

Abstract
Background
Graft-versus-host disease (GVHD) is a major barrier to successful allogeneic hematopoietic stem-cell transplantation (HSCT). The chemokine receptor CCR5 appears to play a role in alloreactivity. We tested whether CCR5 blockade would be safe and limit GVHD in humans.
Methods
We tested the in vitro effect of the CCR5 antagonist maraviroc on lymphocyte function and chemotaxis. We then enrolled 38 high-risk patients in a single-group phase 1 and 2 study of reduced-intensity allogeneic HSCT that combined maraviroc with standard GVHD prophylaxis.
Results
Maraviroc inhibited CCR5 internalization and lymphocyte chemotaxis in vitro without impairing T-cell function or formation of hematopoietic-cell colonies. In 35 patients who could be evaluated, the cumulative incidence rate (±SE) of grade II to IV acute GVHD was low at 14.7±6.2% on day 100 and 23.6±7.4% on day 180. Acute liver and gut GVHD were not observed before day 100 and remained uncommon before day 180, resulting in a low cumulative incidence of grade III or IV GVHD on day 180 (5.9±4.1%). The 1-year rate of death that was not preceded by disease relapse was 11.7±5.6% without excessive rates of relapse or infection. Serum from patients receiving maraviroc prevented CCR5 internalization by CCL5 and blocked T-cell chemotaxis in vitro, providing evidence of antichemotactic activity.
Conclusions
In this study, inhibition of lymphocyte trafficking was a specific and potentially effective new strategy to prevent visceral acute GVHD. (Funded by Pfizer and others; ClinicalTrials.gov number, NCT00948753.)
Acute graft-versus-host disease (GVHD) is a major cause of death and complications after allogeneic hematopoietic stem-cell transplantation (HSCT). The condition occurs in 30 to 50% of patients receiving HLA-matched transplants from a related donor and in 50 to 70% of those receiving transplants from an unrelated donor.1 The pathogenesis of GVHD is multifactorial, but ultimately, donor-derived T cells recognize recipient antigens as foreign, resulting in activation, expansion, and cytokine release and leading to destruction of host tissues.2 Current therapies for GVHD target T cells and cytokines, often antagonize T-cell–mediated graft-versus-tumor responses, and delay immune reconstitution.3 Preventing GVHD without intensive immune suppression would represent a major advance for HSCT recipients.
Recruitment of lymphocytes into tissues plays a major role in GVHD.4 This process starts early after transplantation, allowing T cells to interact with antigen-presenting cells of host and donor origin.5-7 The organ-specific fate of lymphocytes is determined by a combination of adhesion molecules and chemokines that interact with specific receptors on the lymphocyte.8
Chemokine (C-C motif) receptor 5 (CCR5) and its natural ligands CCL3, CCL4, and CCL59 have been implicated in the pathogenesis of GVHD and solid-organ rejection.10,11 CCR5 is critical for lymphocyte recruitment to tissues involved in GVHD,12-15 and in certain murine models, the migration of CCR5+CD8+ cells into the liver and gut is markedly reduced by the use of an anti-CCR5 antibody, resulting in protection against GVHD.16,17 Although studies in mice that are genetically deficient for Ccr5 have shown conflicting results,15,17-19 genetic evidence in humans shows that certain CCR5 polymorphisms are protective against GVHD20,21 and correlate with survival in patients undergoing allogeneic HSCT.22
Maraviroc (Selzentry, Pfizer and ViiV Healthcare) is the first drug in the class of CCR5 antagonists. It is a noncompetitive, slowly reversible small-molecule antagonist that prevents signaling by all three ligands23 and is used as part of combination antiretroviral therapy for patients infected with CCR5-tropic human immunodeficiency virus (HIV) (i.e., a subtype that uses only the CCR5 co-receptor to enter cells).24,25 The effects of maraviroc on chemotaxis and alloreactivity have not yet been explored. We hypothesized that CCR5 blockade with maraviroc early after allogeneic HSCT might inhibit lymphocyte trafficking and decrease the incidence of acute GVHD.
Methods
In Vitro Evaluation Of Maraviroc
We determined the effects of maraviroc on the internalization of CCR5, CCR2, and CCR1 on T-cell subsets and on T-cell chemotaxis in normal donors and donors who were homozygous for the CCR5 Δ32 allele (CCR5-Δ32) and thus were CCR5-deficient. We also assessed the effects of maraviroc on functional T-cell assays and hematopoietic colony formation. Detailed methods are described in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Patients and Treatment
We performed a phase 1 and 2 single-group clinical trial to study the role of maraviroc when it was added to conventional GVHD prophylaxis after reduced-intensity conditioned HSCT for patients with hematologic cancers. The phase 1 portion was designed to confirm the safe dose level that was established in patients with HIV and to confirm consistent target drug levels in vivo. This dose was then used in the phase 2 portion of the study. Study participants included adult candidates for HSCT who met protocol eligibility criteria. We calculated the HSCT comorbidity index for each patient, as described previously.26
From June 2009 through March 2011, we enrolled 38 patients. All eligible patients at our institution were offered the chance to participate. Enrollment was held during the analysis of safety and pharmacokinetic data in the phase 1 portion. All patients received a uniform conditioning regimen of intravenous fludarabine (120 mg per square meter of body-surface area) and busulfan (6.4 mg per kilogram of body weight), followed by the infusion of granulocyte colony-stimulating factor–mobilized peripheral-blood stem cells from either a related or an unrelated donor. A single-antigen HLA mismatch was allowed. We determined the amount of CD34+ and CD3+ cells in the graft using standard procedures.27
All patients received standard GVHD prophylaxis with oral tacrolimus (0.06 mg per kilogram per day) in two divided doses starting 2 days before transplantation and intravenous methotrexate (15 mg per square meter on day 1 and 10 mg per square meter on days 3, 6, and 11). The dose of tacrolimus was adjusted to a target trough level of 5 to 15 ng per milliliter. All patients received antimicrobial prophylaxis, which typically included voriconazole and acyclovir starting 2 days before transplantation and trimethoprim–sulfamethoxazole starting at the time of engraftment.
Maraviroc was given orally twice daily starting 2 days before transplantation until day 30. Administration was suspended in cases of toxic effects that investigators considered to be related to the study drug or in cases of severe mucositis. Planned infusions of donor lymphocytes were not allowed. Donor lymphocyte infusion or chemotherapy could be administered to treat relapse or decreased chimerism levels at the discretion of the treating physician.
End Points
Clinical Outcomes
Primary end points were safety and the cumulative incidence of grade II to IV acute GVHD by day 100. Secondary end points included rates of grade III or IV acute GVHD by day 100 and day 180, organ-specific acute GVHD, moderate-to-severe chronic GVHD, disease relapse, death that was not preceded by disease relapse (non–relapse-related death), and overall survival. We used the Consensus Conference Criteria28 and the National Institutes of Health Consensus Development Project Criteria29 for the grading of acute and chronic GVHD, respectively. We monitored hematopoietic engraftment using molecular chimerism studies (see the Methods section in the Supplementary Appendix). We used the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0, to analyze safety.
Pharmacodynamic Outcomes
To determine the inhibitory capacity of patients' serum on CCR5 internalization, we used a modified internalization assay (see the Supplementary Appendix).30 In order to show the ex vivo effect on chemotaxis, we tested the capacity of patient serum to inhibit chemotaxis (see the Supplementary Appendix).
Study Oversight
The study was approved by the institutional review board at the University of Pennsylvania, and patients provided written informed consent. Independent oversight was provided by a medical monitor and the clinical trials review committee at the Abramson Cancer Center at the University of Pennsylvania. The study was conducted in adherence to the protocol, available at NEJM.org. Authors' contributions are detailed in the Supplementary Appendix. Pfizer provided the experimental drug, partial research support, and assistance in the interpretation of pharmacokinetic data without a data confidentiality agreement. Pfizer played no additional role in the analysis of the data or in writing or approval of the manuscript. All the authors vouch for the accuracy and completeness of the data and the fidelity of the study to the protocol.
Statistical Analysis
We performed a cumulative incidence analysis to estimate the cumulative risk of acute GVHD, chronic GVHD, relapse, and death not preceded by relapse. We used the Kaplan–Meier method to estimate the probability of survival. Death and donor lymphocyte infusions were considered to be competing risks for GVHD and death not preceded by relapse. Cumulative incidence and survival analyses were conducted in R (R Project for Statistical Computing at www.r-project.org). We used Student's t-test or analysis of variance to perform group comparisons of results on flow cytometry and chemotaxis ratios using Stata software, version 11.1.
Results
In Vitro Effects of Maraviroc
Maraviroc inhibited CCL5- or CCL3-driven internalization of CCR5 at concentrations as low as 10 nM (Fig. 1A and 1B), an effect that was seen on effector CD8+ and CD4+ T cells but not on naive (CCR5-negative) CD8+ and CD4+ T cells. In contrast, maraviroc did not block CCL5- or CCL2-driven internalization of the structurally related receptors, CCR1 and CCR2 (Fig. 1C).
Figure 1 (facing page). Specific Inhibition of CCL5- and CCL3-Induced Internalization of CCR5 and Inhibition of T-Cell Chemotaxis Associated with Maraviroc.
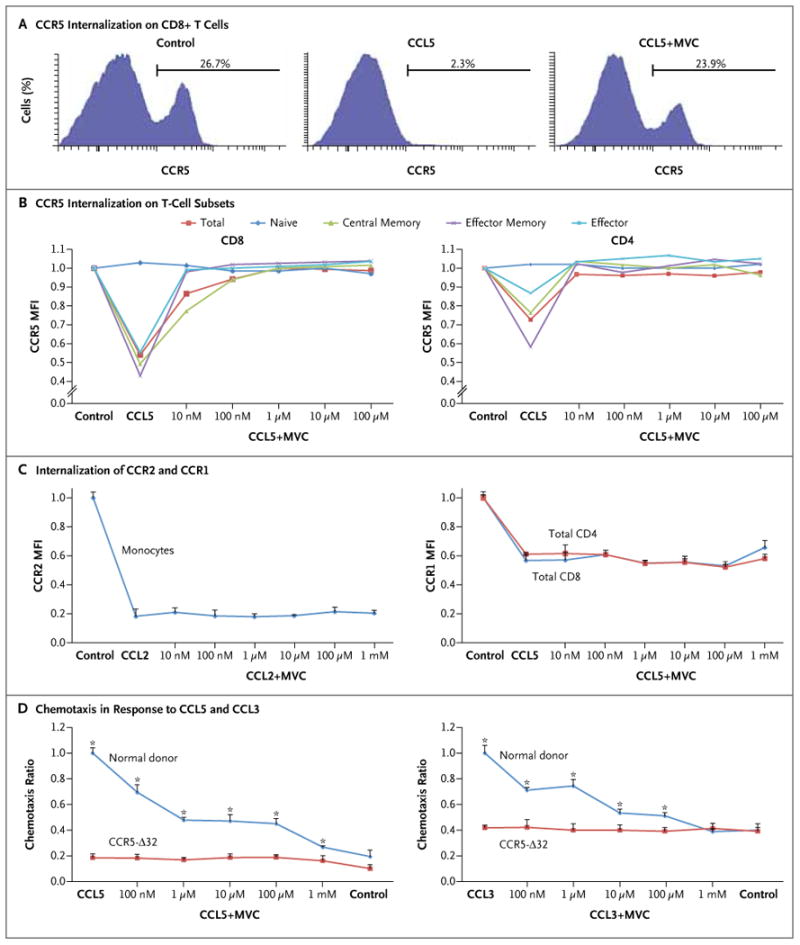
In Panel A, a representative flow-cytometric analysis shows a reduction in surface expression of CCR5 on normal donor CD8+ T cells after incubation with 100 nM of CCL5 for 30 minutes. Preincubation with 1 μM of maraviroc (MVC) for 30 minutes fully abrogated internalization of the receptor. Panel B shows the effect of various concentrations of maraviroc on CCL5-induced internalization of T cells. The mean fluorescence intensity (MFI) of CCR5 was measured by means of flow cytometry after incubation with escalating concentrations of maraviroc and then incubation with 100 nM of CCL5 or CCL3. The surface expression of CD4, CD8, CD45RA, and CCR7 was used to define T-cell subsets. Plots represent means of three different experiments. All T-cell subsets other than naive CD4+ and CD8+ cells showed a significant response to CCL5 and efficient inhibition by maraviroc (P<0.01 by analysis of variance for central memory, effector memory, and effector cells; P>0.05 for naive cells). Similar results were observed with CCL3 (not shown). Panel C shows the lack of effect of maraviroc on CCL5-induced internalization of CCR1 on T cells and CCL2-induced internalization of CCR2 on monocytes (P>0.05 by analysis of variance). Panel D shows the results of chemotaxis assays in the presence of escalating concentrations of maraviroc. Peripheral-blood mononuclear cells from five normal donors and from three donors who were homozygous for the CCR5 Δ32 allele (CCR5-Δ32) were preincubated with maraviroc and then allowed to migrate in a Boyden chamber in response to 100 nM of CCL5 (left panel) or 0.5 nM of CCL3 (right panel) for 3 hours. Migrating cells were stained with surface markers and counted by means of flow cytometry. Chemotaxis ratios represent the number of migrating cells in each experimental condition divided by the number of cells in the positive control sample (chemokine without maraviroc). Means and standard errors (represented by T bars) are shown. P values represent a two-sided t-test between normal donor cells and Δ32 cells for each condition. Asterisks indicate P<0.01.
We tested the effect of maraviroc on T-cell chemotaxis by measuring the migration of lymphocytes from normal donors and CCR5-Δ32 homozygous donors (Fig. 1D). Although Δ32 cells were unresponsive to CCL5- and CCL3-induced chemo-taxis and were unaffected by maraviroc, the drug caused dose-dependent inhibition of chemotaxis of normal donor lymphocytes.
To determine whether CCR5 blockade affected T-cell function, we performed a series of functional assays. Pharmacologic concentrations of maraviroc (0.5 to 5.0 μM) had no effect on T-cell proliferation and cytokine secretion in response to stimulation by cognate viral peptide (cytomegalovirus). Moreover, specific T-cell cytotoxicity against cytomegalovirus peptide-loaded T2 cells was not impaired (Fig. 1A, 1B, and 1C in the Supplementary Appendix). To investigate the effect of maraviroc on hematopoiesis, fresh peripheral-blood mononuclear cells from normal donors were plated in methylcellulose in the presence of appropriate cytokines; formation of myeloid and erythroid colonies was not significantly affected by the presence of maraviroc (Fig. 1D in the Supplementary Appendix).
CCR5 Blockade in Allogeneic HSCT
We added maraviroc to standard GVHD prophylaxis in 38 patients undergoing reduced-intensity conditioned HSCT to assess safety and the effect on the incidence and severity of acute GVHD. A majority of the patients had high-risk features in terms of age, donor–recipient HLA compatibility, and underlying diseases (Table 1). The median follow-up was 19.6 months (range, 13.8 to 34.8).
Table 1. Characteristics of the 38 Patients at Baseline.
| Variable | Value |
|---|---|
| Dose of maraviroc — no. | |
| 150 mg twice daily | 7 |
| 300 mg twice daily | 31 |
| Recipient age | |
| Median — yr | 62 |
| Range — yr | 21–74 |
| >60 yr — no. (%) | 26 (68) |
| Recipient sex — % | |
| Male | 60 |
| Female | 40 |
| Comorbidity index — no. (%) | |
| Low | 21 (55) |
| Intermediate | 13 (34) |
| High | 4 (11) |
| Diagnosis — no. | |
| Acute myeloid leukemia | 15 |
| Myelodysplastic syndromes | 6 |
| Non-Hodgkin's lymphoma | 8 |
| Myeloproliferative disorder or myelofibrosis | 4 |
| Chronic lymphocytic leukemia | 1 |
| Aplastic anemia | 1 |
| Multiple myeloma | 1 |
| Hodgkin's lymphoma | 1 |
| Chronic myeloid leukemia (blast crisis | 1 |
| Donor — no. (%) | |
| Matched related | 13 (34) |
| Matched unrelated | 19 (50) |
| Single-antigen mismatched | 6 (16) |
| Donor age — yr | |
| Median | 38 |
| Range | 20–68 |
| Cytomegalovirus status — no. (%) | |
| Recipient or donor positive (or both | 22 (58) |
| Recipient and donor negative | 16 (42) |
| CD34+ cells/kg ×10−6 — median (range) | 6.6 (1.2–16.1) |
| CD3+ cells/kg ×10−8 — median (range) | 1.9 (0.6–5.4) |
We obtained detailed pharmacokinetic profiles for the first 13 patients at two dose levels (300 mg twice daily in 6 patients and 150 mg twice daily in 7 patients) (Fig. 2 and the Methods section in the Supplementary Appendix). Among patients receiving 150 mg twice daily, 3 of 7 patients did not reach a target level of 100 ng per milliliter, whereas among those receiving 300 mg twice daily, all 6 patients reached the target level. In view of the pharmacokinetic data and the lack of significant drug-related toxicity, the 300-mg twice-daily dose was chosen as the phase 2 dose.
Maraviroc had few documented toxic effects. Administration of the drug was briefly suspended in seven patients because of grade 3 abnormalities on liver-function testing (in two patients) or grade 3 or 4 mucositis (in five). Liver-function abnormalities did not recur when the drug was restarted. The adverse-event profile was similar to the expected toxicity observed in patients undergoing reduced-intensity conditioned HSCT without maraviroc (Table 1 in the Supplementary Appendix).
Clinical Outcomes
Engraftment
Engraftment in recipients of maraviroc was prompt. One patient died from sepsis on day 12 and thus could not be evaluated. In patients who could be evaluated, the median time to neutrophil engraftment (absolute neutrophil count, >500 per cubic millimeter) was 15 days (range, 10 to 27), and the median time to achieve a platelet count of more than 20,000 per cubic millimeter without transfusions was 19 days (range, 9 to 84). One patient continued to require platelet transfusions because of an early relapse of lymphoma. The median percentages of chimerism in whole-blood and T cells at day 100 were 96% (range, 0 to 100) and 81% (range, 0 to 100), respectively (Fig. 3 in the Supplementary Appendix). Patterns of donor chimerism were similar to those in our past experience with HSCT. Three patients had secondary graft losses on days 56, 149, and 331: two cases that were coincident with progressive myelofibrosis and one in a patient with chronic lymphocytic leukemia (CLL) who had autologous recovery of normal hematopoiesis without evidence of CLL.
GVHD
Three patients who received low-dose maraviroc did not reach a predetermined pharmacokinetic target and were excluded from the efficacy analysis. Among 35 patients who could be evaluated, the cumulative incidence rate (±SE) of GVHD at day 100 was 14.7±6.2% for grade II to IV acute disease (the primary end point) and 2.9±2.9% for grade III or IV disease (Table 2 and Fig. 2A). In the first 100 days, there were no cases of acute GVHD involving the liver or gut (Table 2 and Fig. 2C). At day 180, the cumulative incidence of grade II to IV acute GVHD was 23.6±7.4%, and organ involvement remained largely confined to the skin, with low rates of GVHD involving the liver (2.9±2.9%) or gut (8.8±5.0%). At day 180, the cumulative incidence of grade III or IV GVHD was only 5.9±4.1%, largely attributable to low incidence rates of gut and liver GVHD, which were absent before day 100 and remained infrequent up to day 180 (Table 2 and Fig. 2C). In the 11 evaluable patients who received an HLA-matched sibling graft, there were no cases of acute GVHD before day 100 and no grade III or IV GVHD by day 180.
Table 2. Cumulative Incidence of Graft-versus-Host Disease (GVHD)*.
| Variable | Incidence | ||
|---|---|---|---|
| Day 100 | Day 180 percent |
1 Year | |
|
| |||
| Acute GVHD | |||
|
| |||
| Grade II to IV | 14.7±6.2 | 23.6±7.4 | 29.4±8.0 |
|
| |||
| Grade III or IV | 2.9±2.9 | 5.9±4.1 | 14.7±6.2 |
|
| |||
| Liver | 0 | 2.9±2.9 | 8.8±5.0 |
|
| |||
| Gut | 0 | 8.8±5.0 | 8.8±5.0 |
|
| |||
| Skin | 14.7±6.2 | 26.5±7.7 | 29.4±8.0 |
|
| |||
| Chronic GVHD | |||
|
| |||
| Moderate-to-severe | 0 | 2.9±3.0 | 23.6±7.5 |
Plus-minus values are means ±SE.
Figure 2. Clinical Trial Outcomes of Acute GVHD, Moderate-to-Severe Chronic GVHD, and Organ-Specific Acute GVHD.

Shown are cumulative incidence plots of grade II to IV and grade III or IV acute GVHD (Panel A), moderate-to-severe chronic GVHD (Panel B), and organ-specific acute GVHD (in the skin, liver, and gut) (Panel C) in 35 patients undergoing reduced-intensity conditioned hematopoietic stem-cell transplantation with maraviroc added to standard GVHD prophylaxis.
The cumulative incidence of moderate-to-severe chronic GVHD was 23.6±7.5% at 1 year (Table 2 and Fig. 2B). In 10 patients with moderate-to-severe chronic GVHD, organ involvement included skin (in 7 patients), mouth (in 5), eyes (in 4), lungs (in 4), liver (in 3), and gut (in 2). Ten donor lymphocyte infusions were administered to 9 patients to treat dropping chimerism levels (in 3 patients) or relapse (in 7 patients) between days 161 and 433. Immunosuppressive therapy or additional maraviroc were not administered to these patients. After infusions of donor lymphocytes, grade IV acute GVHD developed in 2 patients, and severe chronic GVHD developed in 1 patient.
Death, Infection, Relapse, and Survival
The cumulative incidence of death not preceded by relapse at day 180 was 2.9±2.9%. At 1 year, the cumulative rate of death not preceded by relapse was 11.7±5.6%. Infectious complications in the first 100 days included several cases of bacterial infections at a rate that was expected after reduced-intensity conditioned HSCT (Table 2 in the Supplementary Appendix). There were no viral or fungal infections other than asymptomatic reactivation of cytomegalovirus.
At 1 year, the cumulative incidence of relapse in recipients of maraviroc was 55.9±8.8% which was not higher than expected considering the disease characteristics of the patients and the reduced-intensity conditioning regimen, which is associated with a high risk of relapse. A subset analysis of patients with acute myeloid leukemia (AML) or the myelodysplastic syndromes and lymphoid cancers did not reveal any significant differences in relapse rates (data not shown). The estimated 2-year survival rate was 47.1±8.6%.
Inhibition of Lymphocyte Chemotaxis
To ensure that the antichemotactic effect of maraviroc was preserved in vivo, we tested the capacity of serum from treated patients to inhibit CCR5 internalization and chemotaxis. Serum samples from multiple time points during steady state on day 12 abrogated CCL5-induced internalization of CCR5 on normal donor T cells (Fig. 3A and 3C). As a control, we tested each patient's serum at 60 days (30 days after the last dose of maraviroc) and observed full recovery of CCL5-induced CCR5 internalization at that time point.
Figure 3 (facing page). Inhibition of CCL5-Induced CCR5 Internalization and CCL5-Induced T-Cell Chemotaxis by Serum from Patients Receiving Maraviroc.
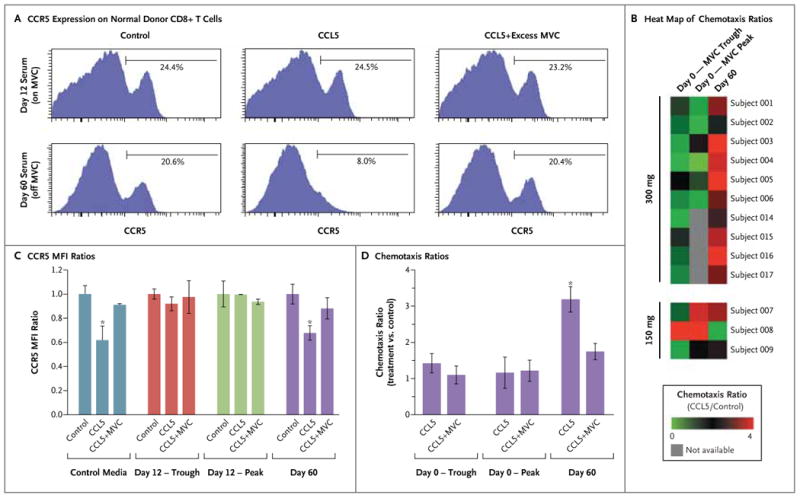
Panels A and C show the loss of responsiveness of CD8+ T cells to CCL5 after incubation with serum obtained from patients receiving maraviroc (MVC) at a dose of 300 mg twice daily on day 12 after transplantation; serum obtained on day 60 (30 days after the last dose of maraviroc) still allowed the cells to respond to CCL5. Panel A includes representative results on flow cytometry that show the expression of CCR5 on normal donor CD8+ T cells after incubation with serum for 1 hour and stimulation by 100 nM of CCL5, 100 nM of CCL5 plus excess maraviroc (1 mM), or control media. CCR5 internalization was reversed by adding excess maraviroc to the day 60 sample but not to the day 12 sample. Panel C shows the ratios of mean fluorescent intensities (MFI) of CCR5 on normal donor CD8+ T cells, expressed as the MFI ratio between each experimental condition and its media control. Serum obtained from six patients on day 12 at trough levels (before drug administration) or peak levels (3 or 4 hours after administration) abrogated the internalization of CCR5 by CCL5. Serum obtained on day 60 and control media did not have an inhibitory effect on the internalization of CCR5 by CCL5, which was efficiently blocked by adding excess maraviroc (1 mM). The asterisks indicate P<0.01 in a paired two-sided t-test. Similar results were seen with CD4+ cells. Panels B and D show the results of chemotaxis experiments in which normal donor peripheral-blood mononuclear cells were suspended for 1 hour in serum from patients obtained on day 0 (maraviroc trough or peak levels) and on day 60 (off maraviroc). The cells were then allowed to migrate in response to 100 nM of CCL5 or control media. Chemotaxis ratios represent the number of migrated CD3+ cells with CCL5 stimulation divided by the number of cells in the control sample. Panel B shows a heat map of chemotaxis ratios on day 0 and day 60, showing consistent inhibition of CCL5-induced chemotaxis in patients receiving maraviroc (at a dose of 300 mg twice daily). Panel D shows the chemotaxis ratios for the first six patients who received maraviroc, showing inhibition of CCL5-induced chemo-taxis at maraviroc peak and trough levels, as compared with those on day 60. The asterisk indicates P<0.01 in a paired two-sided t-test. The addition of 1 mM of maraviroc decreased the chemotaxis ratio only on day 60. The I bars indicate standard errors.
Next, we tested the capacity of serum from treated patients obtained on day 0 to inhibit in vitro lymphocyte chemotaxis. By incubating normal donor T cells in patients' serum and then allowing the cells to migrate in a Boyden chamber, we found that CCL5-induced chemotaxis was significantly impaired by serum obtained on day 0 as compared with day 60 (Fig. 3B and 3D).
Discussion
In this study, we found that maraviroc was biologically active as an inhibitor of lymphocyte chemotaxis both in vitro and in vivo. The addition of maraviroc to standard GVHD prophylaxis resulted in a low incidence of GVHD in high-risk patients with hematologic cancers after allogeneic HSCT. Although cases of skin GVHD were still observed at expected rates, the addition of maraviroc was associated with an absence of liver and gut GVHD through day 100, leading to a low incidence of severe (grade III or IV) GVHD. CCR5 blockade did not disrupt hematopoietic engraftment or lead to a higher-than-expected incidence of relapse or infectious complications.
The outcomes of this study are especially favorable considering the study population, which included older patients (68% over the age of 60 years) and a high proportion of matched unrelated donors (50%) and HLA-mismatched donors (16%); the anticipated incidence of acute GVHD in similar patients is typically more than 50%.31-33 The patients in our study also had major coexisting illnesses, with an intermediate or high co-morbidity index26 in almost half the patients. At our institution, by day 180 among patients receiving tacrolimus and methotrexate for prophylaxis after reduced-intensity conditioned HSCT, the rates of acute GVHD are 38.5% for grade II to IV disease and 21.9% for grade III or IV disease. During the same time period in our study, the use of a combination of maraviroc, tacrolimus, and methotrexate resulted in cumulative incidence rates of 23.6% for grade II to IV disease and 5.9% for grade III or IV disease. Results of this study also compare favorably with other studies of reduced-intensity conditioned HSCT that used similar conditioning regimens.34-41 For example, in a recent report from the Center for International Blood and Marrow Transplant Research involving 1174 patients who underwent reduced-intensity conditioned HSCT with a peripheral-blood stem-cell graft, by day 100 moderate-to-severe acute GVHD occurred in 35.0% of patients receiving matched related-donor transplants and 57.0% of patients receiving unrelated-donor transplants.41 In a similar study, which focused on patients with AML or the myelodysplastic syndromes, grade II to IV acute GVHD occurred in 46.0% of patients undergoing reduced-intensity conditioned HSCT.42 The inclusion of acute GVHD only through day 100 in these large studies probably led to an underestimation of the true incidence of acute GVHD, which is frequently delayed after reduced-intensity conditioned HSCT.37 Of course, the value of maraviroc in lowering the rate of acute GVHD will need to be assessed in a prospective, randomized trial.
The prevention of primarily visceral GVHD is consistent with murine models in which a blocking CCR5 antibody ameliorated GVHD of the liver and gut.16,17 From a clinical standpoint, liver and gut involvement is reported in more than 50% of patients with GVHD43 and frequently determines severity, which itself predicts response to therapy and survival.44 Our own experience with reduced-intensity conditioned HSCT involving similar patients receiving the same conditioning regimen and tacrolimus plus methotrexate as GVHD prophylaxis has shown that among patients with acute GVHD, 44.0% had liver involvement and 39.0% had gut involvement (unpublished data). Although it is unclear why CCR5 blockade would limit visceral GVHD specifically, murine models suggest that tissue distribution may be related to the intensity of responses in type 1, type 2, and type 17 helper T cells.45-47 It is possible that organspecific protection is related to the preferential expression of CCR5 on type 1 helper T cells.48 Other chemokine-signaling mechanisms may be involved in skin infiltration of lymphocytes.
The administration of maraviroc only until day 30 after transplantation resulted in low rates of GVHD up to 6 months after transplantation. Data from murine models suggest that interactions between T cells and antigen-presenting cells that set the stage for GVHD occur early after HSCT.6 It is therefore possible that early and brief CCR5 blockade is sufficient to reduce subsequent GVHD. Conversely, the emerging cases of acute and chronic GVHD after the first 6 months suggest that a longer treatment course of maraviroc may have a beneficial effect.
In summary, a 33-day course of maraviroc, an inhibitor of T-cell chemotaxis, appeared to reduce the incidence of acute visceral GVHD without major adverse events.
Supplementary Material
Acknowledgments
Supported by Pfizer; a Specialized Center of Research grant (to Drs. Porter and Vonderheide) and a Special Fellow in Clinical Research Award (to Dr. Reshef) from the Leukemia and Lym-phoma Society; grants (P30-CA16520, to Drs. Vonderheide, Stadtmauer, and Heitjan and Ms. Mick; K24-CA117879, to Dr. Porter; and U01-HL069286, to Drs. Porter and Stadtmauer) from the National Institutes of Health; a Pilot Project Grant from the Abramson Cancer Center (to Drs. Reshef, Vonderheide, and Porter); and an American Society of Hematology Scholar Award and an American Society of Clinical Oncology Young Investigator Award (both to Dr. Reshef). Dr. Reshef is a fellow of the Institute for Translational Medicine and Therapeutics, University of Pennsylvania.
We thank Marcelo Pasquini of the Center for International Blood and Marrow Transplant Research and Hernan Valdez and Manoli Vourvahis of Pfizer for their helpful discussions; Oren Litvin of Columbia University for help with preparation of the figures; Grace Jeschke for help with colony-formation assays; Amy Marshall and Kristin Coffan for regulatory support; and Pavel Vassilev for data management.
Footnotes
Presented in part at the 2010 American Society of Hematology Annual Meeting, Orlando, FL, December 4–7, 2010; the 2011 American Society of Hematology Annual Meeting, San Diego, CA, December 10–13, 2011; and the 2011 Blood and Marrow Transplantation Tandem Meeting, Honolulu, February 17–21, 2011
Dr. Frey reports receiving lecture fees from Pfizer; Dr. Stadt-mauer, consulting fees from Pfizer; Dr. Vonderheide, grant support from Pfizer; and Dr. Porter, consulting fees from Bristol-Myers Squibb and lecture fees from Celgene and Millennium. No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org
Contributor Information
Ran Reshef, The Abramson Cancer Center and the Division of Hematology and Oncology, the Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Selina M. Luger, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Elizabeth O. Hexner, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Alison W. Loren, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Noelle V. Frey, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Sunita D. Nasta, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Steven C. Goldstein, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Edward A. Stadtmauer, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Jacqueline Smith, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Sarah Bailey, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Rosemarie Mick, The Department of Biostatistics and Epidemiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Daniel F. Heitjan, The Department of Biostatistics and Epidemiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Stephen G. Emerson, The Abramson Cancer Center and the Division of Hematology and Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
James A. Hoxie, The Abramson Cancer Center and the Division of Hematology and Oncology, the Center for AIDS Research, Perelman School of Medicine, University of Pennsylvania, Philadelphia
Robert H. Vonderheide, The Abramson Cancer Center and the Division of Hematology and Oncology, the Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia
David Porter, The Abramson Cancer Center and the Division of Hematology and Oncology, the Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia.
References
- 1.Johnston L. Acute graft-versus-host disease: differing risk with differing graft sources and conditioning intensity. Best Pract Res Clin Haematol. 2008;21:177–92. doi: 10.1016/j.beha.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara JL, Levy R, Chao NJ. Pathophysiologic mechanisms of acute graft-vs.-host disease. Biol Blood Marrow Transplant. 1999;5:347–56. doi: 10.1016/s1083-8791(99)70011-x. [DOI] [PubMed] [Google Scholar]
- 3.Weisdorf D. GVHD the nuts and bolts. Hematology Am Soc Hematol Educ Program. 2007:62–7. doi: 10.1182/asheducation-2007.1.62. [DOI] [PubMed] [Google Scholar]
- 4.Sackstein R. A revision of Billingham's tenets: the central role of lymphocyte migration in acute graft-versus-host disease. Biol Blood Marrow Transplant. 2006;12:2–8. doi: 10.1016/j.bbmt.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Louboutin JP, Zhu J, Rivera AJ, Emerson SG. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J Clin Invest. 2002;109:1335–44. doi: 10.1172/JCI14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–5. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 7.Matte CC, Liu J, Cormier J, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med. 2004;10:987–92. doi: 10.1038/nm1089. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 9.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 10.Fischereder M, Luckow B, Hocher B, et al. CC chemokine receptor 5 and renal-transplant survival. Lancet. 2001;357:1758–61. doi: 10.1016/s0140-6736(00)04898-4. [DOI] [PubMed] [Google Scholar]
- 11.Heidenhain C, Puhl G, Moench C, Lautem A, Neuhaus P. Chemokine receptor 5Delta32 mutation reduces the risk of acute rejection in liver transplantation. Ann Transplant. 2009;14:36–44. [PubMed] [Google Scholar]
- 12.Choi SW, Hildebrandt GC, Olkiewicz KM, et al. CCR1/CCL5 (RANTES) receptor-ligand interactions modulate allogeneic T-cell responses and graft-versus-host disease following stem-cell transplantation. Blood. 2007;110:3447–55. doi: 10.1182/blood-2007-05-087403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serody JS, Burkett SE, Panoskaltsis-Mortari A, et al. T-lymphocyte production of macrophage inflammatory protein-1alpha is critical to the recruitment of CD8(+) T cells to the liver, lung, and spleen during graft-versus-host disease. Blood. 2000;96:2973–80. [PubMed] [Google Scholar]
- 14.New JY, Li B, Koh WP, et al. T cell infiltration and chemokine expression: relevance to the disease localization in murine graft-versus-host disease. Bone Marrow Transplant. 2002;29:979–86. doi: 10.1038/sj.bmt.1703563. [DOI] [PubMed] [Google Scholar]
- 15.Wysocki CA, Burkett SB, Panoskalt-sis-Mortari A, et al. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pre-transplant conditioning. J Immunol. 2004;173:845–54. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- 16.Murai M, Yoneyama H, Harada A, et al. Active participation of CCR5(+)CD8(+) T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J Clin Invest. 1999;104:49–57. doi: 10.1172/JCI6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai M, Yoneyama H, Ezaki T, et al. Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat Immunol. 2003;4:154–60. doi: 10.1038/ni879. [Erratum, Nat Immunol 2003;4:497.] [DOI] [PubMed] [Google Scholar]
- 18.Welniak LA, Kuprash DV, Tumanov AV, et al. Peyer patches are not required for acute graft-versus-host disease after myeloablative conditioning and murine allogeneic bone marrow transplantation. Blood. 2006;107:410–2. doi: 10.1182/blood-2004-11-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welniak LA, Wang Z, Sun K, et al. An absence of CCR5 on donor cells results in acceleration of acute graft-vs-host disease. Exp Hematol. 2004;32:318–24. doi: 10.1016/j.exphem.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Bogunia-Kubik K, Duda D, Suchnicki K, Lange A. CCR5 deletion mutation and its association with the risk of developing acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Haematologica. 2006;91:1628–34. [PubMed] [Google Scholar]
- 21.Ma Q, Gooley TA, Storb RF. CCR5 expression on cells from HLA-matched unrelated marrow donors and graft-versus-host disease. Biol Blood Marrow Transplant. 2010;16:132–3. doi: 10.1016/j.bbmt.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDermott DH, Conway SE, Wang T, et al. Donor and recipient chemokine receptor CCR5 genotype is associated with survival after bone marrow transplantation. Blood. 2010;115:2311–8. doi: 10.1182/blood-2009-08-237768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorr P, Westby M, Dobbs S, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–32. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fätkenheuer G, Nelson M, Lazzarin A, et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N Engl J Med. 2008;359:1442–55. doi: 10.1056/NEJMoa0803154. [DOI] [PubMed] [Google Scholar]
- 25.Gulick RM, Lalezari J, Goodrich J, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359:1429–41. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106:2912–9. doi: 10.1182/blood-2005-05-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherland DR, Anderson L, Keeney M, Nayar R, Chin-Yee I. The ISHAGE guidelines for CD34+ cell determination by flow cytometry: International Society of Hematotherapy and Graft Engineering. J Hematother. 1996;5:213–26. doi: 10.1089/scd.1.1996.5.213. [DOI] [PubMed] [Google Scholar]
- 28.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15:825–8. [PubMed] [Google Scholar]
- 29.Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11:945–56. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Fätkenheuer G, Pozniak AL, Johnson MA, et al. Efficacy of short-term mono-therapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 31.Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117:3214–9. doi: 10.1182/blood-2010-08-302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beatty PG, Anasetti C, Hansen JA, et al. Marrow transplantation from unrelated donors for treatment of hematologic malignancies: effect of mismatching for one HLA locus. Blood. 1993;81:249–53. [PubMed] [Google Scholar]
- 33.Nash RA, Antin JH, Karanes C, et al. Phase 3 study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood. 2000;96:2062–8. [PubMed] [Google Scholar]
- 34.Slavin S, Nagler A, Naparstek E, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood. 1998;91:756–63. [PubMed] [Google Scholar]
- 35.Giralt S, Thall PF, Khouri I, et al. Melphalan and purine analog-containing preparative regimens: reduced-intensity conditioning for patients with hematologic malignancies undergoing allogeneic progenitor cell transplantation. Blood. 2001;97:631–7. doi: 10.1182/blood.v97.3.631. [DOI] [PubMed] [Google Scholar]
- 36.McSweeney PA, Niederwieser D, Shizuru JA, et al. Hematopoietic cell transplantation in older patients with hematologic malignancies: replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood. 2001;97:3390–400. doi: 10.1182/blood.v97.11.3390. [DOI] [PubMed] [Google Scholar]
- 37.Mielcarek M, Martin PJ, Leisenring W, et al. Graft-versus-host disease after nonmyeloablative versus conventional hematopoietic stem cell transplantation. Blood. 2003;102:756–62. doi: 10.1182/blood-2002-08-2628. [DOI] [PubMed] [Google Scholar]
- 38.Schetelig J, Kröger N, Held TK, et al. Allogeneic transplantation after reduced conditioning in high risk patients is complicated by a high incidence of acute and chronic graft-versus-host disease. Haematologica. 2002;87:299–305. [PubMed] [Google Scholar]
- 39.Ringdén O, Labopin M, Ehninger G, et al. Reduced intensity conditioning compared with myeloablative conditioning using unrelated donor transplants in patients with acute myeloid leukemia. J Clin Oncol. 2009;27:4570–7. doi: 10.1200/JCO.2008.20.9692. [DOI] [PubMed] [Google Scholar]
- 40.Alyea EP, Kim HT, Ho V, et al. Comparative outcome of nonmyeloablative and myeloablative allogeneic hematopoietic cell transplantation for patients older than 50 years of age. Blood. 2005;105:1810–4. doi: 10.1182/blood-2004-05-1947. [DOI] [PubMed] [Google Scholar]
- 41.Jagasia M, Arora M, Flowers ME, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296–307. doi: 10.1182/blood-2011-06-364265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luger SM, Ringdén O, Zhang MJ, et al. Similar outcomes using myeloablative vs reduced-intensity allogeneic transplant preparative regimens for AML or MDS. Bone Marrow Transplant. 2012;47:203–11. doi: 10.1038/bmt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alousi AM, Weisdorf DJ, Logan BR, et al. Etanercept, mycophenolate, denileu-kin, or pentostatin plus corticosteroids for acute graft-versus-host disease: a randomized phase 2 trial from the Blood and Marrow Transplant Clinical Trials Network. Blood. 2009;114:511–7. doi: 10.1182/blood-2009-03-212290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine JE, Logan B, Wu J, et al. Graft-versus-host disease treatment: predictors of survival. Biol Blood Marrow Transplant. 2010;16:1693–9. doi: 10.1016/j.bbmt.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest. 2000;105:1289–98. doi: 10.1172/JCI7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yi T, Chen Y, Wang L, et al. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–12. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009;113:1365–74. doi: 10.1182/blood-2008-06-162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391:344–5. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.