

Significance
Misincorporation of dUTP into DNA is detrimental to eukaryotes, prokaryotes, and viruses. This study reveals the fate of uracilated HIV-1 DNA in human cells. Early stages of the viral life cycle are unaffected, but integration of uracilated viral DNA into the host genome is prevented. This effect is wholly dependent on the presence of UNG2, a nuclear enzyme that excises uracil from DNA. This study establishes that UNG2 is a restriction factor for uracilated HIV-1 DNA and explains why this pathway is not fully engaged in CD4+ T cells and macrophages.
Keywords: base excision repair, raltitrexed
Abstract
HIV-1 reverse transcriptase discriminates poorly between dUTP and dTTP, and accordingly, viral DNA products become heavily uracilated when viruses infect host cells that contain high ratios of dUTP:dTTP. Uracilation of invading retroviral DNA is thought to be an innate immunity barrier to retroviral infection, but the mechanistic features of this immune pathway and the cellular fate of uracilated retroviral DNA products is not known. Here we developed a model system in which the cellular dUTP:dTTP ratio can be pharmacologically increased to favor dUTP incorporation, allowing dissection of this innate immunity pathway. When the virus-infected cells contained elevated dUTP levels, reverse transcription was found to proceed unperturbed, but integration and viral protein expression were largely blocked. Furthermore, successfully integrated proviruses lacked detectable uracil, suggesting that only nonuracilated viral DNA products were integration competent. Integration of the uracilated proviruses was restored using an isogenic cell line that had no detectable human uracil DNA glycosylase (hUNG2) activity, establishing that hUNG2 is a host restriction factor in cells that contain high dUTP. Biochemical studies in primary cells established that this immune pathway is not operative in CD4+ T cells, because these cells have high dUTPase activity (low dUTP), and only modest levels of hUNG activity. Although monocyte-derived macrophages have high dUTP levels, these cells have low hUNG activity, which may diminish the effectiveness of this restriction pathway. These findings establish the essential elements of this pathway and reconcile diverse observations in the literature.
Because many DNA polymerases cannot discriminate between dUTP and dTTP (1), uracil can be directly incorporated opposite to dA during DNA replication (2, 3). Although the resulting U/A pairs are not inherently mutagenic, stringent control on uracil incorporation is required because high levels of uracil in DNA can lead to genomic instability (4–7). The primary barrier to dUTP incorporation is the enzyme dUTPase, which hydrolyzes dUTP to dUMP and pyrophosphate (8). Subsequent reductive methylation of dUMP by thymidylate synthase (TS) generates dTMP, which is then phosphorylated to produce dTTP. Thus, dUTPase serves two key roles: ensuring a low dUTP/dTTP ratio and providing dUMP for the de novo pathway for dTTP synthesis. When uracil does enter DNA, it must then be removed via uracil base excision repair (UBER). This process is initiated by the enzyme uracil DNA glycosylase (UNG), which cleaves the N-glycosidic bond of dUrd, generating the free uracil base and an abasic site in DNA. Downstream BER enzymes then excise the abasic nucleoside and fill in the gap with the correct nucleotide (dTTP). Not surprisingly, dUTPase and UNG are coordinately regulated throughout the cell cycle of dividing cells, with the highest expression levels during the G1 and S phases (9, 10). However, in quiescent cells such as macrophages and neurons, dUTP can accumulate to high levels, and mRNA expression of both enzymes is barely detectable (11–13).
A long-standing question is whether quiescent immune cells use their intrinsically high dUTP levels as an innate immunity defense mechanism against infection by pathogenic viruses (1, 14, 15). Phylogenetic evidence for a dUTP-mediated host defense pathway is obtained from inspection of the genomes of β-retroviruses, nonprimate lentiviruses, and endogenous retroviruses, each of which has independently captured a host dUTPase gene during viral evolution (16, 17). Given the parsimonious genomes of these viruses, these findings would strongly suggest that this enzymatic activity provides a survival advantage during infection of at least some cell types (18–21). Biochemical evidence for this innate immunity pathway is suggested by the observation that dUTPase-deficient mutants of nonprimate lentiviruses cannot infect nondividing cells (where host cell dUTPase expression is low and dUTP levels are high) but are fully competent for infection of dividing cells (18–21). Interestingly, HIV-1 does not encode a dUTPase yet is still able to infect human macrophages (albeit with reduced efficiency), even though the dUTP:dTTP ratio in these cells is as high as 60:1 (12).
What effects could uracilation of HIV-1 DNA have on viral infection? Experiments performed in vitro with purified RT show that incorporation of dUTP can lead to inappropriate RNaseH cleavage and also disrupt the site of plus strand initiation (22). In turn, this may also compromise the terminal attachment site of the virus and inhibit integration (22). The presence of uracil in the viral cDNA may also induce structural changes that prevent correct interaction with proteins like integrase (23–25), and if uracil persists in the long terminal repeat (LTR) promoter of the integrated provirus, it may affect gene expression from the viral promoter (26, 27). Finally, if the viral cDNA is uracilated to a sufficiently high density (12, 25), it seems inevitable that the host nuclear UNG would excise the uracils, leading to its degradation once it entered the nucleus. There is little or no experimental data available to establish the importance of any of the above possible outcomes in human cells.
To systematically investigate how dUTP affects the viral life cycle, we developed a model system in which the intracellular dUTP:dTTP ratio could be increased incrementally using the TS-specific inhibitor raltitrexed (RTX). As inferred from HIV-1 infections in nondividing cells (25), we found that HIV-1 readily incorporates dUTP during reverse transcription, and dUTP incorporation only produced a small kinetic delay in cDNA synthesis with no effect on the ultimate levels of viral cDNA. However, dUTP incorporation resulted in a sevenfold reduction in viral integration, and this reduction was completely dependent on the removal of uracils by nuclear hUNG2, leading to degradation of the viral genome. A UNG-independent, postintegration effect of dUTP incorporation was also observed, suggesting that the effects also extend to transcription or translation of viral gene products. Biochemical measurements in the model cell line and in primary immune cells establish that this innate immunity pathway against viral integration requires low dUTPase levels, high levels of dUTP incorporation, and abundant levels of nuclear hUNG2. When any of these elements are absent or diminished, the pathway is debilitated. Knowledge of the essential elements of this pathway suggests targets for pharmacological intervention.
Results
RTX Increases the dUTP:dTTP Ratio in HT29 Cells.
Our goal of understanding the mechanistic basis for the effects of dUTP levels on viral infection required the development of an approach and model system where the dUTP:dTTP ratio could be incrementally increased in a predictable manner. With this goal in mind, the HT29 cell line was selected because it is highly responsive to the TS-specific inhibitor raltitrexed (RTX) (28), a sensitivity that has been attributed to its reduced expression of dUTPase (29). Indeed, in the absence of RTX, the dUTP/dTTP ratio was found to be indistinguishable from zero in HT29 cell extracts as measured using a single-nucleotide extension assay (Fig. 1A; Materials and Methods). In contrast, incubation of HT29 cells with 15–150 nM RTX for only 6 h followed by washout increased the dUTP:dTTP ratio to between 1 and 4 (Fig. 1A). To determine how RTX affected the dUTP:dTTP ratio over time, HT29 cells were pulse treated with 50 nM RTX for 6 h, and then the dUTP:dTTP ratio was determined at various time points after drug washout (see dNTP analysis in Scheme 1). At 4 h after washout, the dUTP:dTTP peaked at about 7 and then declined sharply over the next 24 h (Fig. 1B). This peak ratio is within the range reported for macrophages (11, 12) and is therefore relevant for making meaningful comparisons.
Fig. 1.

RTX increases the dUTP:dTTP ratio in HT29 cells. (A) HT29 cells were pulse treated with RTX for 6 h. dUTP and dTTP levels were determined using an SNE assay (n = 3). (B) Pulse treatment with RTX has a time-dependent increase in the dUTP:dTTP ratio. After pulse treatment with 50 nM RTX, HT29 cells were incubated for 0, 4, 10, 28, or 52 h after a washout with PBS. dUTP and dTTP levels were determined at each time point, and the dUTP:dTTP ratio was calculated (n = 3; Materials and Methods).
Scheme 1.
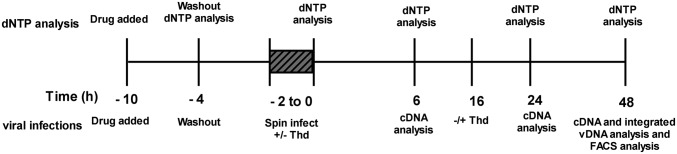
To demonstrate that pulsing with RTX specifically perturbed dUTP and dTTP levels without significantly disrupting other aspects of cell metabolism, we also measured its effect on nucleotide pool levels, cell viability, protein expression, and cell cycle progression. With regard to nucleotide pools, dGTP levels were found to be slightly reduced, as has been seen with other antifolates (30), but the remaining nucleotide levels were unaltered by RTX treatment (Table S1). The short pulse treatment was found to be slightly cytotoxic to HT29 cells, with cell viability of 90% at 24 h and 65% at 48 h as assayed using the neutral red reagent (Fig. S1A). Pulse treatment with RTX had no effect on α-tubulin levels (Fig. S1B), indicating no global perturbation to protein expression (this is further supported by experiments presented below). The short drug exposure produced a small decrease in the G2 population immediately after treatment, and the cell population progressed more slowly through the cell cycle over 48 h after treatment. At 150 nM RTX, the highest concentration used in these studies, the cell population remained arrested in G1 at 48 h, whereas at all lower RTX concentrations, the cells had reentered the cell cycle (Fig. S1C). Extensive experiments presented below unambiguously assign the inhibitory effects of RTX on HIV-1 integration to its perturbation of the dUTP pool, subsequent incorporation of dUTP into viral DNA, and excision of viral uracil by nuclear UNG2. Any other effects of RTX on cell metabolism do not impact viral integration efficiency (vide infra).
High dUTP Levels Induce a Delay in Reverse Transcription and HIV-1 cDNA Becomes Highly Uracilated.
Once the conditions and time course for generating high levels of cellular dUTP were determined, we could address the questions of whether RT incorporates dUTP into cDNA or whether high dUTP levels act as a barrier to reverse transcription. HT29 cells were pulse-treated with several concentrations of RTX and then infected with pseudotyped virus (see viral infections in Scheme 1). At 6 h after infection, a three- to fivefold reduction in late reverse transcripts (cDNAs) was observed in cells treated with 15–150 nM RTX (determined by real-time PCR using late reverse transcript-specific primers; Fig. 2A, white bars). Pretreatment of the isolated viral DNA products with human uracil DNA glycosylase (hUNG)/apurinic/apyrimidinic nuclease 1 (APE1) to degrade any uracilated DNA that may be present did not reduce the real-time PCR amplification products, indicating that the late products appearing at 6 h after infection do not contain detectable uracil (Fig. 2A, black bars). Similar reductions in viral DNA products at 6 h after infection were observed when primer sets specific for early and intermediate stages of reverse transcription were used (Fig. S2). These results indicate that dUTP has a very early effect during reverse transcription that serves to reduce transcript levels but that uracil does not appear in viral DNA products at 6 h after infection. In significant contrast, at 24 h after infection, the levels of late viral DNA products in RTX-treated cells return to the control levels, indicating that the reduction seen at 6 h reflects a delay in reverse transcription and not aborted transcription (Fig. 2B, white bars). Although uracil was not detectable in the viral cDNA at 6 h, it is now prominent in all of the RTX-treated samples at 24 h based on the significant reduction in amplified products when the viral DNA was treated with hUNG and APE1 before the real-time PCR analysis (Fig. 2B, black bars). The profile showed little further change between 24 and 48 h after infection (Fig. 2C). Control experiments established that pulse treatment with RTX specifically directs uracil incorporation into the rapidly replicating viral DNA, because the genomic DNA of the RTX-treated HT29 cells lacked detectable uracil (Fig. S3A).
Fig. 2.

RTX mediates dUTP incorporation into reverse transcribing HIV-1 cDNA and prevents efficient integration and viral protein expression. HIV-1 cDNA levels were measured in RTX-treated HT29 cells at 6 (A), 24 (B), and 48 (C) h after infection. (D) Integrated proviruses were measured at 48 h after infection using an Alu-Gag nested PCR. To determine whether viral DNA contained uracil, PCRs were performed with mock pretreatment of the template (white bars) or pretreatment with 50 nM hUNG and 50 nM APE1 (black bars, A–D), which, when combined, generate strand breaks at uracil sites. (E) Expression of virally encoded eGFP 48 h after infection as analyzed by FACS. (F) Thymidine (1 mM; Thd) was added to the media of infected cells either at the time of infection or 16 h after infection, and the ability to rescue GFP expression was measured by FACS. In all cases, n = 3.
Uracilated Viral cDNA Is Unable to Integrate and Express Viral Protein.
The above studies show that high dUTP levels delayed reverse transcription and that high levels of uracil were incorporated into viral DNA products by 24 h after infection. To determine whether the uracilated cDNA was proficient for integration, total DNA was obtained from cells at 48 h after infection, and the number of integrated proviruses in each sample was determined relative to an integration standard (Materials and Methods). Treatment with RTX induced a dose-dependent reduction in integrated proviruses, with a sevenfold reduction at the highest RTX concentration tested (Fig. 2D, white bars). Although total reverse transcripts at this time point were highly uracilated (Fig. 2C), the integrated proviruses did not contain detectable uracil (Fig. 2D, black bars). This finding requires that the presence of uracil in the HIV-1 cDNA makes it unreactive for integration or renders it unstable in the nucleus.
To evaluate the effects of pulsed RTX treatment on postintegration events, expression levels of virally encoded eGFP were measured (Fig. 2E). A 40-fold reduction in eGFP expression was observed that exceeds the sevenfold effect on integration, suggesting that dUTP also exerts an effect above and beyond the integration effect. We explored the possibility that RTX might have a general effect on transcription or translation. However, this was made unlikely because eGFP expression from a transfected viral plasmid was unaffected when cells were treated with 150 nM RTX (Fig. S1D). This result once again confirms that global protein expression is not affected by RTX treatment but additionally shows that expression from the viral LTR promoter is not compromised by RTX in a nonreplicating plasmid. Taken together, these findings indicate that the antiviral effects of RTX arise from a mechanism that specifically requires dUTP incorporation into viral DNA products.
Thymidine Can Partially Rescue the Antiviral Effect of RTX if Given Before Completion of Reverse Transcription.
Inhibition of TS by RTX blocks the de novo synthesis of dTTP, but dTTP can also be generated through the salvage pathway if thymine and/or thymidine are present in the media. Accordingly, if the antiviral mechanism of RTX is linked to its ability to increase the dUTP:dTTP ratio, then supplementing the media with Thd should antagonize the antiviral effects of RTX. An additional expectation is that Thd supplemention should only be successful if the nucleoside is provided before the completion of reverse transcription to be competitive with dUTP incorporation into viral DNA products. Indeed, when 1 mM Thd is provided at the time of infection, the expression of eGFP is restored to 60% of control expression (no drug), whereas the eGFP expression is only 2% of control when Thd supplementation is not provided (Fig. 2F). When Thd is added at 16 h after infection (when reverse transcription is largely complete) (31), the rescue is significantly diminished, yielding only 16% of control eGFP expression. Combined, these results strongly support a mechanism where the antiviral effects of RTX arise from a perturbation of the dUTP:dTTP pool and subsequent incorporation of dUTP into viral DNA products.
Inhibition of Endogenous hUNG2 Does Not Affect Reverse Transcription.
The observation that only nonuracilated proviruses were stably integrated in HT29 cells prompted an investigation into whether the host nuclear hUNG2 degrades the uracilated viral DNA products before integration. To generate a functional KO of hUNG2, we transfected HT29 cells with a stable transfection vector that constitutively expresses Ugi, a small protein that is a potent inhibitor of hUNG2 (32, 33). This cell line, HT29-Ugi, was confirmed to have no detectable UNG activity (Fig. S3C). As a control, we transfected HT29 cells with the same stable transfection vector, except that it lacked the Ugi coding sequence (HT29-IRES). This cell line had the same UNG activity levels as WT HT29 cells.
We then performed viral infections using the HT29-Ugi and HT29-IRES cells. Both the HT29-Ugi cells (Fig. 3 A–C) and HT29-IRES cells (Fig. S4 A–C) had the same response as WT HT29 cells with respect to accumulation of viral DNA products and uracil incorporation. Therefore, endogenous hUNG2 appears to have no effect on the process of reverse transcription. This result is not surprising, given that reverse transcription occurs in the cytoplasm and hUNG2 is sequestered in the nucleus. We were able to detect a low level of uracil in genomic DNA in RTX treated HT29-Ugi cells, as would be expected in the absence of repair initiated by hUNG2, but this uracil level was significantly less than that detected in viral cDNA (Fig. S3B).
Fig. 3.

Inhibition of hUNG2 completely rescues the ability of uracilated proviruses to integrate, but only slightly rescues viral protein expression. HIV-1 cDNA levels were measured in RTX-treated HT29-Ugi cells at 6 (A), 24 (B), and 48 (C) h after infection. (D) Integrated provirus levels were measured at 48 h after infection using the Alu-Gag nested PCR. The presence of uracil was assessed by pretreatment with hUNG/APE1 as previously described (compare white and black bars). (E) Expression of virally encoded eGFP 48 h after infection as analyzed by FACS. (F) HT29 cells were transfected with UNG2 or AllStars Negative control siRNAs 14 h before RTX treatment and infection. Integrated provirus levels were measured at 48 h after infection. In all cases, n = 3.
Inhibition of hUNG2 Permits Integration of Uracilated Viruses but Does Not Rescue Viral Protein Expression.
We then asked whether host nuclear hUNG2 has an effect on the ability of uracilated viral cDNA to integrate. Total cellular DNA was collected from HT29-Ugi and HT29-IRES cells at 48 h after infection. In HT29-IRES cells treated with RTX, the same dose-dependent decrease in integrated provirus was observed as was seen in WT HT29 cells, and those proviruses that successfully integrated contained no detectable uracil (Fig. S4D). Remarkably, in the HT29-Ugi cells, RTX failed to reduce the levels of integrated provirus, and the integrated proviruses were highly uracilated (Fig. 3D). Two important conclusions can be gleaned from these results: (i) hUNG2 activity is required for the drug effect on integration and (ii) the presence of uracil itself does not prevent a provirus from successful integration. The complete rescue of integration through specific inhibition of UNG2 indicates that backup uracil DNA glycosylases SMUG and TDG do not contribute to this mechanism, consistent with their low activity toward U:A substrates in HT29 extracts (34).
Given that inhibition of hUNG2 activity rendered cells permissive toward integration of uracilated proviruses, it was expected that expression of virally encoded eGFP would be rescued as well. Unexpectedly, the HT29-Ugi cells showed a dose-dependent reduction in eGFP expression (Fig. 3E), although this reduction was eight- and threefold less than WT HT29 (Fig. 2E) and HT29-IRES cells (Fig. S4E), respectively. The postintegration effect of uracil in HT29-Ugi cells may be unrelated to that observed with HT29 and HT29-IRES cells because no uracil was detected in the integrated proviruses in the presence of hUNG2 activity, but large levels of uracil are present in the absence of such activity in the HT29-Ugi line.
dUTP Incorporation into Viral DNA Is Not Highly Mutagenic.
Although U/A base pairs are not expected to result in mutations or coding changes in mRNA or proteins, viral DNA products isolated from HT29-IRES and HT29-Ugi cells after treatment with RTX were sequenced for mutations (Table S2). In the absence of RTX, the viral DNA clones obtained from infections of HT29-IRES and HT29-Ugi cells had on average 1.1 and 0.9 mutations per viral genome, respectively, whereas in the presence of 150 nM RTX, the rate for HT29-IRES increased to 2.9 and that for HT29-Ugi was essentially unchanged at 1.3 mutations per genome. The observed mutations were predominantly G/C→A/T transition mutations, consistent with the natural mutational drift observed in HIV-1 (35). It should also be noted that an increase in transition mutations has been observed in dUTPase-deficient nonprimate lentiviruses (36, 37); therefore, the slight increase in transition mutations on RTX treatment may be the result of uracil misincorporation. However, the inhibitory effects of RTX on viral integration cannot be reasonably attributed to these at most small increases in the mutation frequency, because these effects are completely dependent on the presence of hUNG2 activity.
Partial Knockdown of hUNG2 Yields an Intermediate Phenotype.
An orthogonal approach to inhibiting endogenous hUNG2 was taken to evaluate how the expression level of hUNG2 affects the observed integration block of uracilated proviruses. In this approach, WT HT29 cells were transfected with an siRNA that targets the nuclear isoform of hUNG (hUNG2) or a control AllStars Negative siRNA, followed by drug treatment and infection. hUNG2 was reduced 50–60% at both the time of infection (24 h after transfection of the siRNA) and 48 h after infection (Fig. S5 A and B). These levels of enzyme knockdown are much less than the complete loss of hUNG2 activity measured in the HT29-Ugi strain (Fig. S3C). In the hUNG2 siRNA knockdown cells at 48 h after infection, integration was rescued by twofold relative to cells transfected with the control siRNA (Fig. 3F), and GFP expression was rescued by threefold (Fig. S5C). These results may be compared with the Ugi knockdown, where integration was rescued sevenfold to control levels and GFP was rescued eightfold compared with WT cells (Figs. 2 D and E and 3 D and E). These results confirm the functional KO results obtained with the Ugi overexpression strain and establish that the integration block has a strong hUNG2 dose-response.
Viral Protein R Status of the Infecting Virus Does Not Impact the Antiviral Effects of RTX.
There are several reports that viral protein R (Vpr) recruits UNG2 into HIV-1 particles in producer cells, although the implication of this recruitment is controversial (38–40). To ascertain whether our findings were influenced by the presence of virally packaged hUNG2, we repeated the infection experiments with viruses that encode two defective forms of Vpr. The first construct has a start codon mutation (VprSM) that prevents expression of Vpr, and the second has a point mutation that abrogates the Vpr interaction with UNG2 (W54G). Infection of RTX-treated HT29 cells with either of these viruses resulted in the same reduction in GFP expression as seen with the WT virus (Fig. S6A). Control Western blot analyses confirmed that W54G expressed the mutant Vpr and that VprSM had greatly reduced Vpr loads (<6% of WT; Fig. S6B). Thus, viral packaging of hUNG2 is not required for degradation of uracilated viral DNA, which must be attributed to the nuclear enzyme in infected cells. This conclusion is strongly supported by the experiment that follows below.
Exogenously Uracilated DNA Is Degraded by Nuclear hUNG2 in Cells.
In the previous experiments, uracil was introduced into viral DNA by manipulating the intracellular nucleotide pools. Although the data strongly indicate that the antiviral mechanism initiated by RTX treatment requires uracil incorporation into the viral DNA and also hUNG activity, it is formally possible that other mechanisms could explain the loss of integrated DNA and GFP expression. Therefore, an experiment was designed that eliminates the need for RTX and instead introduces exogenously uracilated DNA directly into the cell and determines its stability and ability to express GFP protein. A linear uracilated expression vector that encodes GFP was constructed by PCR amplification of a 2.5-kb segment of the plasmid pmaxGFP in the presence of dUTP:dTTP ratios of 0, 1.2, or 6.8. The latter conditions mimic the dUTP:dTTP ratios in cells treated with 50 nM RTX at 6 h after infection and at the time of infection, respectively (Fig. 1B). To ascertain that the substrates synthesized in the presence of dUTP contained uracil, they were first analyzed by real-time PCR with or without pretreatment with hUNG/APE1 to degrade uracilated templates. Pretreatment led to a >3-log reduction in copy number for both substrates, indicating the presence of a high degree of uracil (Fig. S7). These substrates were then used to transfect HT29 cells, and after a 48-h incubation, cells were collected for FACS analysis to determine GFP expression and real-time PCR analysis to evaluate DNA levels.
In the control HT29-IRES cell line, transfected cells were found to contain a 6- to 10-fold lower copy number of substrate DNA when the DNA contained uracil compared with the all thymine-containing substrate. This finding suggested that the presence of uracil decreases the stability of the substrate DNA in the cell (Fig. 4A). It is noteworthy that the remaining substrate still contained uracil based on real-time PCR analysis with hUNG/APE1 pretreatment (Fig. 4A; note the difference between white and black bars). These findings suggest that the residual DNA in the HT29-IRES transfection did not encounter endogenous hUNG2. In the HT29-Ugi cell line, transfected cells were found to have the same amount of substrate DNA regardless of its uracil status (Fig. 4B). This finding supports the conclusion obtained from viral infections that hUNG2 alone leads to the degradation of uracilated DNA in the cell.
Fig. 4.

Inhibition of endogenous hUNG2 protects uracilated DNA from degradation. A 2.5-kb fragment of the plasmid pmaxGFP was PCR amplified in the presence of various dUTP:dTTP ratios (0, 1.2, and 6.8). These DNA substrates were nucleofected into HT29-IRES and HT29-Ugi cells. The stability of the uracilated DNA was assessed by qPCR at 48 h after nucleofection in both HT29-IRES (A) and HT29-Ugi (B) cells. To determine whether the substrates still contained uracil at this time point, the in vitro hUNG/APE1 treatment was performed as previously described (compare white and black bars). (C) FACS analysis of HT29-IRES and HT29-Ugi cells 48 h after transfection with the nonuracilatd and uracilated substrates. *Less than 2 × 104 copies of DNA detected. **,***Less than 0.15% and 0.06% GFP positive cells, respectively. In all cases, n = 3.
The ability of the uracilated substrates to express GFP in the HT29-IRES and HT29-Ugi cell lines was then tested. Consistent with the results obtained with uracilated viral DNA, little GFP expression was observed from the uracilated reporter constructs in HT29-IRES cells, whereas GFP expression was modestly reduced (approximately twofold) in HT29-Ugi cells (Fig. 4C). We speculate that the remaining uracilated substrate DNA in the HT29-IRES cells does not produce protein product because it is sequestered in the cytoplasm, making it inaccessible to the transcriptional machinery. It would also not be accessible to nuclear hUNG2, explaining both its persistence and the maintenance of its high uracil content. The twofold reduction in GFP expression with the stable uracilated substrate DNA in the transfected HT29-Ugi line suggests that the presence of unprocessed substrate DNA uracil leads to reduced protein expression by an unknown mechanism. Although speculative, this effect may also explain the UNG-independent reduction in GFP expression observed with integrated uracilated proviruses in the HT29-Ugi line (Fig. 3E).
Do Primary Immune Cells Possess the Required Functional Elements to Support dUTP-Mediated Antiviral Effects?
To determine whether the requisite effector proteins of the antiviral pathway are present in activated CD4+ T cells and monocyte-derived macrophages (MDMs), dUTPase and UNG activities were measured in these cell types and compared with the model cell line HT29. Activated CD4+ T cells had the highest dUTPase activity, approximately fivefold higher than HT29-IRES cells (Fig. 5A), which was not affected by RTX treatment (Fig. S8A). In contrast, lysates from MDMs failed to hydrolyze dUTP even after 2 h of reaction (Fig. 5A). The difference in dUTPase activities between these cell types correlates well with previous measurements of intracellular concentrations of dUTP (11, 12).
Fig. 5.

The natural target cells of HIV-1 do not have the requisite components of the dUTP-mediated restriction pathway. (A) dUTPase activity was measured by the amount of dUTP hydrolysis initiated by 2 μg of CD4+ T cells, MDM, or HT29 protein lysates over 1 h. For CD4+ T cells and MDMs, error bars represent the SD of replicates from two donors. For HT29-IRES cells, two replicates of the same lysate were analyzed. N.D., not detectable. (B) UNG activity was measured for CD4+ T-cell, MDM, and HT29-IRES cell lysates using a uracil-containing molecular beacon. UNG activity was defined as the pmol/min/μg. Error bars represent the SD of replicates from two donors. (C) dUTP and dTTP levels were determined by the SNE assay for CD4+ T cells that were treated for 6 h with 25 nM RTX or DMSO vehicle as previously described (n = 3). (D) HIV-1 cDNA levels were measured in RTX-treated CD4+ T cells at 48 h after infection. The presence of uracil was assessed by pretreatment with hUNG/APE1 as previously described (compare white and black bars; n = 3). (E) Expression of virally encoded eGFP 48 h after infection of RTX-treated CD4+ T cells as analyzed by FACS (n = 3).
With respect to UNG activity, HT29 cells had the highest activity, with activated T cells and MDMs showing 2.5- and 25-fold lower levels, respectively (Fig. 5B). Furthermore, RTX treatment did not affect hUNG2 activity in CD4+ T cells (Fig. S8B). Control experiments showed that nuclease activity was not detectable under these activity assay conditions, and in all samples, the observed UNG activity was completely inhibited by UGI. It was unexpected that MDMs had detectable UNG activity levels given that several studies have demonstrated that hUNG2 mRNA levels are very low in resting cells and that hUNG protein levels in MDMs are too low to be detectable by Western blot (9, 13, 41). However, our UNG activity assay is more sensitive than Western blotting, but it does not discriminate between the nuclear and mitochondrial isoforms of UNG. Therefore, the modest UNG activity observed in MDMs may arise from either mitochondrial hUNG1 or nuclear hUNG2 (9). If the activity does solely arise from the mitochondrial form, then it would be sequestered from the viral DNA products in the cytoplasmic and nuclear compartments and would not participate in the antiviral pathway.
dUTP pool levels have been reported for MDMs and activated peripheral blood mononuclear cells (PBMCs), but to our knowledge the levels in activated CD4+ T cells have not been evaluated (11, 12). [However, Diamond et al. have measured canonical dNTP pools in activated CD4+ T cells, but their assay did not distinguish dUTP from dTTP (42)]. The single nucleotide extension (SNE) assay was used to determine both dUTP and dTTP pools in activated CD4+ T cells collected from donor blood. dTTP was readily detectable at 1.5 pmol/106 cells, whereas dUTP levels were indistinguishable from zero (Fig. 5C). We then attempted to pharmacologically increase the dUTP:dTTP ratio by treating the CD4+ T cells with 25 nM RTX. Although RTX was able to reduce dTTP levels 12-fold, confirming that inhibition of TS was achieved (Fig. 5C), drug treatment did not lead to increased dUTP levels (Fig. 5C). These data suggest that the robust dUTPase activity in CD4+ T cells is sufficient to prevent drug-induced increases in dUTP. Our result differs from a previous report finding that viral DNA from infected CD4+ T cells carried a high level of uracil (>500 uracils/10 kb). Although these uracils were proposed to arise from dUTP incorporation (25), this was not directly determined, and these uracils may instead arise from cytosine deamination given how refractory CD4+ T cells are to increases in the dUTP:dTTP ratio.
If the proposed model for dUTP-mediated viral restriction is correct, then the failure of RTX to increase dUTP levels in CD4+ T cells should also fail to evoke an antiviral response. Accordingly, RTX had no effect on reverse transcript levels at 72 h after infection (Fig. 5D, white bars) and failed to induce uracilation of the viral DNA (Fig. 5D, difference between white and black bars). Expression of virally encoded eGFP was only slightly reduced (Fig. 5E) in contrast to the dramatic reductions seen in RTX-treated HT29 cells (Fig. 2E). These data support the model that dUTP incorporation is a critical component of this antiviral pathway. We attempted to further confirm the hUNG2 requirement of this pathway by overexpressing hUNG2 in MDMs, which already have high dUTP levels. However, this proved to be technically challenging because of the poor transfectability of these cells to plasmid DNA and the large number of positively transfected cells required to adequately address this question.
Discussion
Mechanisms of Uracil-Mediated HIV-1 Restriction.
Immune cells have evolved multiple ways of using uracil as a weapon against invading viruses. One well-characterized mechanism is the enzymatic deamination of cytosine in the minus strand of HIV-1 cDNA by members of the APOBEC3 subfamily (APOBEC3D, -F, -G, and -H) to generate C/G→U/A transition mutations after plus strand synthesis (43, 44). This deamination mechanism leads to lethal hypermutation of the viral genome and potent restriction of viral infection, but there are conflicting reports as to whether UNG2 excision of uracils is part of the restriction mechanism (45–47). In contrast, the dUTP-mediated mechanism elaborated here results in nonmutagenic incorporation of uracil on both strands of the viral DNA and requires hUNG2 to prevent viral integration. The distinct properties of these pathways make them suited for restricting viral infection in different cell types. Although the APOBEC3 pathway is potently restrictive in CD4+ T cells, a dUTP-mediated pathway is likely most relevant for nondividing cells that contain high levels of dUTP (macrophages, resting T cells) (11, 12). Our unique finding that potent dUTP-mediated viral restriction requires both high dUTP levels and abundant nuclear UNG2 establishes the minimal biochemical requirements for operation of this pathway in immune cells.
Roles for Nuclear and Virally Packaged hUNG2 in HIV-1 Infection.
A requirement for hUNG2 in HIV-1 infection has been debated for >15 y. hUNG2 is widely reported to be packaged into HIV-1 particles in producer cells through interactions of its N-terminal domain with the HIV-1 accessory protein Vpr (38–40, 48) [although a packaging interaction with viral integrase has also been reported (49)]. The proposed roles for packaged and nuclear hUNG2 in the viral life cycle have been diverse: (i) the virally packaged enzyme serves to nonenzymatically or enzymatically suppress mutations in the viral genome upon infection of macrophages by positively influencing reverse transcription (38, 39, 41, 50), (ii) the virally packaged UNG2 is completely dispensable for HIV-1 replication of cells with low dUTP levels (45), (iii) virion-associated hUNG2 degrades APOBEC3-edited HIV-1 DNA (46), (iv) virion-associated and nuclear hUNG2 play a selective role in enhancing infection of primary lymphocytes by R5 tropic, but not R4 tropic or pseudotyped HIV-1 (13). This latter role for hUNG2 appears to be entirely independent of dUTP levels, and in addition, is unrelated to our observations because we use a VSV-G pseudotyped HIV-1.
The diversity of results obtained above could arise from highly context-specific effects of uracil and hUNG2 enzymatic activity on viral infectivity. The findings reported here demonstrate that the antiviral effects of dUTP and hUNG2 are both concentration dependent, and depending on what aspect of the viral life cycle is measured, different outcomes could be obtained. For instance, if the role of hUNG2 is studied in the absence of dUTP, no effect would be observed on ultimate levels of reverse transcripts, viral integration efficiency, or viral protein expression. Similarly, if the role of elevated dUTP is studied in the absence of hUNG2 activity, no effect on the ultimate levels of reverse transcripts would be observed, nor would there be a measurable effect on integration of the virus. Although the field has justifiably moved toward the use of primary cells to investigate HIV-1 infectivity, if the component variables required for a given response are not measured (dUTP levels, hUNG2 activity, density of uracils in viral DNA), then it becomes problematic to evaluate the significance of a given result. Moreover, it is difficult to manipulate the required component variables in primary cells due to their poor transfection properties and short culture lifetimes. Accordingly, the linked role of dUTP or hUNG2 in infection is difficult to establish unless the two variables can be manipulated in a predictable way.
Under conditions of abundant nuclear hUNG2 and elevated dUTP, our results indicate that all of the uracilated viral DNA products can be degraded before integration, suggesting that this is a very efficient viral restriction mechanism under these defined conditions. This mechanism fails to reach 100% restriction, as there appears to be a uracil-free population of viral DNA that completes integration unrestricted. Consistent with this view, the fraction of virus that contains detectable uracil in the total reverse transcript population at 48 h (Fig. 2C) is almost identical to the amount of reduction in integration observed at the same time point (Fig. 2D). There are two possible sources for this uracil-free DNA population. First, this may represent viral cDNAs that are initially uracilated, but rapidly repaired. However, efficient repair requires that the uracils be sparse (Fig. 6A), and it is unclear why cDNAs would be repair competent at 6 h after infection (Fig. 2A) when they would have been synthesized during the time frame when dUTP levels were the highest (Fig. 1B; Scheme 1). The second formal possibility is that these uracil-free cDNAs may have been produced in the virion before infection and therefore would have completed reverse transcription before encountering the high intracellular dUTP pools (51). However, virions are believed to have very low dNTP pools, and most reverse transcription that takes place in the virion is aborted at early stages (52). Regardless of the source, attempts to improve on this restriction mechanism will need to minimize the size of this uracil-free pool.
Fig. 6.

The dUTP-mediated restriction mechanism depends on uracil density and hUNG2 activity. (A) Highly uracilated proviruses are proficient for integration in cells lacking hUNG2 activity and maintain their uracil loads. When hUNG2 activity is present, proviruses with low-frequency uracils may be repaired, whereas highly uracilated proviruses are degraded. (B) A 3D representation of the continuum between restriction and integration as it relates to dUTP levels and UNG activity. Warm colors indicate high levels of restriction, whereas cool colors indicate efficient integration. CD4+ T cells and untreated HT29 have undetectable dUTP levels, allowing integration to take place. MDMs have high dUTP levels but lack the hUNG activity required for restriction. HT29 cells treated with high RTX concentrations (R++) have high dUTP levels and high hUNG2 activity, leading to potent restriction. If lower RTX concentrations are used (R+) or if UNG2 activity is compromised by siRNA knockdown (KD), then an intermediate phenotype is obtained from HT29 cells.
The complete rescue of uracilated viral DNA products on inhibition of nuclear hUNG2 suggests that substitution of uracil for thymine in viral cDNA does not inhibit formation of the preintegration complex, nor does it interfere with integrase activity (Fig. 3D). In fact, Yan et al. (25) have proposed that uracilation of viral cDNA aids in integration by discouraging the competing and nonproductive autointegration reaction. Our results now uncover an additional consideration where nuclear hUNG2, if present at high enough levels, would efficiently degrade uracilated DNA.
Our data reveal no detectable role for packaged hUNG2 in the HT29 model system. One basis for this conclusion is the observation that the total levels of cytoplasmic viral DNA products are unaffected in the presence of elevated dUTP and that these products contain high uracil loads (Fig. 2 B and C). This finding strongly suggests that the fraction of the viral DNA pool in the cytoplasmic compartment is not subject to uracil DNA glycosylase activity (i.e., virally packaged enzyme). In contrast, the much smaller pool of uracilated viral DNA products in the nucleus is rapidly degraded by nuclear hUNG2 before integration (although we cannot exclude the possibility that a small amount of packaged hUNG2 might be imported into the nuclear compartment using its N-terminal nuclear targeting region; Fig. 2D). These results may indicate that the endogenous hUNG2 expressed in HEK293 producer cells was not efficiently packaged into virus particles used in these studies. This conclusion is supported by the findings that two Vpr-deficient HIV-1 strains reported to be impaired in hUNG2 packaging produced indistinguishable results from WT HIV-1 (Fig. S6A). Moreover, we were unable to detect endogenous hUNG2 activity in virions isolated from HEK293 cells, which is in line with a previous report (53). Instead, our findings favor hypotheses that Vpr serves to decrease hUNG2 expression either through ubiquitin-mediated degradation or transcriptional silencing to minimize its potential antiviral effects (53–55). If packaged hUNG2 plays a role in viral infectivity on infection of cells that contain elevated dUTP, the outcome would likely depend on the density of the uracils and the levels of the virally packaged and nuclear enzymes (see below).
Postintegration Effects of Viral Uracil.
In addition to the UNG-dependent integration block, there also exists a UNG-independent postintegration block that leads to a reduction in virally expressed eGFP (Fig. 3E). A similar result was found in dUTPase KOs of equine infectious anemia virus (EIAV), which experience a reduction in integration and an even more exaggerated reduction in viral mRNA levels (21). We and others have demonstrated that for HIV-1, these postintegration effects are not due to mutations (41) (Table S2). We speculate that the reduction in viral protein is due to reduced expression from uracilated promoters or other important DNA elements. The absence of the 5-methyl group in uracil has the potential to alter DNA structure and dynamic features that might be important for recognition by transcription factors or other DNA binding proteins (23, 25). For example, uracils localized in the origin of replication in HSV-1 were found to perturb the binding of the HSV-1 origin binding protein (27), and incorporated uracils in the cAMP response element (CRE) of rat stomatostatin gene led to a two- to fourfold decrease in binding of CRE binding proteins from HeLa extracts (26). Our observation of reduced GFP expression from the exogenously uracilated DNA in the HT29-Ugi cell line supports the proposal that the presence of uracil in DNA can lead to reduced gene transcription.
Mechanistic Requirements for dUTP-Mediated HIV-1 Restriction.
A potential key to reconciling conflicting reports as to whether UNG enzymatic activity is protective or destructive to HIV-1 is the frequency of uracils in DNA (which is related to cellular dUTP levels) and the expression levels of the nuclear enzyme (Fig. 6A). Accordingly, studies reporting that packaged UNG is beneficial to viral replication may only apply when uracil densities are significantly low to generate isolated single strand breaks that could be faithfully repaired by host nuclear repair machinery. In contrast, rampant uracil incorporation on both strands is more likely to lead to double-strand breaks and DNA fragmentation as shown here and previously in yeast treated with TS inhibitors (6). It is relevant to note that the dUTP/UNG2-coupled pathway for viral DNA destruction is mechanistically distinct from the A3G-mediated pathway for viral restriction (56). The A3G pathway introduces uracils exclusively into the minus strand of the viral DNA at the preferred triplet sequence CCC, which occurs far less often than the single adenines required for dUTP incorporation. Thus, the restrictive potential of the A3G pathway may be based more on a hypermutation mechanism rather than viral DNA destruction (43, 57).
The findings in the HT29 system and the corresponding biochemical measurements in primary CD4+ T cells and MDMs (Fig. 5) provide relevant measurements to postulate a reasonable model for dUTP-mediated innate immunity (Fig. 6B). The restrictive property of this pathway will require that cells meet three conditions: low dUTPase levels, high dUTP levels, and abundant nuclear hUNG2. The three cell types studied here fall into different regions of a 3D activity space that is defined by these variables, and the position of a given cell in this space will determine the effectiveness of this pathway. Accordingly, RTX-treated HT29 cells are highly restrictive against viral integration because dUTP is elevated, dUTPase is low, and nuclear hUNG2 is highly expressed. Although MDMs meet the requirement of high dUTP (11, 12), these cells have the lowest level of hUNG2 activity and might only experience partial protection based on our measured dose-response with respect to hUNG2 (Fig. 3F; Fig. S5). CD4+ T cells do not benefit from this pathway and are refractory to RTX-induced elevations in dUTP because these cells express high levels of dUTPase (Fig. 5A). These findings suggest that dUTPase inhibitors might be a useful strategy to increase protection of CD4+ T cells (58, 59). The therapeutic index of this approach would depend on the ability to specifically direct uracil into the replicating virus while preserving the integrity of the host genomic DNA (i.e., the relative rates of DNA replication of the viral and human polymerases and the effectiveness of nuclear uracil base excision repair in removing nascent uracils from host genomic DNA). The viral DNA, which is rapidly synthesized in the cytoplasmic compartment, would likely accumulate a much larger total uracil load and be subject to fragmentation on entry into the nuclear compartment, whereas the slowly replicating nuclear DNA would accumulate less uracil and be repaired (Fig. 6A).
Materials and Methods
Cells and Viruses.
HT29 cells were obtained from the ATCC (HTB-38) and maintained in RPMI 1640 + 10% (vol/vol) FBS + 1% Pen/Strep unless otherwise indicated. VSV-G pseudotyped HIV-1 virions were generated as previously described (60). In brief, HEK293T cells were cotransfected with pNL4-3-ΔE-eGFP and pVSV-G. The morning after transfection, media were changed to RFM [(folate-free RPMI 1640 + 10% (vol/vol) dialyzed FBS, 1% Pen/Strep, and 80 nM 5-methyl tetrahydrofolate (5-MeTHF)]. Viral supernatant was collected 60 h after infection, and virions were purified over a 20% (wt/vol) sucrose cushion.
RTX Drug Treatments and Infections.
HT29 cells were plated at ∼250,000 cells per well of a 6-well plate and allowed to grow for 3 d before drug treatment. The media were changed to RFM the night before drug treatment, and fresh RFM media were added immediately before treatment. Cells were incubated with RTX for 6 h. After incubation, cells were washed with PBS, and then fresh RFM was added to each well. For dNTP level determinations, cells were either collected immediately or placed back in the incubator until the given time point. For infections, cells were trypsinized, counted, and plated at a density of 100,000 cells per well using a round bottom 96-well plate. Virus was added to cells, and the plate was spin infected for 2 h at 1,200 × g and 30 °C and then allowed to incubate at 37 °C until the given time points. Expression of virally encoded eGFP was determined by FACS analysis 48 h after infection.
dNTP Extraction, dUTPase Treatment, and SNE Assay.
The extraction of total dNTPs and quantification of dUTP and dTTP from cells were performed as previously described (34) with a few modifications. The amount of dUTPase used was reduced from 1 μM to 50 nM, and the dUTPase reaction was run in 50 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 0.5 mM β-mercaptoethanol (β-ME), and 0.1% BSA. After methanol precipitation of dUTPase, samples were dried down and resuspended in water to perform the SNE reaction (the buffer and salts in the dried pellet were sufficient to support the extension reaction).
Real-Time PCR Analysis of Uracilated Viral DNA Species.
DNA was extracted from infected cells using the DNA mini kit (Qiagen). DNA concentrations were determined on a Nanodrop 2000 (Thermo Scientific), and the same total mass of DNA was used for each sample in a given PCR. Late reverse transcripts were analyzed by real-time PCR using the MH531/MH532 primer set and LRT-p probe as previously described (61). To distinguish uracilated templates from nonuracilated templates, the DNA was first reacted with 50 nM each hUNG/APE1 in 1× TMNB+ buffer (10 mM Tris-HCl, pH 8.0, 20 mM NaCl, 11 mM MgCl2, and 0.002% Brij-35) or mock reacted before real-time PCR amplification. The combined action of hUNG/APE1 generates strand breaks at uracil sites. For convenience, some reactions omitted APE1, because heat is sufficient to cleave the abasic sites generated by hUNG. There are 66 potential sites for uracil incorporation in this amplicon, and at least one site on each strand must be uracilated to prevent amplification of the template. The difference in amplification between the hUNG/APE1 pretreated and mock-treated templates indicates their level of uracilation.
Generation of HT29 Stable Transfectants and Integration Standard.
The pIRESneo3-Ugi plasmid was constructed by cloning the humanized Ugi gene into the NheI and BamHI sites of pIRESneo3 (Clontech). pIRESneo3-Ugi and pIRESneo3 were linearized by NruI and transfected into HT29 cells using Cell Line Kit R (Lonza) and program W017 on a Nucleofector II instrument. Twenty-four hours after transfection, 0.4 mg/mL G418 was added to the media to select for NeoR clones. Resistant cells were expanded and maintained in 0.2 mg/mL G418. The pIRESneo3 stable transfectants were named HT29-IRES and the pIRESneo3-Ugi stable transfectants were named HT29-Ugi. The expression of the UNG-inhibitor Ugi was validated using a fluorescent hairpin reporter of UNG activity (see below) and determined to have no detectable UNG activity.
To generate a stably infected HT29 cell line, a NeoR resistance cassette was inserted into the NL4-3 genome. The synthetic intron (IVS), IRES element, and NeoR gene were amplified from pIRESneo3 and cloned into the NheI site immediately downstream from eGFP in pNL4-3-ΔE-eGFP. The new viral plasmid was named pNL4-3-ΔE-eGFP/NeoR. This plasmid was used to generate virus as described above (Cells and Virus). HT29 cells were then infected with these virions at a low multiplicity of infection to ensure single infection events. Infected cells were selected by treatment with 0.4 mg/mL G418. Resistant cells were cultured for 1 mo to ensure stable infection and were confirmed to contain approximately one provirus per cell. DNA was extracted from these cells and used as an integration standard for real-time PCR. Detection of integrated provirus was performed via the Alu-Gag nested PCR as described previously (62), but using the MH531/532 primer probe set described above for quantitative PCR. An integration standard curve was generated by diluting the integration standard with uninfected HT29 DNA.
siRNA Knockdown of hUNG2.
The nuclear isoform of human uracil DNA glycosylase (hUNG2) was targeted for siRNA knockdown as previously described (63). The siRNA sense sequence was 5′-AUCGGCCAGAAGACGCUCUdTdT-3′ and was purchased from Dharmacon. The AllStars negative control siRNA from Qiagen was used as a negative control: 180 pmol of hUNG2 siRNA or AllStars siRNA was used to nucleofect 5 × 106 HT29 cells in triplicate. After a 14-h incubation, cells were treated with RTX and infected as previously described. The knockdown efficiency was measured with a fluorescence-based UNG activity assay using protein extracts from the transfected cells (see below).
UNG and dUTPase Activity Assays.
A molecular beacon reporter of UNG activity was synthesized on an ABI 394 DNA synthesizer. The molecular beacon, S-pin-18, contains six deoxyuridines, a flexible PEG linker, a FAM fluorophore, and a Dabcyl quencher. Phosphorothioate linkages (indicated by the small “s”) were added to the terminal nucleotide linkages to deter nuclease activity. The sequence of the molecular beacon hairpin is 5′-FAM-GsCsAUUAAGAAGAAG-(PEG)6-CUUCUUAATsGsCs-DAB-3′.
Protein lysates were obtained using CelLytic M reagent (Sigma) according to the manufacturer’s instructions. Reactions were performed using 5 µg of protein lysate and 50 nM S-pin-18 in 10 mM Tris-HCl, pH 7.1, 100 mM NaCl, 1 mM EDTA, and 0.2% Triton X-100. Initial rates were measured on a Fluoro-Max 3 (Horiba Jobin Yvon).
dUTPase activity was assessed by incubating protein lysates with [5-3H] dUTP (Moravek Biochemicals) and determining the fraction of substrate hydrolyzed to dUMP product. Reactions contained 2 µg of protein lysate, 11 µM dUTP, 50 mM Tris-HCl, pH 8.0, 0.5 mM β-ME, 1 mM MgCl2, 0.1% BSA, 5 mM ATP, and 10 mM paranitrophenyl phosphate (p-NPP) at a total volume of 50 µL. ATP was included to inhibit nonspecific phosphatases, and p-NPP was added to prevent the degradation of dUTP by alkaline phosphatase (64). Reactions were incubated at 37 °C for 1 h, after which 2 µL was spotted onto a PEI-cellulose TLC plate. The TLC plates were developed in 0.5 M LiCl to separate dUTP from the product dUMP. Plates were exposed to a tritium sensitive screen overnight and scanned on a Storm Imager.
Generation of a Uracilated Reporter Construct.
To generate a uracilated reporter construct, a 2.5-kb segment of the GFP reporter plasmid pmaxGFP (Lonza) was amplified by PCR in the presence of various dUTP/dTTP ratios using the dU-insensitive Pfu Cx polymerase (Stratagene). This amplicon contains all of the genetic elements necessary for expression of GFP. The T- and U-containing DNAs were transfected into HT29-IRES and HT29-Ugi cells via nucleofection. Expression of GFP was analyzed 48 h after transfection by FACS analysis. Transfected DNA that remained stable in the cell at 48 h was extracted and quantified by real-time PCR using the forward primer 5′-TGAGCTTCAGCTACCGCTAC-3′, reverse primer 5′-TCTTGTCGGTGAAGATCACG-3′, and the probe 5′-/FAM/CGGTGCCCACCACCTTGAAG/TAMRA/-3′.
Primary Cell Studies.
Peripheral blood was obtained from two healthy volunteers. This study was approved by the Johns Hopkins Institutional Review Board, and written informed consent was provided by both donors. PBMCs were obtained from whole blood via Ficoll gradient. CD14+ monocytes were isolated from the PBMC population using the Monocyte Isolation Kit II (Miltenyi Biotec). Monocytes were differentiated into macrophages over 14–15 d using MDM-20 media containing RPMI 1640, 20% (vol/vol) autologous plasma, 1 ng/mL GM-CSF (BD Biosciences), 1× Hepes, and 1× glutamine (Gibco). The CD14− population was cultured for 3 d in folate-free RPMI 1640, 10% (vol/vol) dialyzed FBS, 1% Pen/Strep, 80 nM 5-MeTHF, 1% T-cell growth factor (TCGF), and 100 U/mL IL-2. Cells were activated by the addition of 0.5 μg/mL phytohemagglutinin (PHA). CD4+ T cells were isolated 3 d after activation using the CD4+ T-cell Isolation Kit II (Miltenyi Biotec).
Immediately after isolation, CD4+ T cells were treated with 25 nM RTX or DMSO vehicle for 6 h. Cells were then pelleted and used for protein extraction, dNTP extraction, or infection as described above; for CD4+ T-cell infections, however, cells were plated at a density of 250,000 cells per well and were incubated for 3 d after virus addition before FACS analysis.
Supplementary Material
Acknowledgments
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease, National Institutes of Health: HIV-1 p24 gag monoclonal (24-2) from Dr. Michael H. Malim and HIV-1 Vpr (1-50) antiserum from Dr. Jeffrey Kopp (Fig. S7). This work was supported by National Institutes of Health Grants GM056834 (to J.T.S.) and AI081600 (to R.F.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219702110/-/DCSupplemental.
References
- 1.Priet S, Sire J, Quérat G. Uracils as a cellular weapon against viruses and mechanisms of viral escape. Curr HIV Res. 2006;4(1):31–42. doi: 10.2174/157016206775197673. [DOI] [PubMed] [Google Scholar]
- 2.Brynolf K, Eliasson R, Reichard P. Formation of Okazaki fragments in polyoma DNA synthesis caused by misincorporation of uracil. Cell. 1978;13(3):573–580. doi: 10.1016/0092-8674(78)90330-6. [DOI] [PubMed] [Google Scholar]
- 3.Bessman MJ, et al. Enzymatic synthesis of deoxyribonucleic acid. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acid. Proc Natl Acad Sci USA. 1958;44(7):633–640. doi: 10.1073/pnas.44.7.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.el-Hajj HH, Zhang H, Weiss B. Lethality of a dut (deoxyuridine triphosphatase) mutation in Escherichia coli. J Bacteriol. 1988;170(3):1069–1075. doi: 10.1128/jb.170.3.1069-1075.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gadsden MH, McIntosh EM, Game JC, Wilson PJ, Haynes RH. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 1993;12(11):4425–4431. doi: 10.1002/j.1460-2075.1993.tb06127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seiple L, Jaruga P, Dizdaroglu M, Stivers JT. Linking uracil base excision repair and 5-fluorouracil toxicity in yeast. Nucleic Acids Res. 2006;34(1):140–151. doi: 10.1093/nar/gkj430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kouzminova EA, Kuzminov A. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol Microbiol. 2004;51(5):1279–1295. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 8.McIntosh EM, Ager DD, Gadsden MH, Haynes RH. Human dUTP pyrophosphatase: cDNA sequence and potential biological importance of the enzyme. Proc Natl Acad Sci USA. 1992;89(17):8020–8024. doi: 10.1073/pnas.89.17.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer JA, Muller-Weeks S, Caradonna S. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair (Amst) 2004;3(5):505–513. doi: 10.1016/j.dnarep.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Ladner RD, Caradonna SJ. The human dUTPase gene encodes both nuclear and mitochondrial isoforms. Differential expression of the isoforms and characterization of a cDNA encoding the mitochondrial species. J Biol Chem. 1997;272(30):19072–19080. doi: 10.1074/jbc.272.30.19072. [DOI] [PubMed] [Google Scholar]
- 11.Aquaro S, et al. Macrophages and HIV infection: Therapeutical approaches toward this strategic virus reservoir. Antiviral Res. 2002;55(2):209–225. doi: 10.1016/s0166-3542(02)00052-9. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy EM, et al. Abundant non-canonical dUTP found in primary human macrophages drives its frequent incorporation by HIV-1 reverse transcriptase. J Biol Chem. 2011;286(28):25047–25055. doi: 10.1074/jbc.M111.234047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones KL, et al. X4 and R5 HIV-1 have distinct post-entry requirements for uracil DNA glycosylase during infection of primary cells. J Biol Chem. 2010;285(24):18603–18614. doi: 10.1074/jbc.M109.090126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen R, Wang H, Mansky LM. Roles of uracil-DNA glycosylase and dUTPase in virus replication. J Gen Virol. 2002;83(Pt 10):2339–2345. doi: 10.1099/0022-1317-83-10-2339. [DOI] [PubMed] [Google Scholar]
- 15.Sire J, Quérat G, Esnault C, Priet S. Uracil within DNA: An actor of antiviral immunity. Retrovirology. 2008;5:45. doi: 10.1186/1742-4690-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldo AM, McClure MA. Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J Virol. 1999;73(9):7710–7721. doi: 10.1128/jvi.73.9.7710-7721.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elder JH, et al. Distinct subsets of retroviruses encode dUTPase. J Virol. 1992;66(3):1791–1794. doi: 10.1128/jvi.66.3.1791-1794.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turelli P, et al. Replication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J Virol. 1996;70(2):1213–1217. doi: 10.1128/jvi.70.2.1213-1217.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Threadgill DS, et al. Characterization of equine infectious anemia virus dUTPase: Growth properties of a dUTPase-deficient mutant. J Virol. 1993;67(5):2592–2600. doi: 10.1128/jvi.67.5.2592-2600.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lichtenstein DL, et al. Replication in vitro and in vivo of an equine infectious anemia virus mutant deficient in dUTPase activity. J Virol. 1995;69(5):2881–2888. doi: 10.1128/jvi.69.5.2881-2888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steagall WK, Robek MD, Perry ST, Fuller FJ, Payne SL. Incorporation of uracil into viral DNA correlates with reduced replication of EIAV in macrophages. Virology. 1995;210(2):302–313. doi: 10.1006/viro.1995.1347. [DOI] [PubMed] [Google Scholar]
- 22.Klarmann GJ, Chen X, North TW, Preston BD. Incorporation of uracil into minus strand DNA affects the specificity of plus strand synthesis initiation during lentiviral reverse transcription. J Biol Chem. 2003;278(10):7902–7909. doi: 10.1074/jbc.M207223200. [DOI] [PubMed] [Google Scholar]
- 23.Bailly C, Crow S, Minnock A, Waring MJ. Demethylation of thymine residues affects DNA cleavage by endonucleases but not sequence recognition by drugs. J Mol Biol. 1999;291(3):561–573. doi: 10.1006/jmbi.1999.2979. [DOI] [PubMed] [Google Scholar]
- 24.Wang T, Balakrishnan M, Jonsson CB. Major and minor groove contacts in retroviral integrase-LTR interactions. Biochemistry. 1999;38(12):3624–3632. doi: 10.1021/bi982124i. [DOI] [PubMed] [Google Scholar]
- 25.Yan N, O’Day E, Wheeler LA, Engelman A, Lieberman J. HIV DNA is heavily uracilated, which protects it from autointegration. Proc Natl Acad Sci USA. 2011;108(22):9244–9249. doi: 10.1073/pnas.1102943108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verri A, Mazzarello P, Biamonti G, Spadari S, Focher F. The specific binding of nuclear protein(s) to the cAMP responsive element (CRE) sequence (TGACGTCA) is reduced by the misincorporation of U and increased by the deamination of C. Nucleic Acids Res. 1990;18(19):5775–5780. doi: 10.1093/nar/18.19.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Focher F, Verri A, Verzeletti S, Mazzarello P, Spadari S. Uracil in OriS of herpes simplex 1 alters its specific recognition by origin binding protein (OBP): Does virus induced uracil-DNA glycosylase play a key role in viral reactivation and replication? Chromosoma. 1992;102(1 Suppl):S67–S71. doi: 10.1007/BF02451788. [DOI] [PubMed] [Google Scholar]
- 28.Horowitz RW, Zhang H, Schwartz EL, Ladner RD, Wadler S. Measurement of deoxyuridine triphosphate and thymidine triphosphate in the extracts of thymidylate synthase-inhibited cells using a modified DNA polymerase assay. Biochem Pharmacol. 1997;54(5):635–638. doi: 10.1016/s0006-2952(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 29.Canman CE, et al. Induction of resistance to fluorodeoxyuridine cytotoxicity and DNA damage in human tumor cells by expression of Escherichia coli deoxyuridinetriphosphatase. Cancer Res. 1994;54(9):2296–2298. [PubMed] [Google Scholar]
- 30.Wilson PM, et al. A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 2011;39(17):e112. doi: 10.1093/nar/gkr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7(5):631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 32.Karran P, Cone R, Friedberg EC. Specificity of the bacteriophage PBS2 induced inhibitor of uracil-DNA glycosylase. Biochemistry. 1981;20(21):6092–6096. doi: 10.1021/bi00524a027. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Mosbaugh DW. Uracil-DNA glycosylase inhibitor gene of bacteriophage PBS2 encodes a binding protein specific for uracil-DNA glycosylase. J Biol Chem. 1989;264(2):1163–1171. [PubMed] [Google Scholar]
- 34.Grogan BC, Parker JB, Guminski AF, Stivers JT. Effect of the thymidylate synthase inhibitors on dUTP and TTP pool levels and the activities of DNA repair glycosylases on uracil and 5-fluorouracil in DNA. Biochemistry. 2011;50(5):618–627. doi: 10.1021/bi102046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69(8):5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lerner DL, et al. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc Natl Acad Sci USA. 1995;92(16):7480–7484. doi: 10.1073/pnas.92.16.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turelli P, Guiguen F, Mornex JF, Vigne R, Quérat G. dUTPase-minus caprine arthritis-encephalitis virus is attenuated for pathogenesis and accumulates G-to-A substitutions. J Virol. 1997;71(6):4522–4530. doi: 10.1128/jvi.71.6.4522-4530.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mansky LM, Preveral S, Selig L, Benarous R, Benichou S. The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 In vivo mutation rate. J Virol. 2000;74(15):7039–7047. doi: 10.1128/jvi.74.15.7039-7047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen R, Le Rouzic E, Kearney JA, Mansky LM, Benichou S. Vpr-mediated incorporation of UNG2 into HIV-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J Biol Chem. 2004;279(27):28419–28425. doi: 10.1074/jbc.M403875200. [DOI] [PubMed] [Google Scholar]
- 40.Bouhamdan M, et al. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J Virol. 1996;70(2):697–704. doi: 10.1128/jvi.70.2.697-704.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Priet S, et al. HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol Cell. 2005;17(4):479–490. doi: 10.1016/j.molcel.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 42.Diamond TL, et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279(49):51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris RS, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113(6):803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 44.Harris RS, Hultquist JF, Evans DT. The restriction factors of human immunodeficiency virus. J Biol Chem. 2012;287(49):40875–40883. doi: 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80(2):875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang B, Chen K, Zhang C, Huang S, Zhang H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J Biol Chem. 2007;282(16):11667–11675. doi: 10.1074/jbc.M606864200. [DOI] [PubMed] [Google Scholar]
- 47.Langlois M-A, Neuberger MS. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J Virol. 2008;82(9):4660–4664. doi: 10.1128/JVI.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selig L, et al. Uracil DNA glycosylase specifically interacts with Vpr of both human immunodeficiency virus type 1 and simian immunodeficiency virus of sooty mangabeys, but binding does not correlate with cell cycle arrest. J Virol. 1997;71(6):4842–4846. doi: 10.1128/jvi.71.6.4842-4846.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willetts KE, et al. DNA repair enzyme uracil DNA glycosylase is specifically incorporated into human immunodeficiency virus type 1 viral particles through a Vpr-independent mechanism. J Virol. 1999;73(2):1682–1688. doi: 10.1128/jvi.73.2.1682-1688.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guenzel CA, et al. Recruitment of the nuclear form of uracil DNA glycosylase into virus particles participates in the full infectivity of HIV-1. J Virol. 2011;86(5):2533–2544. doi: 10.1128/JVI.05163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mougel M, Houzet L, Darlix J-L. When is it time for reverse transcription to start and go? Retrovirology. 2009;6:24. doi: 10.1186/1742-4690-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, et al. Reverse transcription takes place within extracellular HIV-1 virions: Potential biological significance. AIDS Res Hum Retroviruses. 1993;9(12):1287–1296. doi: 10.1089/aid.1993.9.1287. [DOI] [PubMed] [Google Scholar]
- 53.Schröfelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79(17):10978–10987. doi: 10.1128/JVI.79.17.10978-10987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahn J, et al. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J Biol Chem. 2010;285(48):37333–37341. doi: 10.1074/jbc.M110.133181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langevin C, et al. Human immunodeficiency virus type 1 Vpr modulates cellular expression of UNG2 via a negative transcriptional effect. J Virol. 2009;83(19):10256–10263. doi: 10.1128/JVI.02654-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 57.Schumacher AJ, Haché G, Macduff DA, Brown WL, Harris RS. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J Virol. 2008;82(6):2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miyahara S, et al. Discovery of a novel class of potent human deoxyuridine triphosphatase inhibitors remarkably enhancing the antitumor activity of thymidylate synthase inhibitors. J Med Chem. 2012;55(7):2970–2980. doi: 10.1021/jm201628y. [DOI] [PubMed] [Google Scholar]
- 59.Miyahara S, et al. Discovery of highly potent human deoxyuridine triphosphatase inhibitors based on the conformation restriction strategy. J Med Chem. 2012;55(11):5483–5496. doi: 10.1021/jm300416h. [DOI] [PubMed] [Google Scholar]
- 60.Zhang H, et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol. 2004;78(4):1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mbisa JL, Delviks-Frankenberry KA, Thomas JA, Gorelick RJ, Pathak VK. Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol Biol. 2009;485:55–72. doi: 10.1007/978-1-59745-170-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Doherty U, Swiggard WJ, Jeyakumar D, McGain D, Malim MH. A sensitive, quantitative assay for human immunodeficiency virus type 1 integration. J Virol. 2002;76(21):10942–10950. doi: 10.1128/JVI.76.21.10942-10950.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischer JA, Muller-Weeks S, Caradonna SJ. Fluorodeoxyuridine modulates cellular expression of the DNA base excision repair enzyme uracil-DNA glycosylase. Cancer Res. 2006;66(17):8829–8837. doi: 10.1158/0008-5472.CAN-06-0540. [DOI] [PubMed] [Google Scholar]
- 64.Williams MV, Parris DS. Characterization of a herpes simplex virus type 2 deoxyuridine triphosphate nucleotidohydrolase and mapping of a gene conferring type specificity for the enzyme. Virology. 1987;156(2):282–292. doi: 10.1016/0042-6822(87)90408-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.