

Abstract
Cytochrome b5 (cyt b5) is one of the key components in the microsomal cytochrome P450 monooxygenase system. Consensus has not been reached on the underlying mechanism of cyt b5 modulation of CYP catalysis. Both cyt b5 and apo b5, are reported to stimulate the activity of several P450 isoforms. In the present study, the surface interactions of both holo and apo b5 with CYP3A4 were investigated and compared for the first time. Chemical cross-linking coupled with mass spectrometric analysis was used to identify the potential electrostatic interactions between the protein surfaces. Subsequently, the interaction models of holo/apo b5 with CYP3A4 were built using the identified interacting sites as constraints. Both cyt b5 and apo b5 were predicted to bind to the same groove on CYP3A4 with close contacts to the B-B’ loop of CYP3A4, a substrate recognition site (SRS). Mutagenesis studies further confirmed that the interacting sites on CYP3A4 (Lys96, Lys127 and Lys421) are of functional importance. Mutation of these residues reduced or abolished cyt b5 binding affinity. The critical role of Arg446 on CYP3A4 in binding to cyt b5 and/or cytochrome P450 reductase (CPR) was also discovered. The results indicated that electrostatic interactions on the interface of the two proteins are functionally important. The results indicate that the apo cyt b5 can dock with CYP3A4 in a manner analogous to holo cyt b5 so electron transfer from cyt b5 is not required for its effects.
Keywords: CYP3A4, cyt b5, apo b5, interaction, chemical cross-linking, MS, mutagenesis, PDB: 1TQN, 1CYO, 1I87
Microsomal cytochrome P450s (CYPs) catalyze biotransformation of a wide variety of chemically and structurally diverse compounds. These reactions account for approximately 85–90% of therapeutic drug metabolism (1). Each reaction cycle of microsomal CYPs requires sequential input of two electrons, which activates a molecule of oxygen to complete the reaction (1). In microsomal systems, electrons are transferred from NADPH, through CPR and/or cyt b5 to CYPs. CPR is an indispensable component for the catalytic cycle because the initial reduction of CYP (the introduction of the first electron) is catalyzed predominantly by CPR (1). In contrast, the mechanism of the second electron transfer and the role of cyt b5 in P450 catalytic cycle are not completely understood.
Cyt b5 exhibits complex effects on CYP-catalyzed reactions. The effects are both CYP isoform- and substrate-dependent. It can stimulate, or inhibit, or have no effect on CYP-catalyzed reactions. Cyt b5 may act as an obligate component, or a modifier, of a reaction. It has been reported that the activity of over 20 CYP isoforms can be modulated by cyt b5, including the majority of the human drug-metabolizing CYPs (2, 3). For example, cyt b5 is absolutely required for the metabolism of methoxyflurane, prostaglandins A1, E1, and E2 by CYP2B4 (4), 7-ethoxycoumarin, chlorzoxazone, aniline, and N-nitrosodimethylamine by CYP2E1 (5), p-nitrophenetole O-deethylation by CYP2B1 (6), and arachidonate by CYP4A7 (7). On the other hand, some CYPs show higher activity in the presence of cyt b5 than in the absence of cyt b5, such as CYP2A6-catalyzed coumarin 7-hydroxylation, CYP3A4-catalyzed testosterone (TST) 6β-hydroxylation, CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation (8, 9), etc.. Reactions inhibited by cyt b5 include CYP2B4-catalyzed benzphetamine demethylation (10). In contrast, cyt b5 shows no effect on CYP1A2- and CYP2D6-catalyzed reactions (8), as well as benzo[a]pyrene hydroxylation by CYP2B4 (11).
Cyt b5 affects CYPs by several possible mechanisms (3). One asserts that cyt b5 facilitates a fast second electron transfer, which is putatively the rate-limiting step in the CYP catalytic cycle, and therefore stimulates some CYP activities such as in CYP2E1 and rabbit CYP2B4 (3, 12, 13). Cyt b5 has also been suggested to decrease the uncoupling of the monooxygenase reaction through its interaction with CYPs and to result in an increased catalytic efficiency. A third proposed mechanism suggests that a heterodimeric complex of CYP-cyt b5 (3) is formed, which is able to accept two electrons from CPR to form a two-electron-reduced complex species (14). This complex facilitates the active peroxo P450 species formation as it excludes the need of interactions twice with CPR. An additional suggested mechanism is the allosteric effect, in which cyt b5 leads to conformational changes of CYPs, resulting in a modulated catalytic efficiency. This mechanism continues to be supported by recent data (8, 9, 15) but its importance is unclear (5, 10, 16). Among these mechanisms, the two basic roles of cyt b5 are proposed to be electron transfer or an allosteric effect via conformational change induced by cyt b5 on CYP. There is no consensus on whether electron transfer from cyt b5 is required, or whether allosteric effects of cyt b5 are involved. Because the effects of cyt b5 on CYPs require complex formation, identification of protein interacting regions and protein orientation in the redox complexes can be important for understanding the mechanisms and effects of cyt b5 (18).
We have previously defined the structural interaction between CYP2E1 and cyt b5 (19). In the CYP2E1-cyt b5 complex model, the cyt b5 heme group protrudes towards the surface of CYP2E1, to which the buried heme of CYP2E1 is in closest proximity (19). CYP2E1 is a CYP isoform whose reactions are highly stimulated by cyt b5 (2, 9, 20), but not by apo b5 (cyt b5 devoid of heme) (9, 20). The requirement of the cyt b5 heme group for CYP2E1-catalyzed reaction supports the mechanism of facilitating electron transfer rather than only causing an allosteric effect for this particular isoform. Inter-molecular electrostatic interactions are the main stabilizing forces for CYP-cyt b5 interactions (3) and contribute to the proper orientations of the heme prosthetic groups. Overall, this inter-molecular electrostatic interaction can result in a change in dielectric constant and subsequent facilitation of the electron-transfer process (21). Unlike its effects on CYP2E1 reactions, cyt b5 mainly exerts an allosteric effect on CYP3A4 reactions as suggested by previous studies (22, 23). Apo b5 has also been shown to stimulate some of CYP3A4-catalyzed reactions, such as testosterone 6β-hydroxylation and nifedipine oxidation, to a slight less extent than holo cyt b5 (natural cyt b5) (24). NMR studies have confirmed that removal of the heme group from cyt b5 only has minimal influence on its secondary structure (25–27). The structural effects of heme removal are highly localized, so the stimulatory effect of apo b5 on CYP3A4 indicates an allosteric mechanism. In addition, cyt b5 changes product regioselectivity and turnover kinetics of some CYP3A4 reactions, such as triazolam and pyrene oxidation (28, 29), suggesting topological changes in the active site of CYP3A4 (30). These observations provide additional evidence for an allosteric mechanism of cyt b5 on CYP3A4.
Extending these studies to the surface interactions between cyt b5 and CYP3A4 can further elucidate the complicated mechanism of cyt b5 in stimulation of CYPs. Chemical cross-linking and mass spectrometry were used to structurally characterize the interacting sites between cyt b5 and CYP3A4 because of the advantage of requiring small amounts of sample and high accuracy in determining the structures; site-directed mutagenesis and metabolic activity assays were used to confirm the functional importance of the identified interacting sites. The identified cyt b5-CYP3A4 interacting sites allowed construction of models for the holo/apo b5-CYP3A4 interactions by computational calculation. The results of the present study indicate that an allosteric effect of cyt b5 contributes to modulation of CYP3A4 reactions.
EXPERIMENTAL PROCEDURES
Materials
Plasmid (His)4HMWHuman-cyt b5 was kindly provided by Dr. Richard J. Auchus, University of Michigan (31, 32). Restriction enzymes and other DNA-modifying enzymes were from New England BioLabs (Beverly, MA). Platinum Pfx DNA polymerase, T4 DNA ligase, Histidine-tagged recombinant human cyt b5 and Vivid™ 3A4 substrate DBOMF were from Invitrogen (Carlsbad, CA). QuikChange® site-directed mutagenesis kit was from Stratagene (La Jolla, CA). Bactotryptone, bactopeptone, and bactoyeast extract were from BD Biosciences Clontech (Palo Alto, CA). Emulgen 911 was from Kao Chemicals (Tokyo, Japan). IPTG (Isopropyl β-D-1-thiogalactopyranoside), δ-ALA (5-aminolevulinic acid), thiamine, imidazole hydrochloride, protease inhibitor cocktail, sodium cholate and dithiothreitol were from Sigma-Aldrich (St. Louis, MO). CHT ceramic hydroxyapatite, type I, 40 μm particle size was from Bio-Rad (Hercules, CA). L-α-dilauroyl-sn-glycero-3-phosphocholine (DLPC), L-α-dioleoyl-sn-glycero-3-phosphocholine (DOPC), L-α-dilauroyl-sn-glycero-3-phospho- serine (DLPS) were from Avanti Polar-lipids, Inc. (Alabaster, AL). Ni-NTA Superflow was from Qiagen (Valencia, CA). The cross-linking reagent 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) was from Pierce Biotechnology, Inc. (Rockford, IL). Sequencing grade modified trypsin was from Roche Applied Science (Indianapolis, IN). 18O-labeled water (99 atom % 18O) was from Isotec (Miamisburg, OH). HPLC solvents were of the HPLC-grade. All other reagents were analytical grade.
Site-Directed Mutagenesis of CYP3A4
For site-directed mutagenesis of CYP3A4, the oligonucleotide primers used in the generation of CYP3A4 mutations K96A, K127A, K421A and R446A were as follows (mismatches indicated by the underlined bases): K96A forward, 5′ CAA AAC AGT GCT AGT GGC AGA ATG TTA TTC TGT CTT C3′; K96A reverse, 5′ GAA GAC AGA ATA ACA TTC TGC CAC TAG CAC TGT TTT G 3′. K127A forward, 5′ GCT GAG GAT GAA GAA TGG GCG AGA TTA CGA TCA TTG C3′; K127A reverse, 5′ GCA ATG ATC GTA ATC TCG CCC ATT CTT CAT CCT CAG C 3′. K421A forward, 5′ CTC CCT GAA AGA TTC AGC GCG AAG AAC AAG GAC AAC 3′; K421A reverse, 5′ GTT GTC CTT GTT CTT CGC GCT GAA TCT TTC AGG GAG 3′. R446A forward, 5′-CCA GAA ACT GCA TTG GCA TGG CGT TTG CTC TCA TG-3′; R446A reverse, 5′-CAT GAG AGC AAA CGC CAT GCC AAT GCA GTT TCT GG -3′. For construction of a CYP3A4 triple mutation K96A/K127A/K421A, a CYP3A4 double mutation K96A/K421A was first constructed using the K421A single mutation plasmid as the template and the K96A forward and reverse primers; afterwards, the triple mutation K96A/K127A/K421A was constructed using the K96A/K421A plasmid as the template and the K127A forward and reverse primers. The mutagenesis was performed using QuikChange® site-directed mutagenesis kit according to the manufacturer’s protocol. The full-length cDNAs of CYP3A4 sequence containing the desired mutations were analyzed at University of Washington Sequencing Facility.
Protein Expression and Purification
N-terminus truncated CYP3A4 and mutations were produced in E. coli C41 cells using the expression vector pCWhum3A4 (His)6. Growth and induction of E. coli C41 cells were performed as described previously (33). Solubilized membranes were prepared and CYP3A4 was purified on the Ni-NTA agarose column as described in (34). The column was equilibrated and loaded with equilibration buffer (50 mM KPi, pH 7.4, 20% glycerol, 0.05% sodium cholate, 5 mM imidazole and 50 UM testosterone), and then washed with 20 column volumes of wash buffer (50 mM KPi, pH 7.4, 20% glycerol, 0.05% sodium cholate, 40 mM imidazole, 100 mM glycine, 0.3 M sodium chloride, 0.2% Emulgen 911, 20 mM β-mercaptoethanol, and 50 UM testosterone). The protein was eluted with a minimal volume of elution buffer (50 mM KPi, pH 7.4, 20% glycerol, 350 mM imidazole and 0.02% sodium cholate) and then dialyzed against hydroxyapatite (HA) equilibration buffer (10 mM KPi, 2 mM BME, 0.2% cholate, and 20% glycerol). The protein was then loaded onto the HA column, washed with HA wash buffer 25 mM KPi, 2 mM BME, and 20% glycerol (pH 7.4), and eluted with 400 mM KPi and 20% glycerol (pH 7.4) and dialyzed into storage buffer (100 mM KPi, pH 7.4, 20% glycerol, 0.5 mM EDTA and 0.1 mM DTT). CYP3A4 content was determined by reduced carbon monoxide difference spectra (34).
Apo b5 was prepared from human cyt b5 according to a previously published protocol (5, 35, 36). Human cyt b5 expression and purification was according to previously described protocols (35, 36). The reconstituted holo b5 activity was comparable with commercial cyt b5 from Invitrogen (data not shown), and was used for equilibrium binding assay. In all other holo b5 assays commercial holo b5 was used. Expression and purification of rat CPR were performed as previously described (37, 38).
Cross-linking Reactions
Electrostatic interactions are considered to be the major driving force in cyt b5-CYP3A4 interactions (39–41). Therefore, EDC, a water-soluble cross-linking reagent, was chosen to “trap” the interaction between cyt b5 and CYP3A4, as EDC can cross-link basic (Lys) and acidic (Asp or Glu) residues that come into very close proximity (42). It generates amide bonds, with “zero-length” linker in between. The “zero-length” linker limits the number of orientations that must be considered in building structural models based on cross-linking data. CYP3A4 and cyt b5 were reconstituted at a molar ratio of 1:1 in 100 uL 50 mM KPi buffer in the presence of liposome (15, 43) (lipids mixture DLPC/DOPC/DLPS, 1/1/1, w/w/w, with final concentration of 10 UM for each enzyme. The solution was gently stirred for 10 min and held at room temperature for 2 hours. Then the chemical cross-linking reagent EDC was added to a final concentration of 10 mM from a 100 mM stock. The reaction was allowed to proceed at room temperature for 2 hours followed by dialysis against 50 mM KPi buffer and 50 mM ammonium bicarbonate buffer to remove EDC and isourea that was generated.
Proteolytic Digestion and Mass Spectrometric Analysis
Digestion of the cross-linked protein complex was performed as previously described (44). Subsequent mass spectrometric analysis for holo b5 and CYP3A4 cross-linking was carried out as described previously (44) using 18O labeling. Mass spectrometric analysis of apo b5 and CYP3A4 cross-linking was using a recently published method (45) which has the advantage of not requiring stable isotopic labeling. An LTQ-Orbitrap (ThermoFisher, San Jose, CA) was used to acquire all MS data at high mass accuracy (typically 0.1–3.0 ppm) because this limits false positives from the large number of possible cross-linked peptide candidates. Cross-linked peptides analyzed by ESI tend to have twice the charge of their linear constituents. This allows the data-dependent ion selection process for collision-induced dissociation (CID) to be focused only on precursors with charge state of 3 and above, thus focusing data acquisition on peptide more likely to be cross-linked. The data were analyzed with the open-modification algorithm Popitam (45). Tandem mass spectra were further analyzed and sequence matches confirmed and assigned with the MS3D collaboratory portal (46, 47) and University of Washington Proteomics Center web tools as references.
Testosterone 6β-hydroxylation and fluorogenic Vivid™ 3A4 substrate metabolism catalyzed by CYP3A4 wild-type and mutations
Working buffers for CYP3A4 incubations were prepared following a published protocol (48). Basically, final reaction buffer contains 0.2 μM CYP3A4, 0.4 μM NADPH-dependent cytochrome P450 reductase (CPR), 0.2 μM cyt b5, 0.1 mg/mL CHAPS, 0.02 mg/mL of lipids mixture, 3 mM GSH, and 30 mM MgCl2 in 50 mM potassium HEPES buffer at pH 7.4. The testosterone 6β-hydroxylation assay was carried out according to a previously described procedure (48) except that 1μg 11α-hydroxyprogesterone per reaction was used as internal standard. Vivid Green™ assay was carried out according to the supplier’s protocol. Both assays were conducted at a fixed enzymes ratio of 1:2:1 for CYP3A4/CPR/cyt b5 unless otherwise noted.
Equilibrium binding
For cyt b5-CYP3A4 affinity assay, experiments were conducted using the method described earlier with buffer conditions slightly modified (16, 49). Briefly, the sample cuvette contained 0.2 μM CYP3A4 in reaction buffer (pH 7.4) as used for activity assays. The reference cuvette contained only reaction buffer (pH 7.4). The difference spectra of the two cuvettes were recorded. Next, cyt b5 (0–3 μM) was titrated into both sample and reference cuvettes, and both were scanned from 340 to 500 nm to monitor the peak heights of the low- (418 nm) and high-spin (390 nm) Soret bands. The changes of the difference between the two peaks in the absolute spectra were plotted against cyt b5 concentration to estimate the binding affinity of cyt b5 to CYP3A4. The dissociation constant (Kd) was determined using the following Equation as described in (50):
| Equation 1 |
The ΔA is the change of absorbance difference between 390 nm and 418 nm, and Amax is the maximal absorbance change; [CYP3A4] is 200 nM in the assay; [b5] is the concentration of holo b5 or apo b5; and Kd is the spectral dissociation constant of the CYP3A4-b5 complex.
For TST binding affinity assay, 1 μM CYP3A4 was used in the buffer of 50 mM KPi, 20% glycerol. The change in absorbance difference between 390 nm and 418 nm ( A) was plotted as a function of TST concentration (51, 52). Apparent dissociation constants were estimated using the Hill equation (Equation 2) with GraphPad software:
| Equation 2 |
The Amax is the maximal absorbance change; S is the TST concentration; Kd(app) is the apparent dissociation constant (the substrate concentration that gives an absorbance change of 50% of Amax); and n is the Hill coefficient.
Molecular Docking and Energy Minimization
High resolution X-ray crystallographic structure of human CYP3A4 (PDB “1TQN”) (53), human holo cyt b5 homology model made from bovine cyt b5 (PDB “1CYO”) crystal structure (54), and rat apo cyt b5 NMR solution structure (PDB “1I87”) were used to construct the model of the CYP3A4-holo/apo b5 complexes. Manual docking of the structures was accomplished by positioning the structures, using DS Viewer Pro 6.0 (Accelrys Software Inc.) and the mass spectrometry cross-linking data as constraints. The distance between the residue pair in each identified cross-linked peptide was minimized. Some side chains of the residues on the protein interacting surfaces were reoriented using the DS Viewer Pro 6.0 program to avoid overlap of the side chains. The complex model was energy minimized using the software package GROMACS (Groningen MAchine for Chemical Simulation) as previously described (55–57). Molecular graphics and analyses were performed with the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera). The intermolecular H-bonds revealed are identified through the use of the FindHBond function in the Chimera program (58). Because of the ambiguous sites on apo b5 in cross-linking with CYP3A4, ZDOCK SERVER (http://zdock.umassmed.edu/) was used to predict apo b5-CYP3A4 interaction model with identified sites as constraints (59). The complex with the lowest energy was selected as the interaction model.
Construction of Membrane Models of Holo/Apo b5-CYP3A4 Interaction
The orientations of CYP3A4 and holo/apo b5 relative to membranes was modeled according to (60). The angle between CYP3A4 heme plane and the membrane was adjusted to 58°, a value that is midway between 38° and 78° according to (60). The holo and apo b5 linker domain conformation was made flexible using the software package Chimera as suggested by (50) and the NMR solution structure of human cyt b5 (PDB: 2I96).
RESULTS
Chemical Cross-linking
Treatment of an equimolar mixture of CYP3A4 and human cyt b5 or apo b5 with EDC resulted in the formation of cross-linked protein complexes, with the major complexes being a binary complex (cyt b5-CYP3A4), the molecular mass of which is ~73 kDa (Fig. 1). The band directly above it with molecular weight of ~90 kDa is likely to be (cyt b5)2-CYP3A4 and other faint bands could be complexes with multiple CYPs.
FIGURE 1.
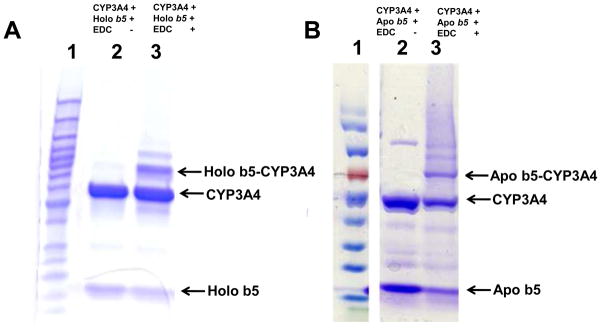
SDS-PAGE analysis of the cross-linking reactions of human cyt b5 and CYP3A4 using EDC. Holo and apo b5 were cross-linked with CYP3A4 at 1:1 mole ratio in the absence and presence of EDC. A. The holo b5 and CYP3A4 reaction. Lane 1, protein ladder; lane 2, holo b5 + CYP3A4 without EDC; lane 3, holo b5 + CYP3A4 with EDC. B. The apo b5 and CYP3A4 reaction. Lane 1, protein ladder; lane 2, apo b5 + CYP3A4 with EDC; lane 3, apo b5 + CYP3A4 without EDC.
Holo B5 and CYP3A4 Interactions by Mass Spectrometric Analysis
Three inter-molecular cross-linked peptides of holo b5-CYP3A4 were identified with five precursor ions at different charge status (Table 1) through mass spectrometric analysis. The sequences of the three cross-linked peptide candidates were fully confirmed by the tandem mass spectra with the cross-linked residue pairs successfully located (Fig. 2). In the cross-linked peptides cyt b5: 48EQAGGDATENFEDVGHSTDAR58 - CYP3A4: 92TVLVKECYSVFTNR105 and cyt b5: 48EQAGGDATENFEDVGHSTDAR58 - CYP3A4: 116SAISIAEDEEWKR128, cross-linked sites were unambiguously determined to be Glu56 (cyt b5) - Lys96 (CYP3A4) and Glu56 (cyt b5)-Lys127 (CYP3A4), respectively (cyt b5 numbering is according to 1CYO). For example, on the tandem MS spectrum of the 1st cross-linked peptide (Fig. 2A), the observance of cyt b5 linear fragment ions such as b8 and a series of y ions including the longest y12 ions indicates that only Glu56 can be the cross-linking residue. In addition, all the cross-linking fragments indicated with signal “x” agree with and further support this assignment. For the cross-linked peptide cyt b5: 35FLEEHPGGEEVLR47 - CYP3A4: 419FSKK422, two possibilities of cross-linking the sites were suggested by the spectrum (Fig. 2C), Glu37 (cyt b5) - Lys421 (CYP3A4) and Glu43 (cyt b5) - Lys421 (CYP3A4). Glu37 and Glu43 are adjacent surface residues on the structure of cyt b5 (61) and predicted to be equally accessible to Lys421 on CYP3A4.
Table 1.
Inter-molecular cross-linked peptides in the complex of holo b5-CYP3A4. The peptide digests were analyzed on an API-US quadrupole mass spectrometer (Micromass, Manchester, UK) as previously described (44).
| Measured Mono-isotopic Peak (m/z) | Char ge State | Measured Peptide Mass (Da) | Calculated peptide Mass (Da) | Mass Matches to Inter-molecular Cross-linked Peptides |
|---|---|---|---|---|
| 781.40 | +5 | 3902.00 | 3901.78 | (holo b5: 48EQAGGDATENFEDVGHSTDA R68)-(CYP3A4: 92TVLVKEC(Carbamidomethyl)Y SVFTNR105) |
| 976.51 | +4 | 3902.04 | ||
| 744.97 | +5 | 3719.85 | 3719.66 | (holo b5: 48EQAGGDATENFEDVGHSTDA R68)-(CYP3A4: 116SAISIAEDEEWKR128) |
| 930.95 | +4 | 3719.80 | ||
| 501.29 | +4 | 2001.16 | 2001.04 | (holo b5: 35FLEEHPGGEEVLR47) -(CYP3A4: 419FSKK422) |
FIGURE 2.



Tandem mass spectra for three cross-linked peptide precursor ions identified from cyt b5-CYP3A4 complex. Fragment ions with superscript “x” represent the cross-linked fragment ions. Ions marked with subscript α are from cyt b5, and ions with subscript β are from CYP3A4. Identified cross-linked residue pairs are linked by bold black lines. A. Ion [M+H]4+=976.4534 for holo b5: 48EQAGGDATENFEDVGHSTDAR68 - CYP3A4: 92TVLVKECYSVFTNR105. B. Ion [M+H]4+=930.9229 for holo b5:48EQAGGDATENFEDVGHSTDAR68-CYP3A4: 116SAISIAEDEEWKR128 . C. Ion [M+H]4+=501.2670 for holo b5: 35FLEEHPGGEEVLR47- 3A4: 419FSKK422. The measurement errors of the three ions are 0.2, 0.9, and 2.4 ppm, respectively.
The two peptides on cyt b5 identified in cross-linking with CYP3A4 contain α2-loop-α3 and α4-loop-α5 (3), which form the hydrophobic core to which the heme group binds. On the other hand, the peptides on CYP3A4 contain B’, C helices, K”-L loop and L helix. All three cross-linked sites are found to be on the proximal surface of CYP3A4, which has been predicted to be the redox partner binding surface for CYPs (62, 63).
Apo B5 and CYP3A4 Interactions by Mass Spectrometric Analysis
Two cross-linked peptide complexes between apo b5 and CYP3A4 were successfully identified (Table 2). The sequences of the cross-linked peptide candidates were confirmed by their corresponding tandem mass spectra (Fig. 3). The cross-linked sites on the peptide complex apo b5: 35FLEEHPGGEEVLR47 - CYP3A4: 116SAISIAEDEEWKR128 were determined to be Glu37 on apo b5 and Lys127 on CYP3A4. On peptide apo b5: 48EQAGGDATENFEDVGHSTDAR68 - CYP3A4: 71VWGFYDGQQPVLAITDPDMIKTVLVK96, Lys91 on CYP3A4 is identified to be the cross-linked site, while further differentiation between the residues Glu48, Asp53, Glu56, Glu59 and Asp60 on cyt b5 involved in cross-linking is difficult due to the complication of the spectra and lack of sufficient fragmentation.
Table 2.
Inter-molecular cross-linked peptide candidates in the complex of apo b5-CYP3A4.
| Measured Mono-isotopic Peak (m/z) | Charge State | Measured Peptide Mass (Da) | Calculated peptide Mass (Da) | Mass Matches to Inter-molecular Cross-linked Peptides |
|---|---|---|---|---|
| 757.3763 | +4 | 3026.4814 | 3026.485 4 |
(apo b5: 35FLEEHPGGEEVLR47)-(CYP3A4: 116SAISIAEDEEWKR128) |
| 606.1026 | +5 | 3026.4812 | ||
| 1024.8989 | +5 | 5120.4627 | 5120.458 2 |
(apo b5: 48EQAGGDATENFEDVGHSTDAR 68)-(CYP3A4: 71VWGFYDGQQPVLAITDPDMIK TVLVK96) |
FIGURE 3.


Tandem mass spectra for two cross-linked peptide precursor ions identified from apo b5-CYP3A4 complex. Fragment ions with superscript “x” represent the cross-linked fragment ions. Ions marked with subscript α are from cyt b5, and ions with subscript β are from CYP3A4. Identified cross-linked residue pairs are linked by bold black lines. A. Ion [M+H]4+=757.3763 for apo b5: 35FLEEHPGGEEVLR47-CYP3A4: 116SAISIAEDEEWKR128. B. Ion [M+H]5+=1024.8989 for apo b5: 48EQAGGDATENFEDVGHSTDAR68-CYP3A4:71VWGFYDGQQPVLAITDPDMIKTVLVK96. The measurement errors for the two ions are 1.3 and 0.8 ppm, respectively.
Interestingly, the peptide segments identified on apo b5 involved in cross-linking with CYP3A4 are the same as those on holo b5 with CYP3A4, including α2-loop-α3 and α4-loop-α5 (Table 1 and 2). On CYP3A4, the Lys127 residue was identified to be cross-linked with both holo and apo b5; the Lys91 and Lys96 residues that were identified to be cross-linked with holo/apo b5 are very close in three-dimension structure of CYP3A4.
Modeling of Holo B5-CYP3A4 and Apo B5-CYP3A4 Complex
Using the cross-linking sites between holo/apo b5 and CYP3A4 as constraints and minimizing the energy of the interactions by protein docking programs, the docked models of holo/apo b5-CYP3A4 complexes were constructed (Fig. 4). For apo b5-CYP3A4 docking calculation, ZDOCK server was able to calculate different combinations with the cross-linking sites as constraints, though through the tandem mass spectrum no differentiation can be made between the acidic residues 48, 53, 56, 59 and 60. The model shown in Fig. 4 has the lowest energy according to the docking calculation.
FIGURE 4.



Holo/apo b5-CYP3A4 complex models. CYP3A4 is white; b5 is green; the heme group of CYP3A4 is red; the heme group of holo b5 is orange. The interacting residues on CYP3A4 and cyt b5 are magenta and blue, respectively. Protein regions on CYP3A4 far away from the interacting surfaces are truncated. A. Top view of holo b5-CYP3A4 model. B. Top view of apo b5-CYP3A4 model. C. R446A (golden) illustrated in the holo b5-CYP3A4 model.
In both interaction models, the interacting surfaces on holo/apo b5 include the segments α2-loop-α3 and α4-loop-α5, with additionally one propionate group of the heme group in holo b5 contributing to the interaction with CYP3A4. Holo and apo b5 were predicted to bind to the same groove on CYP3A4 despite the different orientations. The contact regions on CYP3A4 for holo b5 are predicted to be the B helix, the B-B’ loop, the C-D loop, the D, J’, and K helix, the K”-L meander region, the β-bulge and the L helix (Fig. 4A). Those for apo b5 are the B helix, B-B’ loop, C helix, C-D loop, D helix, K”-L meander region, and the β-bulge (Fig. 4B). Holo and apo b5 both approach the B-B’ loop region and the C helix of CYP3A4 via the α4 and α5 helix, respectively. In holo b5-CYP3A4 model, the CYP3A4 and cyt b5 heme groups are nearly perpendicular, and the shortest distance between the heme groups is ~11 Å.
Site-directed Mutagenesis of CYP3A4
To confirm the biological function of the residues on CYP3A4 identified by the cross-linking study, site-directed single-point and multiple substitutions of the three Lys residues on CYP3A4 (Lys96, Lys127 and Lys421) to Ala were carried out. In addition, Arg446 was included in the mutagenesis study, because this residue was at the interface of holo b5-CYP3A4 complex model, and therefore was predicted to be of significance for cyt b5 interaction. It was postulated that the interactions between CYP3A4 and cyt b5 initiated by electrostatic interaction would be affected by substitution of the basic Lys residues to neutral Ala.
The purified CYP3A4 single-point mutations K96A, K421A, R446A and the triple mutation K96A/K127A/K421A all showed homogenous 450 nm peaks in the reduced carbon monoxide binding difference spectrum (Fig. S1). The expression level of K127A was extremely low for unknown reasons, so it was not further studied. To determine whether their active sites had been affected by the mutations, a testosterone binding affinity assay was done (Fig. S2). Analysis of the data using the Hill equation yielded Kd(app) (or S50) values of 25 ± 12 μM (N=1.53, R2=0.99), 71 ± 28 μM (N=1.45, R2=0.99), 108 ± 34 μM (N=1.07, R2=0.995), 30 ± 8 μM (N=1.34, R2=0.997), and 42± 25 μM (N=1.31, R2=0.99) for CYP3A4 wild-type, K96A, K421A, R446A and triple mutation, respectively. The wild-type apparent dissociation constant is comparable to that previously reported (51, 52). All mutated proteins bind to testosterone with apparent Kds in a range of 1–4 fold of the wild-type, suggesting that the active sites of the mutations were modestly altered by the mutations. Also, the data indicate that the TST induces spin state changes of decreased magnitude for the mutations compared to wild type. Presumably, these mutations affect the conformation of either the substrate free enzyme or the ligand-bound complexes and thus have an impact on the energetics and solvent effects of substrate binding. Specifically for K421A, the mutation site is at the K-L loop, partly involved in channel 5 of CYP3A4’s substrate access/egress channel; however, this channel 5 (between K, K’ to the active site) has been found not to open during the computer dynamic simulations of CYP3A4 (64, 65). An important result of these studies is that the mutations do not significantly impair TST binding, which can, therefore, be used to probe the effects of these residues on cyt b5 binding and catalysis in the presence of CPR.
Effect of the Mutations on Catalytic Activities of CYP3A4
TST 6β-hydroxylation, a probe reaction of CYP3A4, was used to measure the activities of wild-type CYP3A4 and the mutations. The stimulatory effects of cyt b5 on these enzymes were compared, as shown in Table 3 and Fig. 5 for a single concentration of cyt b5, with a 1:2:1 ratio of CYP:CPR:cyt b5. The catalytic efficiency (Vmax/Km) of wild-type CYP3A4, K421A and K96A was increased by cyt b5 2.7-fold, 1.7-fold and 1.3-fold, respectively, under these conditions. The Vmax ratios of +b5/- b5 are 1.7, 1.3 and 2 respectively. The results indicate a complex effect of the Cyt b5 dependence on both Vmax and Km terms for TST hydroxylation. These results indicate that the Lys96 residue likely plays a more important role than Lys421 in the functional interaction of CYP3A4 with cyt b5. Interestingly, the triple mutation K96A/ K127A/K421A displayed very low catalytic activity both in the absence and presence of cyt b5. That is, simultaneous substitution of the three sites depleted the activity of CYP3A4. It is remarkable that R446A also completely abolished CYP3A4 activity. Another probe of CYP3A4, fluorogenic Vivid™ 3A4 substrate DBOMF, was also tested for activities of wild-type CYP3A4 and its mutations (Fig. 6). The results were very consistent with testosterone 6β-hydroxylation activities. Overall, the mutagenesis study confirmed the functional importance of the ion pairs identified by the cross-linking study.
Table 3.
Effect of the Mutations on CYP3A4 Catalytic Activities for testosterone 6β-hydroxylation in the presence and absence of cyt b5. Each point represents the mean ± S.D. (n=3).
| + b5 | − b5 | Ratio of Catalytic Efficiency + b5/− b5 | Ratio of Vmax + b5/− b5 | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Vmax (nmol/nmnolC YP3A4/min) | Km (μM) | Vmax (nmol/nmnolCY P3A4/min) | Km(μM) | |||
| WT | 27.8±2.2 | 152.6±7.5 | 16.4±1.3 | 240.2±48.3 | 2.7 | 1.7 |
| K96A | 16.0±1.3 | 436.8±76.0 | 12.6±2.0 | 470.1±149.0 | 1.3 | 1.3 |
| K421A | 16.6±1.7 | 366.1±81.4 | 8.3±0.8 | 313.9±73.7 | 1.7 | 2 |
| R446A | N/A | N/A | N/A | N/A | ||
| Triple | N/A | N/A | N/A | N/A | ||
FIGURE 5.

NADPH-dependent formation of 6β-hydroxytestosterone by CYP3A4 and its mutations. Reactions were carried out using 40 pmol CYP3A4, 80 pmol CPR and 40 pmol b5, in 200 uL reaction volume for 10 min. Each point represents the mean ± S.D. (n=3).
FIGURE 6.

NADPH-dependent formation of Vivid Green® from Vivid DBOMF by CYP3A4 and its mutations. Reactions were carried out using 40 pmol CYP3A4/mutation, 80 pmol CPR, and 40 pmol b5, in 200 uL reaction volume for 15 min. (A) CYP3A4. (B) K96A. (C) K421A. (D) R446A. Each point represents the mean ± S.D. (n=3).
Effect of the Mutations on CYP3A4 Binding to Cyt B5
The catalytic experiments do not explicitly distinguish between effects of the mutations on cyt b5 binding vs. cyt b5-dependent interactions with TST. To further examine the effect of the mutations on the binding of cyt b5 to CYP3A4, the affinity of cyt b5 to wild-type CYP3A4 and its mutations was measured using a spectral titration method (66, 67). As shown in Fig. 7, cyt b5 bound to CYP3A4 wild-type, K96A and K421A with estimated Kd values of 140 ± 37 nM, 346 ± 198 nM and 287 ± 37 nM. This suggests that effects of mutation observed in functional studies may in part be due to altered affinity of the protein interactions. The studies do not, however, clarify any specific molecular mechanism by which mutation affects KM vs. Vmax in the functional studies. The binding affinity of cyt b5 with R446A was drastically decreased as indicated by the binding assay, with a Kd value estimated greater than 1000 UM.
FIGURE 7.

Titration of cyt b5 to wild-type CYP3A4 and mutations and apo b5 to wild-type CYP3A4. The spectral titration experiment was carried out using 200 nM CYP3A4/mutations and titration with cyt b5 or apo b5. The magnitude of the absorbance change at 390 minus 418 nm was measured following each addition of b5 at room temperature.
It has been reported that the ratio of cyt b5/CYP can affect the CYP catalytic rate, as cyt b5 competes with CPR for the partially overlapping binding sites on CYPs (15, 68, 69). Results in Fig. 8 revealed a ratio-dependent effect of cyt b5 on CYP3A4 activities, with a stimulatory effect at low ratios, but an inhibitory effect with increasing b5/CYP3A4 ratios. Both wild-type CYP3A4 and K421A have their maximal activities at the ratio of 2:1. K96A reached its highest activity at the b5/CYP3A4 ratio of 3:1, which is consistent with previous cyt b5 binding assays with the mutated protein. Expectedly, both the triple mutation and R446A showed very low activity either in the presence or absence of cyt b5.
FIGURE 8.

Effect of cyt b5/CYP ratio on formation of 6β-hydroxytestosterone by CYP3A4 and its mutations. Reactions were carried out using varying concentrations of cyt b5 and constant ratio of CYP3A4 (40 pmol) /CPR (80 pmol) for 10 min in 200 uL reaction volume. Each point represents the mean ± S.D. (n=3).
Effect of the Mutations on CYP3A4 Interaction with CPR
Because CPR is expected to share sites of interaction on CYP with cyt b5, the effects of the mutations on CYP3A4 activity in the presence of varying concentrations of CPR were determined. TST 6β-hydroxylation, dependent on CPR concentrations, exhibited Michaelis-Menten kinetics, therefore apparent K’m,CPR was used to illustrate the affinity of CYP3A4-CPR interaction (49, 70). As shown in Fig. 9, wild-type CYP3A4, K96A and K421A have comparable binding K’m,CPR values (41 ± 10, 91 ± 21, 70 ± 12 nM, respectively). CPR binding to R446A and the triple mutation was abolished. The apparent binding of CPR to CYP3A4 and mutated CYP3A4s agrees with the earlier proposal that cyt b5 and CPR have overlapped binding sites on P450s and they compete with each other for binding to P450s (63, 69, 71).
FIGURE 9.

Effect of CPR/CYP ratio on formation of 6β-hydroxytestosterone by CYP3A4 and its mutations. Reactions were carried out using varying concentrations of CPR, constant ratio of CYP3A4 (40 pmol) / cyt b5 (40 pmol) for 10 min in 200 uL reaction volume. Each point represents the mean ± S.D. (n=3).
DISCUSSION
The current investigation of the role of inter-surface residues in the holo/apo cyt b5-CYP3A4 complex provides information of the structural organization of the complexes, and supports the allosteric roles of holo/apo cyt b5 in modulating catalytic efficiency of CYP3A4. Apo b5 and holo cyt b5 interact with overlapping sites on CYP3A4, with different orientations. Both directly contact with the B-B’ loop region of CYP3A4, one of a major substrate recognition sites (SRSs) of CYP3A4 (72). Previous studies have shown that lapachenole, a substrate of CYP3A4, covalently binds to this loop region (73). This loop region is also found to be highly flexible by computer dynamic simulation studies (64, 74). Thus, it is reasonable to postulate that the contact of cyt b5 with CYP3A4 can induce the conformational changes of this structural element. The induction of conformational changes of CYP enzymes has been observed in P450cam (CYP101) upon cyt b5 binding, whose structural perturbations happened not only to the proximal surface structural elements including B, C helices, but also to those in the distal surface, including regions for substrate access and orientation (75). Consequently, it is reasonable to extrapolate that association CYP3A4 with cyt b5, even in the absence of effects on rates of reduction of intermediates in the CYP reaction cycle, can give rise to the allosteric effect on it. This allosteric effect is further supported by apo b5 exhibiting stimulatory effect on CYP3A4 (Table 4, and (8, 9)).
Table 4.
Comparison of modulation effects of holo b5 and apo b5 on CYP3A4 catalytic activities. Each point represents the mean ± S.D. (n=3).
| CYP3A4 | Vmax (nmol/nmnolCYP3A4/min) |
|---|---|
| + holo b5 | 22.7±0.4 |
| + apo b5 | 17.7±2.6 |
| − b5 | 12.9±1.0 |
The interaction sites of holo/apo b5-CYP3A4 are also found consistent with their membrane-associated models under physiological conditions as in Fig. 10. CYPs are embedded in the membrane by an N-terminal membrane anchor and the F-G loop (60, 76), with 38 – 78° angles between the heme plane and the membrane plane (60) depending on the CYP isoform. Cyt b5 has a flexible 15 amino acid linker domain between its cytosolic functional domain and the C-terminal trans-membrane domain, which facilitates appropriate positions of the functional domain to interact with CYPs effectively (50, 77). The relative positions of CYP3A4 and cyt b5 shown in Fig. 10 indicates that under physiological conditions, holo/apo cyt b5 are able to adopt orientations consistent with the models based on our crosslinking constraints to interact with CYP3A4 effectively.
FIGURE 10.

Orientations of CYP3A4 and holo/apo b5 in membranes. The angle between CYP3A4 heme (red) plane and the membrane slab (blue) was adjusted to 58°, a middle value between 38° and 78° according to (60). Holo and apo b5 linker domain conformation was made flexible according to (50), as well as the human and rabbit cyt b5 NMR structure (PDB: 2I96). CYP3A4 is light purple; holo b5 is green; apo b5 is cyan. (A) Holo b5-CYP3A4 interaction model position relative to the membrane; (B) Apo b5-CYP3A4 interaction model position relative to the membrane.
A comparison of the structural elements on CYP2E1 that interacts with cyt b5 is useful. Our laboratory previously characterized holo b5-CYP2E1 interaction and it is enlightening to compare the current holo b5-CYP3A4 model with it. In the holo b5-CYP2E1 interaction model, the heme groups of cyt b5 and CYP2E1 that were involved in the contacts were predicted to be in an orientation consistent with its electron transfer role for CYP2E1 catalysis (44). It is worth noting that the acidic residues on cyt b5 at the interacting surface, which are negatively charged under physiological conditions, are conserved across species (3). In contrast, the distribution of charged residues on the surfaces of CYP2E1 and CYP3A4 varies because of the diversity of CYP isoforms. Thus, driven by electrostatic forces, cyt b5 can position itself differently in binding to CYP3A4 compared with CYP2E1 (44). This different orientation of cyt b5 in binding to different CYPs could account for its CYP isoform-dependent effect. Comparison of the cyt b5-CYP3A4 and cyt b5-CYP2E1 complex (44) reveals very significant difference in binding features. The J’ helix found in the CYP2E1-cyt b5 interaction does not exist in the cyt b5-CYP3A4 complex. On the other hand, the interacting helices B, D, K” of CYP3A4 were not seen in cyt b5-CYP2E1 complex. Most significantly, the B-B’ loop of CYP3A4, a well-recognized SRS, contacts with the helices α3 and α4 surrounding the heme group of cyt b5. This specific interaction between cyt b5 and CYP2E1 B-B’ loop does not exist in the model. Therefore, cyt b5 does not exert a major allosteric effect on CYP2E1 as it does on CYP3A4.
Functional studies (Fig. 5 and 6) using both a classical substrate testosterone and a fluorogenic Vivid™ CYP3A4 substrate confirmed that the cross-linking sites identified by mass spectrometry study are important for cyt b5 and CYP catalysis. Mutation of Lys96 had a greater impact on the cyt b5 stimulatatory effect on CYP3A4 than that of Lys421, in parallel with the lower binding affinity of cyt b5 with K96A than that of K421A. The affinity of apo b5-CYP3A4 is similar to holo b5-CYP3A4 (Fig. 7), consistent with the similar stimulatory effects of holo b5 and apo b5 on CYP3A4 activity, which can be explained by our models of their interactions; the models showed that holo/apo b5-CYP3A4 both bind to the same groove on CYP3A4, albeit with a different orientation.
The experiment with varying cyt b5/CYP3A4 ratio suggests that cyt b5 and CPR have overlapping binding sites on CYPs, and compete with each other to bind to CYPs (71) (Fig. 8). At higher ratio of cyt b5 to CYP3A4, CYP3A4 activity was decreased compared with stimulated at lower ratio. Similar observations were also reported by Locuson et al. that at high cyt b5 to CYP2C9 ratio (>4), the oxidase activity was decreased (15) and the author proposed that competition between CPR and cyt b5 at very high concentrations of cyt b5 would diminish the essential binding of CPR, and the transfer of electrons from NADPH, therefore resulting in a decrease in overall oxidase activity. In our study, when varying the cyt b5/CYP3A4 ratio, at a 3:1 ratio K96A showed a maximal catalytic activity. The stimulatory effects on K96A catalytic efficiency are slightly lower than for K421A, possibly due to the higher Kd value of cyt b5 with K96A than that with K421A (Fig. 7), since CPR has a comparable affinity with both K96A and K421A. Noting that there is still strong binding of cyt b5 to the single-point mutation, the study also suggests that the sum of different kinds of interactions, e.g., electrostatic and weak hydrophobic interactions, altogether make decisive contributions to functional cyt b5-CYP3A4 interaction.
The triple (K96A/K127A/K421A) mutation exhibited only minimal activity in metabolizing either TST or Vivid™ 3A4 substrate. The triple mutation was, therefore, not considered in further functional studies. As mentioned previously, both K96A and K421A retained CPR binding capability and catalytic activity for both substrates; we therefore suspected that Lys127 might be more important in CPR-CYP3A4 binding. Alternatively the three sites synergistically play a pivotal role in CPR-CYP3A4 binding. R446A showed dramatic decrease in its activity, indicating the importance of this residue in interacting with CPR, an essential coenzyme of P450. In the holo b5-CYP3A4 interaction models, Arg446 residue appeared to be in close contact with the binding partners. The Arg446 was found to be conserved among human CYP species such as 2A6, 2C19, 2D6, 2E1, 3A5, and 2J2. Earlier studies on rabbit CYP2B4 has shown that Arg443 (equivalent to Arg446 in CYP3A4) on CYP2B4 contributes to CPR binding (66).
In conclusion, in the present study, we identified the interaction surfaces of both holo and apo cyt b5 with CYP3A4 using chemical cross-linking coupled with mass spectrometric analysis, and determined the functional importance of the interacting residues on CYP3A4 using site-directed mutagenesis and metabolic assays. Computer models of both the holo/apo cyt b5-CYP3A4 complexes were constructed for the first time at the atomic level, which illustrated the similarity and difference of holo- and apo cyt b5 in interacting with CYP3A4. To the extent that the apo-cyt b5 binds at the same surface as holo cyt b5, and it activates CYP3A4 activity, the results support the possibility that cyt b5 plays an allosteric role in CYP3A4 catalytic activity, in addition to possible electron transfer by the holo protein. The critical role of Arg446 residue on CYP3A4 in interaction with both cyt b5 and CPR is also suggested. These findings provide further insight into the complex mechanisms of cyt b5 modulation of CYPs.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH/NIGMS Program Project Grant GM32165. This work was also supported in part by the University of Washington’s Proteomics Resource (UWPR95794).
We thank Dr. Priska D. von Haller, Dr. Michael J. Dabrowski for their kindly help; the Collaboratory for MS3D Portal sponsored by the National Institutes of Health and the National Science Foundation; and Chimera package developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. Finally, this work is dedicated to the Memory of Professor Sid Nelson, who will be missed.
ABBREVIATIONS
The abbreviations used are
- CYP
cytochrome P450
- cyt b5 or holo b5
Cytochrome b5
- apo b5
cyt b5 devoid of heme
- SRS
substrate recognition sites
- CPR
cytochrome P450 reductase
- EDC
reagent 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- δ-ALA
5-aminolevulinic acid
- IAM
2-Iodoacetamide
- DTT
Dithiothreitol
- DLPC
L-α-dilauroyl-sn-glycero-3-phosphocholine
- DOPC
L-α-dioleoyl-sn-glycero-3-phosphocholine
- DLPS
L-α-dilauroyl-sn-glycero-3-phosphoserine
- GSH
glutathione
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- BME
β-mecaptomethanol
- TST
testosterone
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Mass shift of precursor ions with 18O labeled. CYP3A4 mutations CO-binding difference spectra and TST binding spectra. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 2005. [Google Scholar]
- 2.Porter TD. The roles of cytochrome b5 in cytochrome P450 reactions. Journal of biochemical and molecular toxicology. 2002;16:311–316. doi: 10.1002/jbt.10052. [DOI] [PubMed] [Google Scholar]
- 3.Schenkman JB, Jansson I. The many roles of cytochrome b5. Pharmacol Ther. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 4.Vatsis KP, Theoharides AD, Kupfer D, Coon MJ. Hydroxylation of prostaglandins by inducible isozymes of rabbit liver microsomal cytochrome P-450. Participation of cytochrome b5. J Biol Chem. 1982;257:11221–11229. [PubMed] [Google Scholar]
- 5.Yamazaki H, Johnson WW, Ueng YF, Shimada T, Guengerich FP. Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4. Characterization of a reconstituted cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies with apocytochrome b5. J Biol Chem. 1996;271:27438–27444. doi: 10.1074/jbc.271.44.27438. [DOI] [PubMed] [Google Scholar]
- 6.Kuwahara S, Omura T. Different requirement for cytochrome b5 in NADPH-supported O-deethylation of p-nitrophenetole catalyzed by two types of microsomal cytochrome P-450. Biochem Biophys Res Commun. 1980;96:1562–1568. doi: 10.1016/0006-291x(80)91352-2. [DOI] [PubMed] [Google Scholar]
- 7.Loughran PA, Roman LJ, Miller RT, Masters BS. The kinetic and spectral characterization of the E. coli-expressed mammalian CYP4A7: cytochrome b5 effects vary with substrate. Arch Biochem Biophys. 2001;385:311–321. doi: 10.1006/abbi.2000.2136. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki H, Gillam EM, Dong MS, Johnson WW, Guengerich FP, Shimada T. Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: effects of cytochrome P450-P450 and cytochrome P450-b5 interactions. Arch Biochem Biophys. 1997;342:329–337. doi: 10.1006/abbi.1997.0125. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki H, Nakamura M, Komatsu T, Ohyama K, Hatanaka N, Asahi S, Shimada N, Guengerich FP, Shimada T, Nakajima M, Yokoi T. Roles of NADPH-P450 reductase and apo- and holo-cytochrome b5 on xenobiotic oxidations catalyzed by 12 recombinant human cytochrome P450s expressed in membranes of Escherichia coli. Protein Expr Purif. 2002;24:329–337. doi: 10.1006/prep.2001.1578. [DOI] [PubMed] [Google Scholar]
- 10.Morgan ET, Coon MJ. Effects of cytochrome b5 on cytochrome P-450-catalyzed reactions. Studies with manganese-substituted cytochrome b5. Drug Metab Dispos. 1984;12:358–364. [PubMed] [Google Scholar]
- 11.Gruenke LD, Konopka K, Cadieu M, Waskell L. The stoichiometry of the cytochrome P-450-catalyzed metabolism of methoxyflurane and benzphetamine in the presence and absence of cytochrome b5. J Biol Chem. 1995;270:24707–24718. doi: 10.1074/jbc.270.42.24707. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrandt A, Estabrook RW. Arch Biochem Biophys. 1971;143:66–79. doi: 10.1016/0003-9861(71)90186-x. [DOI] [PubMed] [Google Scholar]
- 13.Bonfils C, Balny C, Maurel P. J Biol Chem. 1981;256:9457–9465. [PubMed] [Google Scholar]
- 14.Schenkman JB, Voznesensky AI, Jansson I. Arch Biochem Biophys. 1994;314:234–241. doi: 10.1006/abbi.1994.1435. [DOI] [PubMed] [Google Scholar]
- 15.Locuson CW, Wienkers LC, Jones JP, Tracy TS. CYP2C9 protein interactions with cytochrome b(5): effects on the coupling of catalysis. Drug Metab Dispos. 2007;35:1174–1181. doi: 10.1124/dmd.107.014910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilep AA, Guryev OL, Usanov SA, Estabrook RW. Apocytochrome b5 as an indicator of changes in heme accessability: preliminary studies with cytochrome P450 3A4. Journal of inorganic biochemistry. 2001;87:237–244. doi: 10.1016/s0162-0134(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 17.Tamburini PP, MacFarquhar S, Schenkman JB. Evidence of binary complex formations between cytochrome P-450, cytochrome b5, and NADPH-cytochrome P-450 reductase of hepatic microsomes. Biochem Biophys Res Commun. 1986;134:519–526. doi: 10.1016/s0006-291x(86)80451-x. [DOI] [PubMed] [Google Scholar]
- 18.Rodgers KK, Sligar SG. J Mol Biol. 1991;221:1453–1460. doi: 10.1016/0022-2836(91)90945-3. [DOI] [PubMed] [Google Scholar]
- 19.Gao Q, Doneanu CE, Shaffer SA, Adman ET, Goodlett DR, Nelson SD. Identification of the interactions between cytochrome P450 2E1 and cytochrome b5 by mass spectrometry and site-directed mutagenesis. J Biol Chem. 2006;281:20404–20417. doi: 10.1074/jbc.M601785200. [DOI] [PubMed] [Google Scholar]
- 20.Chen W, Koenigs LL, Thompson SJ, Peter RM, Rettie AE, Trager WF, Nelson SD. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol. 1998;11:295–301. doi: 10.1021/tx9701687. [DOI] [PubMed] [Google Scholar]
- 21.Di Primo C, Deprez E, Sligar SG, Hui Bon Hoa G. Origin of the photoacoustic signal in cytochrome P-450cam: role of the Arg186-Asp251-Lys178 bifurcated salt bridge. Biochemistry. 1997;36:112–118. doi: 10.1021/bi961508a. [DOI] [PubMed] [Google Scholar]
- 22.Yamazaki H, Shimada T, Martin MV, Guengerich FP. J Biol Chem. 2001;276:30885–30891. doi: 10.1074/jbc.M105011200. [DOI] [PubMed] [Google Scholar]
- 23.Yamazaki H, Nakamura M, Komatsu T, Ohyama K, Hatanaka N, Asahi S, Shimada N, Guengerich FP, Shimada T, Nakajima M, Yokoi T. Protein Expr Purif. 2002;24:329–337. doi: 10.1006/prep.2001.1578. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki H, Nakano M, Gillam EM, Bell LC, Guengerich FP, Shimada T. Biochem Pharmacol. 1996;52:301–309. doi: 10.1016/0006-2952(96)00208-0. [DOI] [PubMed] [Google Scholar]
- 25.Moore CD, al-Misky ON, Lecomte JT. Similarities in structure between holocytochrome b5 and apocytochrome b5: NMR studies of the histidine residues. Biochemistry. 1991;30:8357–8365. doi: 10.1021/bi00098a012. [DOI] [PubMed] [Google Scholar]
- 26.Moore CD, Lecomte JT. Structural properties of apocytochrome b5: presence of a stable native core. Biochemistry. 1990;29:1984–1989. doi: 10.1021/bi00460a004. [DOI] [PubMed] [Google Scholar]
- 27.Moore CD, Lecomte JT. Characterization of an independent structural unit in apocytochrome b5. Biochemistry. 1993;32:199–207. doi: 10.1021/bi00052a026. [DOI] [PubMed] [Google Scholar]
- 28.Schrag ML, Wienkers LC. Adv Exp Med Biol. 2001;500:347–350. [PubMed] [Google Scholar]
- 29.Jushchyshyn MI, Hutzler JM, Schrag ML, Wienkers LC. Arch Biochem Biophys. 2005;438:21–28. doi: 10.1016/j.abb.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi Y, Khan KK, He YA, He YQ, Halpert JR. Drug Metab Dispos. 2004;32:155–161. doi: 10.1124/dmd.32.1.155. [DOI] [PubMed] [Google Scholar]
- 31.Guryev OL, Gilep AA, Usanov SA, Estabrook RW. Biochemistry. 2001;40:5018–5031. doi: 10.1021/bi002305w. [DOI] [PubMed] [Google Scholar]
- 32.Holmans PL, Shet MS, Martin-Wixtrom CA, Fisher CW, Estabrook RW. Arch Biochem Biophys. 1994;312:554–565. doi: 10.1006/abbi.1994.1345. [DOI] [PubMed] [Google Scholar]
- 33.Gillam EM, Baba T, Kim BR, Ohmori S, Guengerich FP. Expression of modified human cytochrome P450 3A4 in Escherichia coli and purification and reconstitution of the enzyme. Arch Biochem Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- 34.Woods CM, Fernandez C, Kunze KL, Atkins WM. Allosteric activation of cytochrome P450 3A4 by alpha-naphthoflavone: branch point regulation revealed by isotope dilution analysis. Biochemistry. 2011;50:10041–10051. doi: 10.1021/bi2013454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmans PL, Shet MS, Martin-Wixtrom CA, Fisher CW, Estabrook RW. The high-level expression in Escherichia coli of the membrane-bound form of human and rat cytochrome b5 and studies on their mechanism of function. Arch Biochem Biophys. 1994;312:554–565. doi: 10.1006/abbi.1994.1345. [DOI] [PubMed] [Google Scholar]
- 36.Naffin-Olivos JL, Auchus RJ. Human cytochrome b5 requires residues E48 and E49 to stimulate the 17,20-lyase activity of cytochrome P450c17. Biochemistry. 2006;45:755–762. doi: 10.1021/bi051623y. [DOI] [PubMed] [Google Scholar]
- 37.Shen AL, Christensen MJ, Kasper CB. NADPH-cytochrome P-450 oxidoreductase. The role of cysteine 566 in catalysis and cofactor binding. J Biol Chem. 1991;266:19976–19980. [PubMed] [Google Scholar]
- 38.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 39.Schenkman JB, Jansson I. Drug Metab Rev. 1999;31:351–364. doi: 10.1081/dmr-100101923. [DOI] [PubMed] [Google Scholar]
- 40.Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. J Biol Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 41.Hlavica P, Schulze J, Lewis DF. J Inorg Biochem. 2003;96:279–297. doi: 10.1016/s0162-0134(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 42.Grabarek Z, Gergely J. Zero-length crosslinking procedure with the use of active esters. Analytical biochemistry. 1990;185:131–135. doi: 10.1016/0003-2697(90)90267-d. [DOI] [PubMed] [Google Scholar]
- 43.Mosher CM, Tai G, Rettie AE. CYP2C9 amino acid residues influencing phenytoin turnover and metabolite regio- and stereochemistry. J Pharmacol Exp Ther. 2009;329:938–944. doi: 10.1124/jpet.109.150706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Q, Doneanu CE, Shaffer SA, Adman ET, Goodlett DR, Nelson SD. Identification of the interactions between cytochrome P450 2E1 and cytochrome b5 by mass spectrometry and site-directed mutagenesis. J Biol Chem. 2006;29:20404–20417. doi: 10.1074/jbc.M601785200. [DOI] [PubMed] [Google Scholar]
- 45.Singh P, Shaffer SA, Scherl A, Holman C, Pfuetzner RA, Larson Freeman TJ, Miller SI, Hernandez P, Appel RD, Goodlett DR. Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal Chem. 2008;80:8799–8806. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schilling B, Row RH, Gibson BW, Guo X, Young MM. MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. Journal of the American Society for Mass Spectrometry. 2003;14:834–850. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 47.Yu ET, Hawkins A, Kuntz ID, Rahn LA, Rothfuss A, Sale K, Young MM, Yang CL, Pancerella CM, Fabris D. The collaboratory for MS3D: a new cyberinfrastructure for the structural elucidation of biological macromolecules and their assemblies using mass spectrometry-based approaches. Journal of proteome research. 2008;7:4848–4857. doi: 10.1021/pr800443f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw PM, Hosea NA, Thompson DV, Lenius JM, Guengerich FP. Reconstitution premixes for assays using purified recombinant human cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b5. Arch Biochem Biophys. 1997;348:107–115. doi: 10.1006/abbi.1997.0378. [DOI] [PubMed] [Google Scholar]
- 49.Kaspera R, Naraharisetti SB, Evangelista EA, Marciante KD, Psaty BM, Totah RA. Drug metabolism by CYP2C8.3 is determined by substrate dependent interactions with cytochrome P450 reductase and cytochrome b5. Biochemical pharmacology. 2011;82:681–691. doi: 10.1016/j.bcp.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clarke TA, Im SC, Bidwai A, Waskell L. The role of the length and sequence of the linker domain of cytochrome b5 in stimulating cytochrome P450 2B4 catalysis. J Biol Chem. 2004;279:36809–36818. doi: 10.1074/jbc.M406055200. [DOI] [PubMed] [Google Scholar]
- 51.Farooq Y, Roberts GC. Kinetics of electron transfer between NADPH-cytochrome P450 reductase and cytochrome P450 3A4. The Biochemical journal. 2010;432:485–493. doi: 10.1042/BJ20100744. [DOI] [PubMed] [Google Scholar]
- 52.Roberts AG, Campbell AP, Atkins WM. The thermodynamic landscape of testosterone binding to cytochrome P450 3A4: ligand binding and spin state equilibria. Biochemistry. 2005;44:1353–1366. doi: 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- 53.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 54.Cunane LM, Chen ZW, Durley RC, Mathews FS. X-ray structure of the cupredoxin amicyanin, from Paracoccus denitrificans, refined at 1.31 A resolution. Acta crystallographica Section D, Biological crystallography. 1996;52:676–686. doi: 10.1107/S0907444996001072. [DOI] [PubMed] [Google Scholar]
- 55.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 56.Berendsen HJC, Van Der Spoel D, Drunen RV. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 57.Lindahl E, Hess B, Van Der Spoel D. J Mol Mod. 2001:306–317. [Google Scholar]
- 58.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 59.Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PloS one. 2011;6:e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohta Y, Kawato S, Tagashira H, Takemori S, Kominami S. Dynamic structures of adrenocortical cytochrome P-450 in proteoliposomes and microsomes: protein rotation study. Biochemistry. 1992;31:12680–12687. doi: 10.1021/bi00165a019. [DOI] [PubMed] [Google Scholar]
- 61.Durley RC, Mathews FS. Refinement and structural analysis of bovine cytochrome b5 at 1.5 A resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:65–76. doi: 10.1107/S0907444995007827. [DOI] [PubMed] [Google Scholar]
- 62.Hlavica P, Schulze J, Lewis DF. Functional interaction of cytochrome P450 with its redox partners: a critical assessment and update of the topology of predicted contact regions. Journal of inorganic biochemistry. 2003;96:279–297. doi: 10.1016/s0162-0134(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 63.Im SC, Waskell L. The interaction of microsomal cytochrome P450 2B4 with its redox partners, cytochrome P450 reductase and cytochrome b(5) Arch Biochem Biophys. 2011;507:144–153. doi: 10.1016/j.abb.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendrychova T, Anzenbacherova E, Hudecek J, Skopalik J, Lange R, Hildebrandt P, Otyepka M, Anzenbacher P. Flexibility of human cytochrome P450 enzymes: molecular dynamics and spectroscopy reveal important function-related variations. Biochimica et biophysica acta. 2011;1814:58–68. doi: 10.1016/j.bbapap.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 65.Hendrychova T, Berka K, Navratilova V, Anzenbacher P, Otyepka M. Dynamics and hydration of the active sites of Mammalian cytochromes p450 probed by molecular dynamics simulations. Current drug metabolism. 2012;13:177–189. doi: 10.2174/138920012798918408. [DOI] [PubMed] [Google Scholar]
- 66.Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 67.Guryev OL, Gilep AA, Usanov SA, Estabrook RW. Interaction of apo-cytochrome b5 with cytochromes P4503A4 and P45017A: relevance of heme transfer reactions. Biochemistry. 2001;40:5018–5031. doi: 10.1021/bi002305w. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H, Hamdane D, Im SC, Waskell L. Cytochrome b5 inhibits electron transfer from NADPH-cytochrome P450 reductase to ferric cytochrome P450 2B4. J Biol Chem. 2008;283:5217–5225. doi: 10.1074/jbc.M709094200. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Myshkin E, Waskell L. Role of cytochrome b5 in catalysis by cytochrome P450 2B4. Biochem Biophys Res Commun. 2005;338:499–506. doi: 10.1016/j.bbrc.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 70.Wen B, Lampe JN, Roberts AG, Atkins WM, David Rodrigues A, Nelson SD. Cysteine 98 in CYP3A4 contributes to conformational integrity required for P450 interaction with CYP reductase. Arch Biochem Biophys. 2006;454:42–54. doi: 10.1016/j.abb.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang H, Im SC, Waskell L. Cytochrome b5 increases the rate of product formation by cytochrome P450 2B4 and competes with cytochrome P450 reductase for a binding site on cytochrome P450 2B4. J Biol Chem. 2007;282:29766–29776. doi: 10.1074/jbc.M703845200. [DOI] [PubMed] [Google Scholar]
- 72.Roussel FKK, Halpert JR. The importance of SRS-1 residues in catalytic specificity of human cytochrome P450 3A4. Arch Biochem Biophys. 2000;374:269–278. doi: 10.1006/abbi.1999.1599. [DOI] [PubMed] [Google Scholar]
- 73.Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Biochemistry. 2005;44:1833–1845. doi: 10.1021/bi048228c. [DOI] [PubMed] [Google Scholar]
- 74.Cojocaru V, Winn PJ, Wade RC. The ins and outs of cytochrome P450s. Biochimica et biophysica acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 75.Rui L, Pochapsky SS, Pochapsky TC. Comparison of the complexes formed by cytochrome P450cam with cytochrome b5 and putidaredoxin, two effectors of camphor hydroxylase activity. Biochemistry. 2006;45:3887–3897. doi: 10.1021/bi052318f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berka K, Hendrychova T, Anzenbacher P, Otyepka M. Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. The journal of physical chemistry A. 2011;115:11248–11255. doi: 10.1021/jp204488j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Banci L, Bertini I, Rosato A, Scacchieri S. Solution structure of oxidized microsomal rabbit cytochrome b5. Factors determining the heterogeneous binding of the heme. European journal of biochemistry / FEBS. 2000;267:755–766. doi: 10.1046/j.1432-1327.2000.01054.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.