

Abstract
Hematopoietic stem cells (HSCs) are the source of all blood lineages, and HSCs must balance quiescence, self-renewal, and differentiation to meet lifelong needs for blood cell development. Transformation of HSCs by the breakpoint cluster region-ABL tyrosine kinase (BCR-ABL) oncogene causes chronic myelogenous leukemia (CML). The E-twenty six (ets) transcription factor GA binding protein (GABP) is a tetrameric transcription factor complex that contains GABPα and GABPβ proteins. Deletion in bone marrow of Gabpa, the gene that encodes the DNA-binding component, caused cell cycle arrest in HSCs and profound loss of hematopoietic progenitor cells. Loss of Gabpα prevented development of CML, although mice continued to generate BCR-ABL–expressing Gabpα-null cells for months that were serially transplantable and contributed to all lineages in secondary recipients. A bioinformatic screen identified the serine-threonine kinase protein kinase D2 (PRKD2) as a potential effector of GABP in HSCs. Prkd2 expression was markedly reduced in Gabpα-null HSCs and progenitor cells. Reduced expression of PRKD2 or pharmacologic inhibition decreased cell cycling, and PRKD2 rescued growth of Gabpα-null BCR-ABL–expressing cells. Thus, GABP is required for HSC cell cycle entry and CML development through its control of PRKD2. This offers a potential therapeutic target in leukemia.
Keywords: LSC, cell cycle control, signal transduction, imatinib
Hematopoietic stem cells (HSCs) are the source of all blood lineages in bone marrow and peripheral blood. HSCs must balance quiescence, growth, and differentiation to meet lifelong demands for blood cell development. HSCs give rise to lineage-committed progenitor cells, yet retain the ability to renew the HSC pool. Transcription factors affect HSC proliferation, survival, and differentiation, and are implicated in leukemic transformation of HSCs (1).
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm (MPN) characterized by infiltration of bone marrow, peripheral blood, and viscera by myeloid cells. In CML, reciprocal translocation of chromosomes 9 and 22 generates the breakpoint cluster region-ABL tyrosine kinase (BCR-ABL) oncogene. CML arises in a leukemic stem cell (LSC), which drives the expansion of granulocytes and their precursors that is the hallmark of this MPN. BCR-ABL is a constitutively active tyrosine kinase that activates downstream signal transduction pathways, and inhibition of BCR-ABL by tyrosine kinase inhibitors (TKIs), such as imatinib, induces hematologic, cytogenetic, and molecular remission in most patients with CML (2). A mouse model of CML recapitulates aspects of human CML, and mice die within weeks of transplantation with BCR-ABL–expressing bone marrow (3).
GA binding protein (GABP) is a tetrameric transcription factor that contains two molecules of GABPα and two molecules of various GABPβ proteins. GABP is unique among more than two dozen mammalian E-twenty six (ets) transcription factors because it is the only obligate multimer, i.e., it is active only when the tetramer is formed. The carboxyl-terminal ets DNA binding domain of GABPα binds to aminoterminal ankyrin-like repeats of GABPβ, whereas the transcription activation domain is encoded in the GABPβ carboxyl terminus. GABP controls expression of genes that are required for innate and acquired immunity, including CD18; Elastase, Neutrophil Expressed (ELANE); and α4 integrin in myeloid cells (4), and interleukin-7 receptor (IL7R) and Pax5 in lymphocytes (5, 6). GABP also controls expression of genes that are required for cell cycle control, and for ribosomal and mitochondrial biogenesis (4).
GABPA is a unique gene in the human and mouse genomes, and its product is the only protein that can recruit GABPβ to DNA. Deletion of mouse Gabpa inactivates the Gabp complex, and was shown to cause embryonic lethality (5, 7, 8). Conditional deletion of Gabpa in mouse embryonic fibroblasts caused profound G1S cell cycle arrest (8). Loss of Gabpα in bone marrow caused myelodysplasia and profound loss of bone marrow progenitor cells, but reports differ regarding the specific effects of Gabpa deletion on HSCs (9, 10).
We show that disruption of Gabpa markedly reduced HSC cell cycle activity, and that Gabpα loss prevented development of CML in BCR-ABL–expressing bone marrow. Rather than developing leukemia, Gabpα-null BCR-ABL+ HSCs continued to generate mature granulocytes for many months. Gabpα-null BCR-ABL+ HSCs were transplantable into secondary recipients and contributed to all hematopoietic lineages. A bioinformatic screen implicated the diacylglycerol- and protein kinase C (PKC)-activated serine-threonine kinase protein kinase D2 (PRKD2) as a potential effector of GABP in CML. Knockdown or pharmacologic inhibition of PRKD2 mimicked the effect of Gabpa disruption on the growth of Gabpα-null HSCs and, conversely, ectopic expression of PRKD2 overcame the growth defect of BCR-ABL–expressing Gabpα-null HSCs. Thus, Gabpα loss and expression of BCR-ABL achieve a standoff of sorts, i.e., the proliferative thrust of BCR-ABL partially overcomes the cell cycle arrest of Gabpα loss, whereas Gabpa disruption prevents BCR-ABL–associated CML. This report describes a cell cycle control mechanism that prevents development of leukemia despite continued production of oncogene-expressing stem cells, and reports PRKD2 as a mediator of BCR-ABL transformation in CML. These findings identify a therapeutic target in CML and strategies to prevent development of leukemia in oncogene-expressing hematopoietic cells.
Results
Gabpa Deletion in Bone Marrow Causes Cell Cycle Arrest in HSCs.
We created mice in which loxP recombination sites flank exons that encode the DNA-binding ets domain (Gabpafl/fl or floxed mice) (8). The Mx1Cre transgene was bred into these mice, and injection with polyinosine-cytosine (pIC) deletes Gabpa in bone marrow (Gabpa KO or simply KO mice) (9). As controls, Gabpafl/fl littermates that lacked the Mx1Cre transgene were injected with pIC. Half of the Gabpa KO mice died within 2 wk after the first pIC injection (Fig. 1A). White blood cells, platelets, and hemoglobin in peripheral blood declined dramatically in KO mice between days 5 and 14 (Fig. 1B), and microscopic examination revealed bone marrow hemorrhage with only rare nucleated cells (Fig. 1C). However, peripheral blood counts in surviving KO mice recovered (Fig. 1B), and these mice lived for months without apparent hematologic defects or other abnormalities. Only Gabpα-replete cells could form in vitro colonies when bone marrow was sampled 3 wk after pIC injection (Fig. S1). Thus, rapid overgrowth of bone marrow by the initially small population of cells that failed to delete Gabpa indicates that Gabpα-replete bone marrow cells have a growth advantage over Gabpα-null cells.
Fig. 1.

Conditional deletion of Gabpa in mouse bone marrow causes pancytopenia in association with reduced HSC cell cycle activity. (A) Kaplan–Meier survival curve and (B) peripheral white blood cell (WBC) and platelet counts per cubic millimeter and hemoglobin (Hgb) concentration (in g/dL) of control and KO mice following injection with pIC. (C) Bone sections of control and KO mice 2 wk after pIC injection, stained with hematoxylin and eosin (H&E) (magnification 400×). (D) Flow cytometry after staining with pyronin Y and Hoechst 33342 for cell cycle analysis, with percentages of G0, G1, and S/G2/M indicated (*P < 0.03).
Bone marrow HSCs are characterized by the absence of lineage markers (Lin−) and expression of Sca-1 and c-Kit (LSK cells), and can be distinguished from myeloid progenitor cells, which do not express Sca-1 (LK cells). Progenitor cells were strikingly decreased in Gabpα-null bone marrow, yet the overall number of HSCs was preserved (9). Cell cycle activity of Gabpα-null HSCs was markedly reduced, and we observed a significant increase in G0 cells (Fig. 1D). In contrast, Yu, et al. described loss of HSCs and increased HSC cell cycle activity following conditional deletion of Gabpa in bone marrow (10). We developed an experimental strategy that enabled us to reconcile these divergent reports, by directly comparing Gabpα-null and Gabpα-replete bone marrow cells from the same mouse. We bred the ROSA26 loxP-STOP-loxP YFP transgene into Gabpafl/fl Mx1Cre mice, and induced Cre recombinase to simultaneously activate expression of yellow fluorescent protein (YFP) (by deletion of the upstream STOP codon) and delete Gabpa (Fig. S2A). This experimental approach permitted isolation of distinct populations of YFP+/Gabpα null HSCs and YFP−/Gabpα replete cells from bone marrow of single animals. One day after the second pIC injection, we sorted YFP+ and YFP− bone marrow cells and confirmed deletion of Gabpa in YFP+ HSCs and retention of the undeleted Gabpafl/fl in YFP− cells (Fig. S2B).
We sought to examine the effect of Gabpa disruption on stem and progenitor cells. We measured the percentage of HSCs and progenitor cells among lineage marker negative (Lin−) cells in the YFP+ and YFP− compartments. As expected in Gabpα-replete (or WT) bone marrow, the YFP− bone marrow pool contains more progenitor cells than HSCs (Fig. S2C; see also Fig. 3C and Fig. S4A). In contrast, the Gabpα-null YFP+ bone marrow pool exhibited a profound loss of progenitor cells and a relative increase in the percentage of HSCs. Disruption of Gabpa caused a significant reduction in cell cycle activity in HSCs (P < 0.01; Fig. S2D). Thus, we confirmed the profound loss of bone marrow progenitor cells, but preservation of primarily quiescent HSCs in Gabpα-null bone marrow.
Fig. 3.
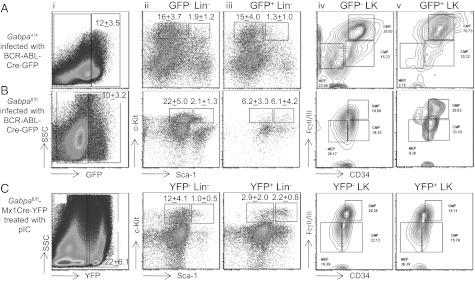
Cell cycle arrest in Gabpα-null HSCs blocks BCR-ABL–induced proliferation of HSCs and expansion of myeloid progenitor cells. Flow cytometry analysis of bone marrow cells from mice transplanted with BCR-ABL-Cre-GFP–infected Gabpa+/+ cells (A) or BCR-ABL-Cre-GFP–infected Gabpafl/fl cells (B). Gabpα-null (YFP+) bone marrow cells from mice that are Gabpafl/flMx1CreYFP after treatment with polyI:C are shown in C. Cell surface markers are as follows: (i) side scatter (SSC; y axis) vs. GFP or YFP (x axis), (ii and iii) c-Kit vs. Sca-1, and (iv and v) FcγRII/III vs. CD34 [Lin, lineage markers; LK, Lin−c-Kit+Sca1− (myeloid progenitors); LSK, Lin−c-Kit+Sca-1+ (HSCs).]
Gabp Is Essential for Development of CML.
In CML, transformation of hematopoietic cells by BCR-ABL increases cellular proliferation and causes massive expansion of the granulocyte pool. Transplantation of mice with bone marrow cells that express BCR-ABL recapitulates many aspects of CML (3). We previously defined LSCs as BCR-ABL–expressing HSCs (11). Because loss of Gabpα reduced HSC cell cycle activity, we sought to determine the effect of Gabpa deletion on development of BCR-ABL–transformed bone marrow. To delete Gabpa in BCR-ABL–expressing cells, we used a tricistronic retrovirus that expresses BCR-ABL, Cre recombinase, and green fluorescent protein (GFP) (12) to infect WT or Gabpafl/fl bone marrow before transplantation (Fig. 2A). Expression of GFP permits identification of cells that both express BCR-ABL and have undergone Cre-mediated deletion of floxed Gabpa. Control mice, i.e., mice transplanted with BCR-ABL-Cre-GFP–infected WT bone marrow, uniformly died with massive visceral infiltration by granulocytes (Fig. 2B). The survival curve for mice that were transplanted with Gabpafl/fl bone marrow was strikingly different. More than two thirds of this cohort of mice appeared healthy for at least 6 mo beyond transplantation, and never developed an MPN.
Fig. 2.
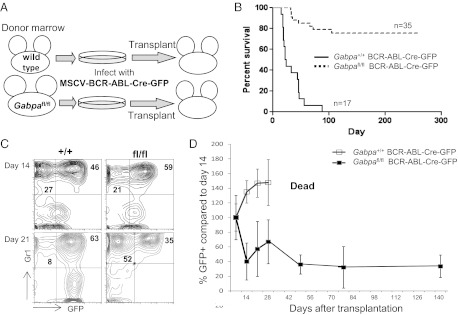
Gabpa deletion prevents development of CML, but production of BCR-ABL–expressing HSCs continues for months. (A) Schema for infecting bone marrow cells from Gabpafl/fl or WT donors with MSCV-BCR-ABL-Cre-GFP virus, followed by bone marrow transplantation into irradiated recipient mice, and (B) the resultant Kaplan–Meier survival curve. (C) Flow cytometry of peripheral blood for Gr1 and GFP expression at days 14 and 21 after transplantation with WT (+/+) or Gabpafl/fl (fl/fl) bone marrow; percentages of cells in relevant quadrants are indicated. (D) Chimerism analysis of GFP+ Gr1+ peripheral blood cells following bone marrow transplantation with BCR-ABL-Cre-GFP–infected WT (+/+) or Gabpafl/fl (fl/fl) cells; values (±SEM) for each individual mouse are expressed relative to the percentage of GFP+ cells in that mouse on day 14, arbitrarily set to 100% (n ≥ 3).
Peripheral blood from mice transplanted with BCR-ABL-Cre-GFP–infected bone marrow contains both GFP+ and GFP− granulocytes. GFP+ BCR-ABL+ granulocytes are derived from bone marrow cells that were infected with the retrovirus, whereas GFP− cells are derived from transplanted bone marrow cells that were not infected with the retrovirus. We examined peripheral blood from individual transplanted mice and directly compared GFP+ BCR-ABL+ granulocytes with GFP− cells in the same mouse. Fourteen days after transplantation of a control mouse with BCR-ABL-Cre-GFP–infected WT bone marrow, the peripheral blood contained twice the number of GFP+ BCR-ABL–expressing granulocytes as normal granulocytes (Fig. 2C), and, by day 21, GFP+ BCR-ABL–expressing cells dominated the peripheral blood, and the mouse died soon thereafter from the MPN.
Transplantation with retrovirus-infected Gabpafl/fl bone marrow yielded a very different pattern (Fig. 2C). Although GFP+ granulocytes predominated on day 14, by day 21, GFP+ cells represented only a minority of granulocytes in the peripheral blood. Indeed, we observed sustained and stable levels of GFP+ granulocytes in peripheral blood for months following transplantation (Fig. 2D). Because each mouse exhibited a distinct level of chimerism of GFP+ and GFP− cells, data for each mouse were expressed relative to its own level of chimerism on day 14 (arbitrarily set to 100%). We confirmed that peripheral blood GFP+ cells were Gabpα-null (Fig. S3A). We also confirmed that the GFP+ Gabpα-null bone marrow cells expressed BCR-ABL transcript mRNA that was comparable to the GFP+, Gabpα-undeleted cells (Fig. S3B). Thus, we observed continued and stable production of GFP+ BCR-ABL+ Gabpα-null granulocytes more than 4 mo after transplantation, without development of CML.
Six weeks after transplantation with BCR-ABL-Cre-GFP–infected Gabpafl/fl cells, bone marrow was retransplanted into secondary recipient mice. The majority of GFP+ cells in secondary recipients were granulocytes, but peripheral blood and bone marrow also contained small populations of GFP+ B and T lymphocytes and monocytes (Fig. S3C). Mice secondarily transplanted with primary BCR-ABL-Cre–transduced WT bone marrow cells develop CML-like disease (13); in contrast, secondary recipients of primary BCR-ABL-Cre–infected Gabpafl/fl cells did not develop CML, even after more than 4 mo of additional observation. The continued production of GFP+ BCR-ABL+ Gabpα-null granulocytes for months after primary transplantation and the generation of GFP+ cells in secondary recipients indicate that long term (LT) HSCs were the source of BCR-ABL+ Gabpα-null peripheral blood cells.
Reduced Cell Cycle Activity in Gabpα-Null Cells Prevents CML Development.
We characterized in greater detail the bone marrow of individual mice following transplantation with BCR-ABL-Cre-GFP–infected WT or Gabpafl/fl bone marrow, as demonstrated in Fig. 3. GFP− cells serve as controls in each mouse and, as expected in normal bone marrow, the GFP− population contained more progenitor cells than HSCs (Fig. 3 A, ii, and B, ii, and Fig. S4A), and comparable numbers of common myeloid progenitor, granulocyte-monocyte progenitor (GMP), and megakaryocyte-erythroid progenitor cells (Fig. 3 A, iv, and B, iv). Similarly, bone marrow of mice transplanted with BCR-ABL-Cre-GFP–infected WT cells exhibited more progenitors than HSCs (Fig. 3 A, iii) and a strong bias of progenitor cells toward GMP (Fig. 3 A, v). These BCR-ABL+ GFP+ WT HSCs showed a marked increase in the percentage of Ki-67+, active cycling cells compared with GFP− HSCs (P < 0.005; Fig. S4B). In contrast, bone marrow of mice transplanted with BCR-ABL-Cre-GFP–infected Gabpafl/fl cells revealed equalization of the progenitor-to-HSC ratio, indicating a significant loss of hematopoietic progenitor cells (Fig. 3 B, iii, and Fig. S4A), which was also seen in Gabpα-null YFP+ bone marrow (Fig. 3 C, iii, and Fig. S4A). The Gabpα-null BCR-ABL+ progenitor cells did not exhibit a bias toward GMP; instead, they showed comparable numbers of GMP, common myeloid progenitor, and megakaryocyte-erythroid progenitor cells (Fig. 3 B, v). These Gabpα-null BCR-ABL+ cells exhibited neither the excess proliferation seen in WT BCR-ABL+ cells nor the cell cycle arrest of Gabpα-null YFP+ cells (Fig. S4B). Furthermore, disruption of Gabpa alone only modestly increased apoptosis in BCR-ABL–transduced LSK cells, but imatinib treatment of Gabpα-null BCR-ABL+ LSK cells markedly increased apoptosis, compared with imatinib-treated Gabpα-undeleted BCR-ABL+ LSK cells (Fig. S4C). Thus, loss of Gabpα sensitizes BCR-ABL+ LSK cells to TKI-induced apoptosis. We conclude that Gabpa disruption reduced cell cycle activity in normal HSCs and in BCR-ABL–transformed HSCs, and thus prevented the rapid expansion of CML myeloid progenitor cells. The cell cycle arrest accounted for the failure to develop CML in Gabpα-null bone marrow.
PRKD2 Is an Essential and Direct Target of GABP in HSCs.
We used a bioinformatic approach to identify candidate targets of GABP that might mediate its effects on cell cycle activity in normal and in BCR-ABL–transformed HSCs. We compared gene expression profiles of BCR-ABL–transformed mouse HSCs to normal mouse HSCs and identified 2,154 genes that were up-regulated at least twofold (Fig. S5A) (13). Similarly, 4,412 genes were up-regulated at least twofold in human CD34+ CML cells compared with normal human CD34+ cells (14). A total of 595 genes were up-regulated in mouse and human CML data sets. Recently, Yu, et al. identified 8,383 genes that were bound by GABP in human CD34+ blood cells (10). Among the Gabpα-bound genes, 115 were among the 595 genes that were up-regulated in mouse and human CML. We particularly focused on CML-associated, GABP-bound genes that are involved in cellular signaling because BCR-ABL transformation is associated with activation of key signal transduction pathways.
PRKD2 is a serine-threonine protein kinase that transmits signals from diacylglycerol and PKC (15). PRKD2 is the major hematopoietic PRKD isoform. We found that PRKD2 is up-regulated in mouse and human CML data sets and is bound by GABP in human HSCs (Fig. S5B). PRKD2 has been identified as a susceptibility locus in chronic lymphocytic leukemia (16), but it has not been implicated in CML or in myeloid cell development. We measured Prkd2 expression in normal and Gabpα-null mouse HSCs by using the YFP selection strategy described in Fig. S2A. Prkd2 mRNA expression was reduced by 60% in Gabpα-null HSCs (P < 0.04), and 80% in Gabpα-null progenitor cells (P < 0.01; Fig. S5C). We conclude from chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq) and gene expression data that Prkd2 is a direct functional target of Gabp in mouse HSCs.
We sought to determine if PRKD2 functionally mediates Gabp effects in HSCs and in CML. In vitro colony formation in the absence of added cytokines is characteristic of BCR-ABL–transformed cells; untransformed hematopoietic cells fail to grow under these conditions. As expected, in the absence of added cytokines, WT bone marrow infected with murine stem cell virus (MSCV)-GFP control virus failed to generate in vitro colonies (Fig. 4A). As positive controls, WT bone marrow infected with BCR-ABL-Cre-GFP and Gabpafl/fl bone marrow infected with BCR-ABL-GFP (which lacks Cre) generated robust colony growth. However, Gabpafl/fl bone marrow infected with BCR-ABL-Cre-GFP yielded only rare colonies. Thus, despite expression of BCR-ABL, Gabpα-null cells failed to generate colonies in the absence of added cytokines.
Fig. 4.
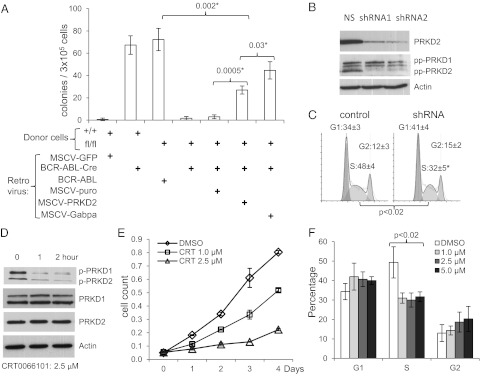
PRKD2 mediates cell cycle effects of BCR-ABL in CML cells. (A) In vitro colony forming assay (in the absence of added cytokines) of WT or Gabpafl/fl mouse bone marrow infected with the indicated viruses. (B and C) Knockdown of PRKD2 by lentiviral infection of K562 cells with scrambled (NS) or two different shRNAs directed against PRKD2, analyzed by (B) immunoblotting with antibodies against PRKD2, phospho-PRKD at S744 and S748 (pp-PRKD), or β-Actin; and (C) cell cycle analysis by propidium iodide staining. (D–F) K562 cells grown with indicated duration and concentration of CRT0066101, analyzed by (D) immunoblotting with antibodies against PRKD1, PRKD2, and phospho-PRKD at S916 (p-PRKD), (E) growth by MTT assay, and (F) cell cycle analysis.
To determine if PRKD2 could restore growth of Gabpα-null cells, we used a second “rescue” virus in this assay. As expected, coinfection with empty MSCV-puro virus failed to restore colony growth (Fig. 4A). Coinfection with MSCV-Gabpa restored colony growth to two thirds of the level of the positive controls. The failure of MSCV-Gabpa to fully restore colony growth of Gabpα-null cells (P < 0.002) indicates that not all Gabpa-deleted cells were successfully coinfected by the rescue virus. Coinfection with MSCV-PRKD2 significantly restored growth compared with MSCV-puro (P < 0.0005), achieved nearly 40% of the colony number of the positive controls, and was two thirds as effective in restoring in vitro colonies as MSCV-Gabpa itself (P < 0.03). We conclude that PRKD2 partially restored growth of Gabpα-null bone marrow cells and thus represents a functionally significant mediator of Gabp effects on BCR-ABL–transformed mouse HSCs.
To further examine the role of PRKD2 in cell cycle control of CML cells, we used shRNA knockdown of PRKD2 in BCR-ABL–expressing K562 erythroleukemia cells (Fig. 4B). PRKD2 protein was reduced by more than 80% by two different lentiviral shRNA constructs, compared with scrambled control shRNA. Although K562 cells express both PRKD1 and PRKD2, shRNA treatment reduced only PRKD2 expression. Knockdown of PRKD2 significantly reduced the percentage of K562 cells in S phase of the cell cycle (P < 0.02; Fig. 4C). Similarly, knockdown of Prkd2 in BCR-ABL–transformed primary mouse bone marrow (Fig. S6A) significantly reduced colony formation (P < 0.003; Fig. S6B), but did not affect normal primary mouse bone marrow (Fig. S6C).
PRKD2 transmits signals from diacylglycerol and protein kinase C, and its phosphorylation is necessary for activation of these pathways (15). The pyrazine benzamide CRT0066101 inhibits phosphorylation of all PRKD isoforms (17). Treatment of K562 cells with CRT0066101 reduced phosphorylation of both PRKD1 and PRKD2, but did not affect the protein level of either isoform (Fig. 4D). Culture of K562 cells in the presence of CRT0066101 caused a dose-dependent reduction of cell growth (Fig. 4E) and reduced the percentage of S phase cells (P < 0.02; Fig. 4F). Similarly, treatment of primary human CML cells with CRT0066101 reduced the percentage of S phase (Fig. S6 D and E) and increased the annexin V+ apoptotic cells, but did not affect apoptosis in normal human blood cells (Fig. S6F). We conclude that PRKD2 is a functionally significant mediator of the effect of GABP on proliferation and apoptosis of BCR-ABL–expressing cells.
Discussion
GABP is an ets transcription factor that plays essential roles in myeloid and lymphoid differentiation and in control of the cell cycle (4). We now show that disruption of mouse Gabpa caused pancytopenia as a result of sharply reduced HSC cell cycle activity and profound loss of hematopoietic progenitor cells. Gabpa disruption prevented development of CML from BCR-ABL–expressing mouse bone marrow, despite ongoing production of Gabpα-null BCR-ABL+ granulocytes. Thus, Gabpa disruption prevented leukemia development in this CML model and, conversely, BCR-ABL partially overcame the cell cycle arrest in HSCs associated with Gabpα loss. BCR-ABL+ HSCs were secondarily transplantable and contributed to all hematopoietic lineages in those recipients. We showed that expression of the serine-threonine kinase PRKD2 was decreased in Gabpα-null HSCs, knockdown and pharmacologic inhibition of PRKD2 reduced cell cycle activity of BCR-ABL transformed HSCs, and reexpression of PRKD2 partially rescued the growth defects of Gabpα-null bone marrow cells. We conclude that Gabp is required for HSC cell cycle activity and that loss of Gabpα blocked development of CML, in part through its previously unrecognized role in regulating the protein kinase PRKD2. Thus, deletion of Gabpa and expression of BCR-ABL in HSCs causes a functional standoff that balances cell cycle arrest with oncogene-induced hyperproliferation and thereby prevents development of leukemia.
Our findings regarding the loss of cell cycle activity in Gabpα-null HSCs differ from those of Yu et al. (10), who described increased HSC cell cycle activity and apoptosis following Gabpa deletion. How can these divergent findings be reconciled? Yu et al. first analyzed bone marrow more than 3 wk after initial Gabpa deletion, and, at that point in time, we found that bone marrow was overgrown by Gabpα-replete cells. It is unclear if their analysis reflected properties of Gabpα-null bone marrow cells alone or represented a mixed population of deleted and undeleted cells. Our present experimental strategy, which used activation of YFP expression to sort Gabpa-deleted and undeleted cells from the same mouse, unambiguously demonstrated that that Gabpα-null HSCs exhibit reduced cell cycle activity.
Mice that were transplanted with BCR-ABL-Cre-GFP–infected Gabpafl/fl bone marrow continued to generate GFP+ granulocytes cells for many months after transplantation. This indicates that LT-HSCs were targets of BCR-ABL-Cre-GFP retrovirus infection, because short-term stem cells do not survive beyond 4 mo. Transplantation into secondary recipients confirmed that, indeed, LT-HSCs were infected by the BCR-ABL retrovirus, and this conclusion was reinforced by the presence of GFP+ cells in granulocytes, monocytes, and B and T lymphocytes of secondary recipients. Despite infection of LT-HSCs, transplanted mice developed neither CML nor other leukemia.
PRKD2 is a member of a family of three serine-threonine kinases that are increasingly implicated in normal cellular functions and in malignant processes. PRKDs are effectors of diacylglycerol-regulated signal transduction pathways and are activated by phosphorylation through protein PKC-dependent and PKC-independent pathways. PRKDs are involved in DNA synthesis, gene expression, chromatin organization, cell survival, differentiation, and other essential cellular functions (15). Overexpression of PRKD1 or PRKD2 enhanced cell cycle progression and DNA synthesis in fibroblasts (18), and PRKD2 was identified as one of the susceptible loci in human chronic lymphocytic leukemia in a genome-wide association study (16). However, a role for PRKDs has not previously been demonstrated in HSCs, myeloid cells, or CML. Herein, we report PRKD2 as a mediator of GABP regulation of cell cycle control and its involvement in BCR-ABL transformation. CRT0066101 is a pyrazine benzamide compound that specifically blocks PRKD1 and PRKD2, but did not suppress PKCα, PKCβ, or PKCɛ; MAPK/ERK; c-Raf; c-Src; or c-Abl. CRT0066101 blocked growth of pancreatic cancer in vivo (17). Targeting multiple kinase pathways in LSCs is essential for improved treatment of BCR-ABL+ leukemia in mice (11), and CRT0066101 may offer a therapeutic opportunity in leukemia and other hematopoietic malignancies.
TKIs induce remissions in the large majority of patients with CML, but LSCs themselves are resistant to TKIs (19). We and others have described regulatory pathways that modify development of CML, and the growth and differentiation properties of LSCs. Some pathways, such as PTEN, act as suppressors of CML progression, whereas others, including HIF1α, are accelerators of CML (20). This suggests signaling pathways that might be manipulated to target CML LSCs despite their intrinsic resistance to TKIs. We previously identified a leukotriene pathway that is required by LSCs, but not by normal HSCs (13), and this pathway offers an opportunity to target LSCs without impairing normal hematopoiesis. Here we showed that Gabpa disruption prevented mouse CML development and that targeting GABP sensitized CML LSCs to imatinib-induced apoptosis, but GABP also plays essential roles in normal hematopoiesis and thus may not be an ideal target in the treatment of human CML. However, we identified PRKD2 as an essential mediator of the effects of GABP on CML cells. Knockdown or inhibition of PRKD2 reduced cell cycle activity and increased apoptosis in mouse or human primary CML cells, yet did not significantly affect normal mouse or human blood cells. Thus, PRKD2 may also offer a novel therapeutic target in CML, with the potential to suppress or eradicate LSCs.
In summary, we showed that Gabp is required for cell cycle activity of HSCs, and that conditional disruption of Gabpa prevented development of CML in a mouse model. Simultaneous expression of BCR-ABL and loss of Gabpα in HSCs resulted in a standoff in which cell cycle arrest associated with Gabpa deletion was overcome but leukemia failed to develop despite ongoing production of differentiated BCR-ABL+ cells. We identified PRKD2 as a mediator of Gabp in HSC cell cycle control, apoptosis, and of BCR-ABL signaling. This suggests that pharmacologic inhibition of PRKD2 may represent a valuable approach to targeting the LSCs in CML.
Materials and Methods
Animal studies were approved by the University of Massachusetts Institutional Animal Care and Use Committee. This use of human material is considered exempt according to University of Massachusetts Investigation Review Board. Mice and bone marrow transplantation, analysis of peripheral blood and mouse tissues, flow cytometry, cell sorting and cell cycle analysis, reverse transcription, real-time quantitative PCR and immunoblotting, cell culture, chemicals and retroviral gene transfer, and data analysis are described in detail in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Xuejun Zhu, Karen Drumea, and James Cormier for technical assistance and Jonathan Licht, Lucio Castilla, and Glen Raffel for helpful discussions. We thank Cancer Research Technology Discovery Laboratories, Wolfson Institute for Biomedical Research, London WC1E 6BT, United Kingdom, for providing CRT0066101. This work was supported by National Institutes of Health Grants R01 HL073945 (to A.G.R.), R01 CA122142 (to S.L.), CA 114199 (to S.L.), and GM 033977 (to M.R.G.). M.R.G. is an investigator of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212904110/-/DCSupplemental.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: An evolving paradigm for stem cell biology. Cell. 2008;132(4):631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talpaz M, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Ilaria RL, Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189(9):1399–1412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosmarin AG, Resendes KK, Yang Z, McMillan JN, Fleming SL. GA-binding protein transcription factor: A review of GABP as an integrator of intracellular signaling and protein-protein interactions. Blood Cells Mol Dis. 2004;32(1):143–154. doi: 10.1016/j.bcmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Xue HH, et al. GA binding protein regulates interleukin 7 receptor alpha-chain gene expression in T cells. Nat Immunol. 2004;5(10):1036–1044. doi: 10.1038/ni1117. [DOI] [PubMed] [Google Scholar]
- 6.Xue HH, et al. The transcription factor GABP is a critical regulator of B lymphocyte development. Immunity. 2007;26(4):421–431. doi: 10.1016/j.immuni.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Ristevski S, et al. The ETS transcription factor GABPalpha is essential for early embryogenesis. Mol Cell Biol. 2004;24(13):5844–5849. doi: 10.1128/MCB.24.13.5844-5849.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang ZF, Mott S, Rosmarin AG. The Ets transcription factor GABP is required for cell-cycle progression. Nat Cell Biol. 2007;9(3):339–346. doi: 10.1038/ncb1548. [DOI] [PubMed] [Google Scholar]
- 9.Yang ZF, et al. GABP transcription factor is required for myeloid differentiation, in part, through its control of Gfi-1 expression. Blood. 2011;118(8):2243–2253. doi: 10.1182/blood-2010-07-298802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu S, et al. GABP controls a critical transcription regulatory module that is essential for maintenance and differentiation of hematopoietic stem/progenitor cells. Blood. 2011;117(7):2166–2178. doi: 10.1182/blood-2010-09-306563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y, et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci USA. 2006;103(45):16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng C, et al. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood. 2010;115(3):626–635. doi: 10.1182/blood-2009-06-228130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41(7):783–792. doi: 10.1038/ng.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radich JP, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA. 2006;103(8):2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rozengurt E. Protein kinase D signaling: Multiple biological functions in health and disease. Physiology (Bethesda) 2011;26(1):23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Bernardo MC, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2008;40(10):1204–1210. doi: 10.1038/ng.219. [DOI] [PubMed] [Google Scholar]
- 17.Harikumar KB, et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2010;9(5):1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. and erratum (2010) 9(7):2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem. 2004;279(16):16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Peng C, Li D, Li S. Molecular and cellular bases of chronic myeloid leukemia. Protein Cell. 2010;1(2):124–132. doi: 10.1007/s13238-010-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119(11):2595–2607. doi: 10.1182/blood-2011-10-387381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.