatide once weekly in Japanese patients with type 2 diabetes. Endocr J. 2009;56(8):951–962. doi: 10.1507/endocrj.k09e-147. [DOI] [PubMed] [Google Scholar]
- 26.Li X, et al. Precise control and prediction of hydrogel degradation behavior. Macromolecules. 2011;44:3567–3571. [Google Scholar]
- 27.Lin CC, Sawicki SM, Metters AT. Free-radical-mediated protein inactivation and recovery during protein photoencapsulation. Biomacromolecules. 2008;9(1):75–83. doi: 10.1021/bm700782c. [DOI] [PubMed] [Google Scholar]
- 28.Lin CC, Metters AT. Hydrogels in controlled release formulations: network design and mathematical modeling. Adv Drug Deliv Rev. 2006;58(12-13):1379–1408. doi: 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Mather B, Viswanathan K, Miller K, Long T. Michael addition reactions in macromolecular design for emerging technologies. Prog Polym Sci. 2006;31(5):487–531. [Google Scholar]
- 30.Devaraj NJ. Advancing tetrazine bioorthogonal reactions through the development of new synthetic tools. Synlett. 2012;23(15):2147–2152. [Google Scholar]
- 31.Houchin ML, Topp EM. Chemical degradation of peptides and proteins in PLGA: A review of reactions and mechanisms. J Pharm Sci. 2008;97(7):2395–2404. doi: 10.1002/jps.21176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.