

Abstract
The critical role of cytotoxic lymphocyte antigen 4 (CTLA-4) in inhibiting antigen-driven T cell responses upon engagement with its ligands, B7-1 and B7-2, and its importance for peripheral T cell tolerance and T cell homeostasis has been studied intensively. The CTLA-4 splice variant, li-CTLA-4 is expressed in naïve and activated T cells and can actively alter T cell signaling despite its lack of a B7 binding domain. To study the effect of li-CTLA-4 in regulating T cell responses in the context of autoimmunity, we engineered a B6.CTLA-4 (floxed-Exon2)-BAC-transgene, resulting in selective expression of li-CTLA-4 upon Cre-mediated deletion of Exon 2. Introducing the B6.BAC into the NOD background, which is genetically deficient for li-CTLA-4, restores mRNA levels of li-CTLA-4 to those observed in B6 mice. Further, re-expressing this ligand non-binding isoform in NOD mice reduced IFN-γ-production in T effector cells accompanied by a significant decrease in insulitis and T1D frequency. However, selective expression of li-CTLA-4 could not fully rescue the CTLA-4KO disease phenotype when bred onto NOD.BDC2.5.CTLA-4KO background due to the requirement of the full-length, B7-binding CTLA-4 molecule on T effector cells. Thus, the li-CTLA-4 form, when expressed at physiologic levels in the CTLA-4 sufficient NOD background can suppress autoimmunity, however, the functionality of the li-CTLA-4 isoform depends on the presence of the full-length molecule to alter effector T cell signaling.
Introduction
The negative regulator cytotoxic T lymphocyte antigen 4 (CTLA-4) has been established to critically affect T cell function and peripheral tolerance (1–3). In humans, the CTLA-4 gene has been reported to contribute to a general susceptibility for autoimmune diseases, especially for endocrine disorders but also SLE, MS and RA (4–6). CTLA-4 deficiency in mice results in a severe form of lymphoproliferative disease accompanied by multiorgan infiltration and death within 2–5 weeks after birth (7,8). The inhibitory function of CTLA-4 is most obvious upon antigen-specific T cell activation, resulting in decreased T cell proliferation and cytokine production, consequently diminishing the T cell response (9–11). Several mechanisms of CTLA-4 mediated suppression have been proposed, such as competition of CTLA-4 with CD28 for binding to their shared ligands B7-1 and B7-2 (1,12–14). In addition, it has been suggested that accumulation of CTLA-4 within the immunological synapse disrupts CD28 localization therein (15–17). Both mechanisms of action have been described to be dependent on the extracellular domain of CTLA-4 (1,15). Thus, many efforts have focused on this ligand interaction to address CTLA-4 function in controlling T cell responses. However, multiple studies have described an additional mechanism for CTLA-4 inhibitory function mediated by its intracellular domain. We and others have shown that the biochemical basis for CTLA-4 function is associated with the interaction of the tyrosine phosphatases, PP2A and SHP-2 (12,18–21) with the cytoplasmic tail of CTLA-4 and that this interaction promotes dephosphorylation of the TCRζ chain as well as other TCR complex components like LAT and ZAP70 (22,23). Moreover, CTLA-4 was demonstrated to inhibit ERK phosphorylation/activation as well as c-JNK and therefore additionally regulates signaling members of the mitogen-associated protein kinase (MAPK) family (24–26). These results describe a mechanism of action by which CTLA-4 can block TCR proximal and distal signaling and consequently attenuates cell cycle progression, cytokine production and proliferation through its cytoplasmic tail. Interestingly, recent work by Singer et al indeed showed that TCR hypo-signaling requires CTLA-4’s internal domain but also demonstrated that this can occur in a co-stimulatory/B7-independent fashion (27). In addition, we have found that CTLA-4 within lipid rafts is able to control TCR signaling events in the absence of B7 engagement (Chikuma S. and Bluestone J.A, unpublished observations). Most significantly in this context is the recent discovery of a naturally occurring CTLA-4 isoform (li-CTLA-4), which acts in a B7 ligand-independent fashion (28). Kuchroo and colleagues reported that over-expression of li-CTLA-4 in T cells reduced proliferation, cytokine production and TCRζ phosphorylation in CTLA-4KO T cells transfected with li-CTLA-4 (29). This isoform of CTLA-4 has been associated with type 1 diabetes (T1D) in humans and the NOD mouse model (28,30,31). In the latter, the CTLA-4 gene was mapped within the T1D susceptibility locus Idd5.1 (32,33) and recent studies suggested that linkage between disease and the CTLA-4 gene is primarily based on a significant reduction in li-CTLA-4, since the NOD Idd5.1 interval produces approximately 70% less li-CTLA-4 than does the B6 allele (28). The molecular basis for this observation has been suggested to a single nucleotide polymorphism (SNP) in Exon 2 of the CTLA-4 gene (28), resulting in altered splicing favoring the inclusion of Exon2, which encodes the extracellular domain of the protein. Recently, Kuchroo’s and Wicker’s group showed that constitutive expression of only li-CTLA-4 resulted in reduced NOD diabetes frequency (34). However, the use of constitutive promoters results in non-physiological transgene expression levels which may have had non-physiologic effects, especially considering the differential expression of li-CTLA-4 among specific T cell subsets and upon T cell activation (29,35,36). Thus, we aimed to study the role of li-CTLA-4 in the context of autoimmune diabetes by changing the physiologic levels of the isoform in NOD mice to B6 levels. In addition, we examined how small changes in levels of the li-CTLA-4 isoform in the presence or absence of full length CTLA-4 altered T cell function. The B6.CTLA-4.BAC was bred onto the NOD background under conditions that delete exon 2 resulting in B6-level expression of the li-CTLA-4 isoform. Increased expression of li-CTLA-4 to B6 levels significantly reduced T1D incidence in NOD mice. Further analyses on the NOD.BDC2.5.CTLA-4KO background showed that protection from T1D development requires full length CTLA-4 expression on T effector versus T regulatory cells. Our results clearly demonstrate the importance of li-CTLA-4 in controlling T1D development in the NOD mouse and describe, for the first time, that this effect strongly depends on the presence of a full-length molecule.
Material and methods
Generation of NOD.B6-CTLA-4 – exon 2 deleting BAC transgenic mice
A B6.BAC (RP24-316C5) containing exclusively the CTLA-4 locus was engineered to express loxP sequences upstream and downstream of the exon 2 of the ctla4 gene, which encodes the entire extracellular domain. The modified BAC was purified using cesium chloride gradient ultracentrifugation and microinjected by the UCSF Transgenic/Targeted Mutagenesis Core Facility into pronuclei of nonobese diabetic (NOD) mouse embryos. Founder mice carrying the B6.CTLA-4(floxed-Exon 2)-BAC-transgene were crossed to the VAV-Cre deleter-strain (termed NOD-Tg-Cre mice) (37). Vav1-Cre transgenic mice were a gift from Dr. Kioussis. In some studies, the mice were further bred onto the NOD.CTLA-4 KO or NOD.BDC2.5.CTLA-4 KO strains, which were generated and maintained in our laboratory (7). All mice were back-crossed and maintained on the NOD background. Mice were housed in a specific pathogen-free facility at the University of California at San Francisco. All experiments complied with the Animal Welfare Act and the National Institutes of Health guidelines for the ethical care and use of animals in biomedical research and were approved by the Institutional Animal Care and Use Committee of the University of California at San Francisco.
Assessment of Diabetes and Insulitis
Diabetes development was assessed weekly by testing either urinary or blood glucose levels with Diastix (Bayer) or a Lifescan glucose meter (One Touch II; Lifescan, Roche), respectively. Mice were considered diabetic after two consecutive measurements of >250 mg/dl. Diabetes was also induced in 8 week old pre-diabetic mice by two intraperitoneal injections of cyclophosphamide (200 mg/kg) on days 0 and 7. Diabetes onset was monitored daily for 36 days. The severity of insulitis in 20–24 week old pre-diabetic mice was analyzed using formalin-fixed (10%) and paraffin-embedded pancreatic tissues. After hematoxylin and eosin staining, individual islets were given a score based on the degree of lymphocytic infiltration as described previously (38).
Flow cytometry and cell sorting
Labeled antibodies specific for CD4 (RM4-5), CD44 (IM7), CD25 (PC61), CD62L (MEL14), GITR (DTA-1), ICOS (7E.17G9), Helios (22F6), CD152 (UC10-4F10), FoxP3 (FJK-16s), and IFN-γ (XMG1.2) were purchased from BD Biosciences or eBioscience. Isolated cells from total or pancreatic lymph nodes were stained with commercially available antibodies, listed above. Intracellular staining for FoxP3, CTLA-4 (CD152) and IFN-γ was performed using a FoxP3 staining buffer set, according to the manufacturer’s instructions (eBioscience). Stained single-cell suspensions were analyzed using a BD LSRII flow cytometer running FACSDiva software (BD Biosciences). Cell sorting experiments were performed with a MoFlo cytometer high-speed cell sorter (Dako) or a FACSAria (BD Biosciences).
Adoptive Transfer experiments
CD4+/CD62Lhi/CD25− naive T cells from NOD.BDC2.5 mice were purified by FACS-sorting and stimulated in vitro for 3 days with soluble anti-CD3 (1μg/ml) and anti-CD28 (1μg/ml) in the presence of 200U/ml IL-2. The number of cells indicated in each experiment was adoptively transferred into full NOD recipient mice by retro-orbital injection. Diabetes development was monitored daily for 30 days as described above. NOD.CD28KO recipients (5 weeks old) were injected with 5×105 primary CD4+/CD62Lhi/CD25+ T regulatory cells, isolated from spleen and lymph nodes of NOD.BDC2.5-transgenic donor mice. Diabetes development was assessed weekly as described above.
Assessment of cytokine production
FACS-sorted CD4+/CD62Lhi/CD25− T naïve cells from lymph nodes of NOD and NOD-Tg-Cre mice were stimulated with soluble anti-CD3 (1μg/ml) and anti-CD28 (1μg/ml) in the presence of 200U/ml IL2. At day 3, cells were stimulation with 50ng/ml PMA, 500μM Ionomycin and 2μM Monensin for 5 hours and IFN-γ production was examined by flow cytometry as described above.
Real-time PCR analysis
RNA was isolated from sorted CD4+ T conventional or CD4+/CD62Lhi/CD25+ regulatory T cells with an RNeasy Kit (Qiagen) and reverse transcribed using a Superscript kit (Invitrogen) according to the manufacturers’ instructions. The amounts of li-CTLA-4 and fl-CTLA-4 cDNA were measured using quantitative real-time PCR analysis (GeneAmp 7900; Applied Biosystems) and were normalized to 18S (Eukaryotic 18S rRNA). The TaqMan primer-probe for 18S was purchased from Applied Biosystems (ID: 4333760F). Primers and probes for li-CTLA-4 and fl-CTLA-4, according to Gerold et al., 2011 (39), were purchased from Integrated DNA Technologies.
Statistical analysis
All statistical analyses were performed using an unpaired two-tailed Mann-Whitney U test. Values of P ≤ 0.05 were considered significant.
Results
Generation of CTLA-4.Exon 2 floxed BAC-transgenic mice and selective expression of li-CTLA-4
CTLA-4 transgenic mice were generated using a Bacterial Artificial Chromosome (BAC) construct that contains the entire B6.CTLA-4 locus. This strategy ensures appropriate expression of both, the full-length and the ligand-independent CTLA-4 isoform by utilizing the gene’s endogenous promoter. The B6.BAC was further engineered to express loxP sequences flanking Exon 2 of the CTLA-4 gene. Exon 2 encodes the extracellular, B7 binding domain of the CTLA-4 molecule and thus, Cre mediated deletion of Exon 2 results in expression of only the ligand-independent isoform from the transgene. First, we introduced the B6.BAC into the full NOD background. Selective expression of li-CTLA-4 from the B6.BAC was achieved by breeding founders to a Vav-Cre deleter strain (Fig 1A). In some experiments, mice expressing the transgene were backcrossed onto the NOD.BDC2.5.CTLA-4KO background resulting in distinct lines that could be used to examine the role of li-CTLA-4 in normal NOD (NOD-Tg-Cre) and T cell receptor transgenic, CTLA-4-deficient, settings (Fig 1B).
Figure 1. Generation of CTLA-4 BAC-transgenic mice and expression of ligand-independent and full-length CTLA-4 isoforms.

(A) Targeting strategy to generate CTLA-4 BAC-transgenic mice. A B6.BAC (RP24-316C5) containing exclusively the CTLA-4 locus was engineered to express 2 loxP sites, flanking Exon2 of the CTLA-4 gene. The modified B6.BAC was introduced into the NOD background and founders were crossed to the VAV-Cre deleter-strain to achieve excision of floxed Exon2, resulting in selective expression of only the B7, ligand non-binding isoform of CTLA-4. (B) Schematic representation of CTLA-4 mutant mice generated by introducing the B6.CTLA-4.BAC in the full NOD and NOD.BDC2.5 CTLA-4KO background. Expression of the full-length and or ligand-independent CTLA-4 isoforms from either the endogenous NOD or the B6-transgene allele is indicated for each mouse. (C) mRNA expression of full-length and ligand-independent CTLA-4 in CD4+ T cells of B6, NOD and NOD-Tg-Cre mice. Expression levels were normalized to 18S and are displayed as relative expression (mean ± SD).
We first evaluated the effects of expressing “B6” levels of the li-CTLA-4 in the li-CTLA-4-deficient NOD background (Fig 1B, left panel). Real time PCR analyses were performed to measure and compare expression levels of ligand-independent and full-length CTLA-4 in FACS sorted CD4+ T cells from B6, wild-type NOD and NOD-Tg-Cre mice. Consistent with previous observations (28,34), there were significantly lower levels of li-CTLA-4 mRNA levels in autoimmune susceptible NOD mice as compared to B6 mice. In contrast, li-CTLA-4 mRNA levels in NOD-Tg-Cre mice resembled that observed in B6 mice, demonstrating successful expression of li-CTLA-4 from the BAC transgene at physiological expression levels (Fig 1C, left panel). Moreover, the levels of full-length CTLA-4 mRNA levels were equivalent when comparing T cells from B6, NOD and NOD-Tg-Cre mice suggesting that the expression of the BAC did not alter full length CTLA-4 expression (Fig 1C, right panel). Specific analyses of CTLA-4 intracellular and cell surface expression suggested that NOD-Tg-Cre T cells expressed slightly higher surface levels of the full-length CTLA-4 molecule in the CD4+/CD44+ T effector/memory compartment; however, those changes did not reach statistical significance (Fig. 2A and suppl. Fig. 1A, left panel).
Figure 2. T cell phenotype in prediabetic NOD mice expressing ligand-independent CTLA-4 at B6 levels.

PancLN cells from 7 week old prediabetic wild-type NOD and NOD-Tg-Cre mice were stained for CD4, CD44, CD62L, CD25, GITR, ICOS and full-length CTLA-4 (fl-CTLA-4) to assess the activation status of CD4+ conventional (A), CD4+FoxP3+ regulatory T cells (B) and T effector/memory populations (C). (D) Total cellularity of pancreatic lymph node (pancLN), axillary lymph node (axLN) and spleen (SP) comparing 7 week old prediabetic wild-type NOD and NOD-Tg-Cre mice.
Expression of increased levels of li-CTLA-4 does not alter the overall T cell numbers or phenotype in prediabetic NOD mice
The autoimmune susceptible strain NOD is genetically deficient in the li-CTLA-4 isoform (28) due to a single nucleotide polymorphism that alters CTLA-4 gene splicing. Increased expression of li-CTLA-4 in NOD-Tg-Cre mice, at levels observed in B6 mice, did not alter the overall immune cell subset distribution and activation state. There were equal protein expression levels of CD25, GITR, and ICOS suggesting that there were no differences in the activation status of T conventional (Tconv) versus T regulatory (Treg) cells (Fig. 2A–C; Suppl. Fig. 1A, right panel). Furthermore, total cellularity of the lymphatic organs, spleen, axillary lymph nodes (axLN) and pancreatic lymph nodes (pancLN) was unaltered (Fig 2D). In addition, comparable numbers of total CD4+ T conventional, as well as effector/memory and naive T cells (suppl. Fig 1B) were observed when comparing the NOD-Tg-Cre to WT-NOD mice.
NOD mice expressing li-CTLA-4 at B6 levels have reduced insulitis and are protected from cyclophosphamide-induced diabetes
Utilizing our NOD.CTLA-4.BAC-transgenic mouse, which selectively expresses li-CTLA-4 at B6 levels, allowed us to address the biological significance of this CTLA-4 isoform in the context of T1D development in the NOD mouse. T1D development in 8-week NOD and NOD-Tg-Cre mice was examined following 2 injections of 200mg/kg Cyclophosphamide (CTX) at d0 and 7, which has been shown to synchronize and accelerate disease onset and has been used to study NOD.Idd5.1-congenic mice. We observed a significant reduction in disease frequency with only 42% incidence in NOD-Tg-Cre mice as compared to 88% in wild-type littermate controls (Fig 3A). The level of protection was comparable to the one observed in CTX-treated NOD.Idd5.1-congenic mice (40). Next, we compared the degree of insulitis in 20–24 week old pre-diabetic NOD and NOD-Tg-Cre mice. The individual islets were scored based on the severity of insulitis (38). The degree of islet infiltration was significantly reduced in NOD-Tg-Cre mice as compared to wildtype littermates (Fig 3B). All mice used were normal glycemic. These results confirm other genetic studies, which suggested that the reduced disease observed in NOD.Idd5.1-congenic mice was due to increased li-CTLA-4 expression (34).
Figure 3. Reduced diabetes frequency in NOD mice expressing li-CTLA-4 at B6 levels.
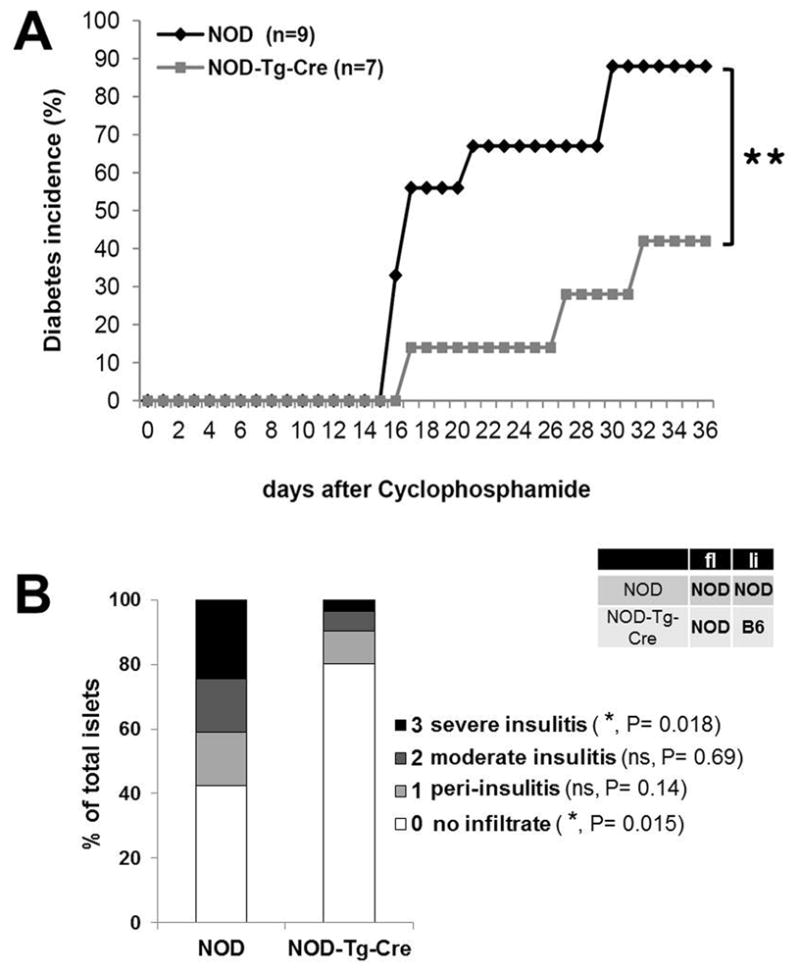
(A) Diabetes incidence in 8 week old NOD and NOD-Tg-Cre mice, injected intra peritoneal with 200mg/kg cyclophosphamide at days 0 and 7. Statistical analysis was performed using the Logrank test (**, P < 0.01). (B) Severity of pancreatic islet infiltration in 20 week old pre-diabetic NOD and NOD-Tg-Cre mice. Insulitis scores are representative for a total of 550 islets per group. Statistical analysis was performed using an unpaired t test.
Expression of li-CTLA-4 in NOD mice effects T effector cells but not T regulatory cells
The clinical result described above would be consistent either with changes in the activation state of the conventional T cells or changes in the Treg population. Therefore, we examined the effect of altered li-CTLA-4 expression on individual T cell subsets. Quantitative PCR was performed to measure expression levels of full-length and ligand-independent CTLA-4 in FoxP3+ T cells in wild type NOD versus NOD-Tg-Cre Treg cells. We identified similar levels of full length (fl)-CTLA-4 mRNA but increased li-CTLA-4 expression in Tregs from NOD-Tg-Cre mice compared to wild-type littermates (Fig. 4A), which reflects the higher mRNA levels of li-CTLA-4 from the B6 allele. These results were similar to what we observed in CD4+ T conventional cells (Fig. 1C). However, there were no detectable differences in the absolute numbers of FoxP3+ T cells or in FoxP3 expression levels on a per cell basis between NOD and NOD-Tg-Cre mice (Fig 4B). These results were consistent with recent studies suggesting that the intracellular domain of CTLA-4 controls some aspects of Treg function (27). Further analyses of the thymically-derived, natural (nTreg) versus peripherally-developing adaptive Treg (aTreg) subsets in the NOD-Tg-Cre mice were performed based on the expression of the transcription factor, Helios (41,42). The percentage and ratio of nTreg (FoxP3+/Helios+) versus aTreg (FoxP3+/Helioslo) cells were not altered when compared to wild-type NOD mice (Fig. 4C). Although there were no changes in the overall Treg cell phenotype under steady state conditions in prediabetic NOD-Tg-Cre mice, we addressed whether increased levels of the li-CTLA-4 protein in Tregs affected their suppressive activity. NOD-Tg-Cre mice were bred with the BDC2.5 T cell receptor (TCR) transgenic mouse strain. The TCR expressed on the T cells in this mouse recognizes an islet autoantigen, Chromogranin A (43). Previous studies have shown that BDC2.5 T effector cells can induce T1D upon adoptive transfer (44) while the transfer of BDC2.5 Tregs can block the development of diabetes when transferred into NOD or highly susceptible Treg-deficient, CD28-deficient NOD mice (45–47). Thus, purified Tregs from NOD.BDC2.5 and NOD.BDC2.5-Tg-Cre mice were adoptively transferred into NOD.CD28KO mice at 5 weeks of age. As shown in Fig.4D, the adoptive transfer of 5×104 BDC2.5.NOD Tregs delayed disease onset and decreased T1D incidence (66% as compared to 100% in untreated recipients). Similar results were observed using BDC2.5 Tregs derived from the NOD-Tg-Cre mice. Thus, expression of li-CTLA-4 at B6 levels in the full NOD background does not alter Treg cell function in vitro (data not shown) or in vivo.
Figure 4. Expression of li-CTLA-4 in NOD mice diminishes T effector cell function but does not affect T regulatory cells.

(A) Relative levels of fl- and li-CTLA-4 mRNA expression normalized to 18S expression in sorted CD4+CD25hiCD62L− Treg cells from NOD and NOD-Tg-Cre mice (mean ± SD). (B) Regulatory T cells numbers displayed as percentage of total pancLN cells (left panel) and FoxP3 protein expression levels (right panel). (C) Percentages of natural (CD4+FoxP3+Helios+) and adoptive (CD4+FoxP3+Helioslow) T regulatory cells in pancLN cells from NOD and NOD-Tg-Cre mice. (D) Diabetes frequency in NOD.CD28KO recipients adoptively transferred with either no or of 5×104 BDC2.5-Treg cells from NOD and NOD-Tg-Cre mice, respectively. Statistical analysis was performed using the Logrank test (*, P < 0.05; **, P < 0.01). (E) Percentage of CD4+/CD44+ activated T effector cells in pancLN and pancreas of NOD and NOD-Tg-Cre mice. (F) Sorted CD4+/CD62Lhi T naïve cells from lymph nodes of NOD and NOD-Tg-Cre mice were activated with anti-CD3 and anti-CD28 in the presence of 200U/ml IL2. At day 3, cells were stimulated with PMA/Ionomycin/Monensin for 5 hours and the percentage of IFN-γ producing CD4+ T cells was examined by flow cytometry. Shown are representative flow plots (F, left panel) and the fold changes (F, right panel) of IFN-γ producing CD4+ T cells from NOD and NOD-Tg-Cre mice. Statistical analyses in E and F were performed using an unpaired t test (*, P < 0.05; **, P < 0.01).
Attention was, therefore, focused on the T conventional populations to determine what affect higher expression of li-CTLA-4 had on the development and function of T effector cells. As seen in Fig 4E, there was a marked decrease in the percent of activated T effector cells in the pancreatic lymph nodes and pancreas of NOD-Tg-Cre mice. In the pancreatic lymph node, the percentage of activated CD4+/CD44+ T cells was reduced from 15 % ± 0.46% in WT to 10.5% ± 1.1% in Tg-Cre mice. Similarly, the percentage of CD4+ T cells infiltrating the pancreas were decreased from 9.6% ± 2% in wild type NOD mice to 3.8% ± 0.65% in Tg-Cre mice. Since we observed that re-expression of li-CTLA-4 in NOD mice does not affect the overall phenotype and function of Treg cells, it was likely that altered T effector cell expansion was an intrinsic effect on this T cell subset and not reflective of changes in Treg activity. In order to directly examine the possibility that li-CTLA-4 negatively affects T cell activation, NOD and NOD-Tg-Cre CD4+ T cells were stimulated with anti-CD3 and anti-CD28 for 3 days and IFN-γ production was examined by flow cytometry. Under these conditions, there was significantly reduced induction of IFN-γ producing CD4+ T cells in NOD-Tg-Cre mice as compared to wild-type littermates (Fig. 4F) suggesting that increased expression of the li-CTLA-4 isoform altered T cell activation due to the increased ability of the cytoplasmic domain of CTLA-4 to alter T cell signal transduction.
Selective expression of the li-CTLA-4 isoform in the full length CTLA-4-deficient background does not protect from T1D development
Previous studies by our group and others have shown that ligand independent forms of CTLA-4 can alter the progression of the hyperproliferation observed in the CTLA-4-deficient setting (34,48). However, those studies did not address the potential importance of expression of the full-length CTLA-4 in the context of antigen-specific T cell responses in the T1D setting. Therefore, we bred the NOD-Tg and NOD-Tg-Cre mice onto the NOD.BDC2.5.CTLA-4KO background, as Mathis and colleagues had previously shown that breeding the CTLA-4 KO to the NOD.BDC2.5-TCR transgenic mouse rescued the systemic autoimmune phenotype observed in straight CTLA-4 KO mice. Moreover, they observed that these mice exhibited accelerated diabetes development between 6–8 weeks of age (11). This allowed us to distinguish the effects of the li-CTLA-4 isoform in a selective antigen-specific TCR Tg background from effects on the overall disruption of immune homeostasis seen in CTLA-4 KO mice that leads to death in 2–5 weeks. Mice on the BDC2.5.CTLA-4KO background expressing both the full-length and li-CTLA-4 isoforms from the B6-BAC in the absence of the VAV-Cre transgene are referred to as BDC-KO-Tg mice. For comparison, BDC2.5.CTLA-4KO mice expressing the VAV-Cre transgene and thus only express li-CTLA-4 from the B6-BAC are referred to as BDC-KO-Tg-Cre mice (Fig 1B, right panel). First, we compared the level of expression the both CTLA-4 isoforms in CD4+ T cells isolated from NOD.BDC2.5.CTLA-4 WT, KO, KO-Tg or KO-Tg-Cre mice. As expected, only the li-CTLA-4 isoform was observed in the BDC-KO-Tg-Cre T cells, indicating that the expression of the Cre resulted in a highly efficient recombination of floxed Exon 2 (Fig. 5 A). In contrast, similar mRNA and protein levels of fl-CTLA-4 could be detected in BDC-WT and BDC-KO-Tg mice (Fig. 5 A and suppl. Fig. 2 A). There was increased expression of li-CTLA-4 in BDC-KO-Tg and BDC-KO-Tg-Cre mice as compared to BDC-WT NOD mice consistent with the inherent genetic deficiency of the ligand independent form in this genetic setting. Importantly, the expression of both the full-length and the ligand-independent isoform from the full BAC transgene in BDC-KO-Tg mice completely rescued the mice from increased lymph node cellularity (Fig 5B) as well as the development of a highly activated T cell phenotype and increase in CD25+/FoxP3+ regulatory T cells detected in BDC.CTLA-4KO mice at 6 weeks of age (Fig 5C). The physiologic expression of only the li-CTLA-4 isoform in BDC-KO-Tg-Cre mice rescued the highly increased lymph node cellularity. This resulted in a significant but not total reduction of CD4+/CD44+ T effector cells in BDC.CTLA-4KO mice (Fig. 5 B–C and suppl. Fig 2 B). Similarly, the percentage and total number of CD4+ FoxP3+ Tregs in BDC-KO-Tg-Cre mice was comparable to those observed in BDC-KO-Tg and WT littermates (Fig. 5 C and suppl. Fig 2 B).
Figure 5. Selective expression of li-CTLA-4 partially rescues the T cell phenotype observed in NOD.BDC2.5.CTLA-4KO mice.

Relative levels of fl- and li-CTLA-4 mRNA expression in CD4+ T cells (A) and lymph node cellularity (B) in 6 week old NOD.BDC2.5.CTLA-4 WT, KO, KO-Tg or KO-Tg-Cre mice. (C) Representative flow plots, showing the frequencies of activated CD4+/CD44+/CD62L- T effector and resting CD4+/CD44-/CD62Lhi naïve T cells (upper panel) as well as CD4+/CD25+/FoxP3+ regulatory T cells (lower panel) in lymph node cells from 6 week old mice. (D) Quantification of frequencies of CD44+ T effector and FoxP3+ regulatory T cells determined in (C) and expressed as percentage of CD4+ T cells. Statistical analyses were performed using an unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.0001).
Next, we determined the effect of the different isoforms on diabetes incidence by comparing NOD.BDC2.5.CTLA-4 WT, KO, KO-Tg and KO-Tg-Cre mice (Fig. 6A). We found that BDC-KO-Tg mice, expressing fl- and li-CTLA-4 from the BAC-transgene, had a lower incidence of T1D than WT NOD.BDC2.5 littermates (Fig 6A). Although the difference in disease incidence did not reach statistical significance, these results corroborate those described in the conventional NOD-Tg-Cre mice, in which the higher li-CTLA-4 expression introduced by the B6 allele significantly ameliorates disease (Fig. 3). The results support a key role of the li-CTLA-4 isoform in controlling T cell autoreactivity independent of TCR specificity. However, when the li-CTLA-4 isoform was examined in the absence of the full-length molecule a different outcome was observed. The NOD.BDC2.5-KO-Tg-Cre mice expressing only the B6-BAC-derived li-CTLA-4 isoform developed diabetes with the same kinetics observed in straight NOD.BDC2.5.CTLA-4KO mice. Thus, despite the partial rescue of the generalized activated T cell phenotype in BDC-KO-Tg-Cre mice, expression of the ligand non-binding CTLA-4 isoform did not confer protection from autoimmunity in the absence of the full-length molecule. Next, we examined whether the requirement of full-length CTLA-4 in conjunction with li-CTLA-4 isoform was essential on T effector and or Treg cells. Adoptive transfer experiments were performed in which NOD.BDC2.5.CTLA-4 WT, NOD.BDC2.5.CTLA-4 KO and NOD.BDC2.5.CTLA-4 KO-Tg-Cre Tregs were transferred into NOD.CD28KO recipients to compare their relative in vivo suppressive activity. As depicted in figure 6B, adoptive transfer of Tregs from NOD.BDC2.5.CTLA-4 WT, NOD.BDC2.5.CTLA-4 KO or NOD.BDC2.5.CTLA-4 KO-Tg-Cre mice display equal suppressive activity, similarly reducing diabetes incidence. Thus, it appears that the absence of the fl-CTLA-4 molecule in BDC-KO-Tg-Cre Tregs did not account for the loss of disease protection in those mice. Finally, we examined the potential of NOD.BDC2.5.CTLA-4 WT, NOD.BDC2.5.CTLA-4 KO and NOD.BDC2.5.CTLA-4 KO-Tg-Cre T effector cells to cause diabetes in an adaptive transfer model. One million in vitro activated T effector cells were transferred into full NOD recipients. The BDC2.5 T effector cells expressing only the li-CTLA-4 isoform induced disease more rapidly as compared to BDC-WT cells expressing both the truncated and full-length CTLA-4 molecules. Moreover, disease kinetics was comparable when transferring BDC2.5-T effectors expressing only the li-CTLA-4 isoform and T effectors completely deficient for CTLA-4 (Fig. 6C). In addition, the adoptive transfer of 5×105 WT-BDC2.5 T effectors caused disease in only 33% of full NODs, whereas the same number of NOD.BDC2.5.CTLA-4 KO or NOD.BDC2.5.CTLA-4 KO-Tg-Cre T effectors induced diabetes in 100% and 75% of recipients, respectively (suppl. Fig. 3A). Similar results were observed following the adoptive transfer of 1×106 naive BDC2.5 T cells in NOD.RAG KO mice (data not shown). The differences in disease incidence (100% versus 75%) upon transfer of 5×105 CTLA-4KO versus KO-Tg-Cre T effector cells could be due to a decrease in IFN-γ producing T effector cells in KO-Tg-Cre mice as compared to CTLA-4KO littermates (suppl. Fig. 3 B). Chikuma et al. (48) reported a similar finding of intermediate IFN-γ production in T cells over-expressing li-CTLA-4 when compared to WT and CTLA-4KO T cells. Thus, T cells from KO-Tg-Cre mice are more potent in transferring disease as compared to WT T cells but appear to have slightly diminished T effector function when compared to T cells from CTLA-4 deficient animals. It should be noted that we were unable to examine selective expression of li-CTLA-4 in Treg cells only using the FoxP3-Cre-deleter strain (49), since we could not obtain homozygous CTLA-4KO mice carrying the FoxP3-Cre-Tg. It appeared that the insertion site of the FoxP3-Cre-Tg is in close proximity to the endogenous CTLA-4 locus, thus preventing independent heredity/transmission of both alleles. However, taken together, the data indicate that despite the presence of the li-CTLA-4 isoform, the absence of a full-length, B7 binding, CTLA-4 molecule results in a more aggressive T effector population and that even low numbers of antigen-specific li-CTLA-4 T effectors can confer disease development. These results are summarized in Table 1.
Figure 6. Selective expression of li-CTLA-4 in the NOD.BDC2.5.CTLA-4KO background does not protect from T1D development.

(A) Diabetes incidence comparing NOD.BDC2.5.CTLA-4 WT, KO, KO-Tg or KO-Tg-Cre mice. (B) Diabetes frequency in NOD.CD28KO recipients adoptively transferred with either no or of 5×104 BDC2.5-Treg cells from CTLA-4 WT, KO, KO-Tg or KO-Tg-Cre mice. (C) Diabetes development in full NOD recipients after adoptive transfer of 1×106 in vitro activated BDC2.5-T effector cells from CTLA-4 WT, KO or KO-Tg-Cre mice, respectively. Statistical analyses were performed using the Logrank test (*, P < 0.05; **, P < 0.01; ***, P < 0.0001).
Table 1.
Comparative summary of results obtained upon selective expression of li-CTLA-4.
| WT | KO | KO-Tg | KO-Tg-Cre | ||||
|---|---|---|---|---|---|---|---|
| fl-CTLA-4 (NOD) | li-CTLA-4 (NOD) | fl-CTLA-4 | li-CTLA-4 | fl-CTLA-4 (B6) | li-CTLA-4 (B6) | fl-CTLA-4 | li-CTLA-4 (B6) |
| + | + | − | − | + | + | − | + |
| Compared to WT | |||||||
| T1D development | exacerbated | ameliorated | exacerbated | ||||
| Treg suppressive function | normal | n.d | normal | ||||
| T effector function | increased | n.d | increased compared to WT | ||||
| reduced compared to KO | |||||||
Discussion
CTLA-4 is known to control important checkpoints for T cell function and has been demonstrated to be essential for peripheral T cell tolerance in the autoimmune setting and critically affects homeostatic proliferation in neonates (1–3,7,8). The biology of CTLA-4 has been complicated by the recent discovery of a truncated form of CTLA-4, li-CTLA-4, which does not express an extracellular domain and thus cannot bind its ligands, B7-1 and B7-2 (28). Over-expression of li-CTLA-4 inhibits T-cell activation in vitro (29) and can function in vivo to delay CTLA-4 KO disease onset and progression (34,48). Significantly, one of the major genetic loci of diabetes susceptibility in NOD mice, Idd5, is linked to a 70% reduction of this truncated, ligand non-binding form of the molecule (28,32,33). In this study, we addressed the biological significance of physiologic levels of li-CTLA-4 in the context of type 1 diabetes development in the NOD mouse. We generated a novel mutant mouse by introducing a floxed B6-CTLA-4-BAC transgene into the NOD background. We observed physiological expression of li-CTLA-4 at levels observed in B6 mice as well as the selective deletion of the B6-CTLA-4 BAC derived full-length CTLA-4 isoform. Using this approach, we showed that increasing li-CTLA-4 expression to levels observed in B6 mice decreases insulitis in the replete NOD mouse and significantly reduced diabetes incidence in the cyclophosphamide model of T1D to levels comparable to those observed in NOD.Idd5.1-congenic mice (40). These results are consistent with a recent study suggesting that disease protection conferred by the B10-Idd5.1 allele is exclusively mediated by li-CTLA-4 (34). Moreover, these studies extended the previous observations in several aspects including the demonstration that expression of li-CTLA-4 at B6 levels in the NOD background does not alter the overall immune homeostasis with regard to the activation state of naïve T conventional and T regulatory cell subsets under steady state conditions. In addition, we showed that disease protection, when increasing the expression of li-CTLA-4 in the full NOD background, is unlikely to be based on improved Treg function but rather a direct consequence of enhanced negative signaling in T effector cells, resulting in reduced cytokine production and diminished T cell activation.
In the past, several studies by us, and others, suggested that li-CTLA-4 is not only critical for tonic T cell signaling but is also involved in negatively regulating TCR dependent T cell activation. Along those lines, ectopic expression of li-CTLA-4 in a CTLA-4 deficient setting resulted in reduced T cell proliferation, diminished cytokine production and less ERK activation following T cell stimulation (34,48). In this study, the finding that CD4+ T cells from NOD-Tg-Cre mice showed greatly reduced IFN-γ production upon anti-CD3 stimulation as compared to cells isolated from wildtype littermates demonstrated that even in the presence of a full-length molecule, appropriate levels of li-CTLA-4 are critical to control T cell activation resulting in reduced disease frequency. We speculate that li-CTLA-4 stabilizes surface expression of the full-length molecule in the context of T cell activation, thus enhancing ligand engagement and consequently diminishing T effector function. This hypothesis is consistent with the observation that increased expression of li-CTLA-4 in NOD mice results in a slight increase of surface expression of fl-CTLA-4. In addition, our previous observations that li-CTLA-4 can be detected together with fl-CTLA-4 in the lipid-raft fraction of activated T cells (9,12) as well as the direct demonstration of heterodimer-formation of li-CTLA-4 with the full-length molecule (29) supports this hypothesis.
To address the role of li-CTLA-4 in the context of T1D development, in the absence of the full-length molecule, we crossed our BAC-transgenic mice onto the NOD.BDC2.5.CTLA-4KO background. Mathis and colleagues demonstrated that CTLA-4 KO mice on the BDC2.5-transgenic background are protected from CTLA-4 KO disease, namely lymphocyte hyperproliferation and multiorgan infiltration, but develop accelerated T1D by the age of 6–8 weeks (11). We observed that expression of only the li-CTLA-4 isoform in the NOD.BDC2.5.CTLA-4 KO background significantly reduced the number of activated effector/memory T cells and almost completely rescues the highly increased lymph node cellularity observed in CTLA-4 KO littermates. These results point to an important intrinsic function of CTLA-4 independent of B7 engagement. However, selective expression of li-CTLA-4 failed to rescue T1D development observed in CTLA-4 KO mice. We speculate that the partial rescue of the highly activated T cell phenotype reflects an important role of li-CTLA-4 in regulating tonic TCR signaling in which the biochemical effects known to be mediated by the intracellular portion of the molecule raise the activation threshold of naïve T cells. However, following acute antigenic stimuli, T cells require CTLA-4 ligand, B7-1- and B7-2, engagement to control autoimmunity in this antigen-specific model of T1D. This is likely considering the differential expression of both isoforms, with li-CTLA-4 preferentially expressed in naïve T cells and fl-CTLA-4 first upregulated upon T cell activation (29). Thus, we propose that the two CTLA-4 isoforms have distinct functions in different T cell subsets and that CTLA-4 regulates T cell homeostasis, immune activation and tolerance by distinct intrinsic versus extrinsic mechanisms.
The fact that expression of only the ligand independent form of CTLA-4 results in a significant decrease of activated T cells in three different transgenic mouse models (34,48) compared to a CTLA-4KO background (7,8), we hypothesized that this phenotype might be a consequence of improved Treg function by li-CTLA-4. To directly evaluate the relative role of full-length versus ligand independent CTLA-4 in Treg function, we used an in vivo adaptive transfer model of T1D and identified that BDC2.5-transgenic li-CTLA-4 Tregs confer disease protection comparable to wild-type Tregs. Interestingly, CTLA-4 KO Tregs also suppressed diabetes development in this setting. These results might be surprising given the numerous reports linking CTLA-4 and especially extrinsic CTLA-4 signaling with Treg suppressive function (50). In particular the recent study by Sakaguchi and colleagues concludes a Treg-specific role of CTLA-4, as mice lacking CTLA-4 only in Tregs still develop lymphoproliferative disease (51). Moreover, recent studies by Singer and colleagues demonstrated the requirement of extracellular CTLA-4 on Treg cells to control T effector functions in a model of inflammatory bowel disease (27). However, the results reported herein are consistent with previous studies from our laboratory demonstrating that CTLA-4 is dispensable on Treg function in animal models of T1D ((52) and Bluestone and Tang unpublished observation). The reasons for the differential role of CTLA-4 in different autoimmune and hyper-proliferative models remains unclear but may reflect the multitude of documented suppressive mechanisms meditated by this specialized T cells subset including the production of multiple suppressive factors such as TGF-δ, IL-10 and IL-35 (52–54). In this regard, we previously demonstrated that CTLA-4 KO Tregs express high levels of TGF-β that is involved in Treg suppressive function in a model of T1D (52). Thus, it is likely that Treg cells selectively expressing li-CTLA-4 suppress through these bystander mechanisms. Nevertheless, despite normal Treg suppressive activity, NOD.BDC2.5.CTLA-4 KO-Tg-Cre mice expressing only the li-CTLA-4 isoform develop diabetes with kinetics comparable to NOD.BDC2.5.CTLA-4 KO mice. This suggests a major role of CTLA-4 on effector T cells in this setting. Performing adoptive transfer experiments, we observed that BDC2.5-transgenic T effector cells expressing only li-CTLA-4 were more potent in transferring disease in full NOD recipients compared to wildtype cells. Disease kinetics were identical with those observed when transferring CTLA-4 KO cells. Notably, low numbers of antigen-specific T effector cells selectively expressing li-CTLA-4 induced T1D development in 75% of recipients compared to only 33% when transferring equal numbers of wild-type cells. These results might also provide an explanation for the finding that NOD.BDC2.5.KO-Tg-Cre mice are not protected from diabetes, despite a significant reduction of T effectors in those animals. Thus, we propose that the ectodomain of CTLA-4 and therefore ligand-binding is particularly crucial to control T effector function. This is in line with a recent study demonstrating that selective blockade of extrinsic CTLA-4 on T effector cells increases tumor immunity (55). Overall, our results demonstrate the biological significance of li-CTLA-4 in T1D development but also suggest that li-CTLA-4 function in the context of autoimmunity strictly depends on the presence of the full-length molecule.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Kioussis for providing the VAV-Cre transgenic mice as well as Dr. Nigel Killen from the Mutagenesis Core Facility for transgene injection. We also thank Dr. Abul Abbas for critical reading of the manuscript and Mike Lee for technical help with cell sorting. This work was supported by National Institutes of Health Grant P01 AI35297, U19 AI056388, P30DK63720-06A1
Reference List
- 1.Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev. 2011;241:180–205. doi: 10.1111/j.1600-065X.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 3.Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–155. doi: 10.1111/j.1600-065X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 4.Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. 2005;204:102–115. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- 5.Holmberg D, Cilio CM, Lundholm M, Motta V. CTLA-4 (CD152) and its involvement in autoimmune disease. Autoimmunity. 2005;38:225–233. doi: 10.1080/08916930500050210. [DOI] [PubMed] [Google Scholar]
- 6.Ghaderi A. CTLA4 gene variants in autoimmunity and cancer: a comparative review. Iran J Immunol. 2011;8:127–149. [PubMed] [Google Scholar]
- 7.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 8.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 9.Chikuma S, Imboden JB, Bluestone JA. Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2003;197:129–135. doi: 10.1084/jem.20021646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luhder F, Chambers C, Allison JP, Benoist C, Mathis D. Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc Natl Acad Sci U S A. 2000;97:12204–12209. doi: 10.1073/pnas.200348397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chikuma S, Bluestone JA. CTLA-4 and tolerance: the biochemical point of view. Immunol Res. 2003;28:241–253. doi: 10.1385/IR:28:3:241. [DOI] [PubMed] [Google Scholar]
- 13.Balzano C, Buonavista N, Rouvier E, Golstein P. CTLA-4 and CD28: similar proteins, neighbouring genes. Int J Cancer Suppl. 1992;7:28–32. [PubMed] [Google Scholar]
- 14.Howard TA, Rochelle JM, Seldin MF. Cd28 and Ctla-4, two related members of the Ig supergene family, are tightly linked on proximal mouse chromosome 1. Immunogenetics. 1991;33:74–76. doi: 10.1007/BF00211698. [DOI] [PubMed] [Google Scholar]
- 15.Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21:401–413. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 16.Saito T, Yokosuka T, Hashimoto-Tane A. Dynamic regulation of T cell activation and co-stimulation through TCR-microclusters. FEBS Lett. 2010;584:4865–4871. doi: 10.1016/j.febslet.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 17.Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, Yagita H, Tokunaga M, Saito T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010;33:326–339. doi: 10.1016/j.immuni.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, Collins M, Carreno BM, Madrenas J, Kuchroo VK. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J Immunol. 2002;168:5070–5078. doi: 10.4049/jimmunol.168.10.5070. [DOI] [PubMed] [Google Scholar]
- 19.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ, Thompson CB. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. 2000;13:313–322. doi: 10.1016/s1074-7613(00)00031-5. [DOI] [PubMed] [Google Scholar]
- 21.Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272:1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- 22.Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA. Molecular basis of T cell inactivation by CTLA-4. Science. 1998;282:2263–2266. doi: 10.1126/science.282.5397.2263. [DOI] [PubMed] [Google Scholar]
- 23.Guntermann C, Alexander DR. CTLA-4 suppresses proximal TCR signaling in resting human CD4(+) T cells by inhibiting ZAP-70 Tyr(319) phosphorylation: a potential role for tyrosine phosphatases. J Immunol. 2002;168:4420–4429. doi: 10.4049/jimmunol.168.9.4420. [DOI] [PubMed] [Google Scholar]
- 24.Calvo CR, Amsen D, Kruisbeek AM. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J Exp Med. 1997;186:1645–1653. doi: 10.1084/jem.186.10.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuang E, Lee KM, Robbins MD, Duerr JM, Alegre ML, Hambor JE, Neveu MJ, Bluestone JA, Thompson CB. Regulation of cytotoxic T lymphocyte-associated molecule-4 by Src kinases. J Immunol. 1999;162:1270–1277. [PubMed] [Google Scholar]
- 26.Schneider H, Mandelbrot DA, Greenwald RJ, Ng F, Lechler R, Sharpe AH, Rudd CE. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4+ T cells from receptor/ligand-deficient mice. J Immunol. 2002;169:3475–3479. doi: 10.4049/jimmunol.169.7.3475. [DOI] [PubMed] [Google Scholar]
- 27.Tai X, Van LF, Pobezinsky L, Guinter T, Sharrow SO, Adams A, Granger L, Kruhlak M, Lindsten T, Thompson CB, Feigenbaum L, Singer A. Basis of CTLA-4 function in regulatory and conventional CD4+ T cells. Blood. 2012 doi: 10.1182/blood-2011-11-388918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di GG, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 29.Vijayakrishnan L, Slavik JM, Illes Z, Rainbow D, Peterson LB, Sharpe AS, Wicker LS, Kuchroo VK. An autoimmune disease-associated CTLA4 splice variant lacking the B7 binding domain signals negatively in T cells. Novartis Found Symp. 2005;267:200–212. doi: 10.1002/047002139x.ch13. [DOI] [PubMed] [Google Scholar]
- 30.Howson JM, Dunger DB, Nutland S, Stevens H, Wicker LS, Todd JA. A type 1 diabetes subgroup with a female bias is characterised by failure in tolerance to thyroid peroxidase at an early age and a strong association with the cytotoxic T-lymphocyte-associated antigen-4 gene. Diabetologia. 2007;50:741–746. doi: 10.1007/s00125-007-0603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, Larrad MT, Rios MS, Chow CC, Cockram CS, Jacobs K, Mijovic C, Bain SC, Barnett AH, Vandewalle CL, Schuit F, Gorus FK, Tosi R, Pozzilli P, Todd JA. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet. 1996;5:1075–1080. doi: 10.1093/hmg/5.7.1075. [DOI] [PubMed] [Google Scholar]
- 32.Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes. 2000;49:1744–1747. doi: 10.2337/diabetes.49.10.1744. [DOI] [PubMed] [Google Scholar]
- 33.Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL, Howson JM, Smink LJ, Kingsnorth A, Lyons PA, Gregory S, Rogers J, Todd JA, Peterson LB. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol. 2004;173:164–173. doi: 10.4049/jimmunol.173.1.164. [DOI] [PubMed] [Google Scholar]
- 34.Araki M, Chung D, Liu S, Rainbow DB, Chamberlain G, Garner V, Hunter KM, Vijayakrishnan L, Peterson LB, Oukka M, Sharpe AH, Sobel R, Kuchroo VK, Wicker LS. Genetic evidence that the differential expression of the ligand-independent isoform of CTLA-4 is the molecular basis of the Idd5.1 type 1 diabetes region in nonobese diabetic mice. J Immunol. 2009;183:5146–5157. doi: 10.4049/jimmunol.0802610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, Wang Y, Walunas T, Bluestone J, Listman J, Finn PW. Regulation of CTLA-4 expression during T cell activation. J Immunol. 1996;156:4154–4159. [PubMed] [Google Scholar]
- 36.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 37.de BJ, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 38.Bour-Jordan H, Salomon BL, Thompson HL, Santos R, Abbas AK, Bluestone JA. Constitutive expression of B7-1 on B cells uncovers autoimmunity toward the B cell compartment in the nonobese diabetic mouse. J Immunol. 2007;179:1004–1012. doi: 10.4049/jimmunol.179.2.1004. [DOI] [PubMed] [Google Scholar]
- 39.Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes. 2011;60:1955–1963. doi: 10.2337/db11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamhamedi-Cherradi SE, Boulard O, Gonzalez C, Kassis N, Damotte D, Eloy L, Fluteau G, Levi-Strauss M, Garchon HJ. Further mapping of the Idd5.1 locus for autoimmune diabetes in NOD mice. Diabetes. 2001;50:2874–2878. doi: 10.2337/diabetes.50.12.2874. [DOI] [PubMed] [Google Scholar]
- 41.Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol. 2006;18:1197–1209. doi: 10.1093/intimm/dxl060. [DOI] [PubMed] [Google Scholar]
- 42.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, Marrack P, Mahata SK, Kappler JW, Haskins K. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 45.Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. 2005;175:3053–3059. doi: 10.4049/jimmunol.175.5.3053. [DOI] [PubMed] [Google Scholar]
- 46.Meagher C, Tang Q, Fife BT, Bour-Jordan H, Wu J, Pardoux C, Bi M, Melli K, Bluestone JA. Spontaneous development of a pancreatic exocrine disease in CD28-deficient NOD mice. J Immunol. 2008;180:7793–7803. doi: 10.4049/jimmunol.180.12.7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chikuma S, Abbas AK, Bluestone JA. B7-independent inhibition of T cells by CTLA-4. J Immunol. 2005;175:177–181. doi: 10.4049/jimmunol.175.1.177. [DOI] [PubMed] [Google Scholar]
- 49.Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, Bluestone JA. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bour-Jordan H, Bluestone JA. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev. 2009;229:41–66. doi: 10.1111/j.1600-065X.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 52.Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 53.Collison LW, Vignali DA. Interleukin-35: odd one out or part of the family? Immunol Rev. 2008;226:248–262. doi: 10.1111/j.1600-065X.2008.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.