

Abstract
Our previous studies have suggested that street and fixed rabies viruses (RABV) induce diseases in the mouse model via different mechanisms. In the present study, attempts were made to determine if it is the glycoprotein (G) that is responsible for the observed differences in the pathogenic mechanisms. To this end, an infectious clone from fixed virus B2c was established and used as a backbone for exchange of the G from street viruses. The rate of viral replication, expression of viral proteins, and the induction of innate immune responses were compared in cells or in mice infected with each of the viruses. Furthermore, the infiltration of inflammatory cells into the CNS and the enhancement of blood-brain-barrier (BBB) permeability were also compared. It was found that fixed viruses induced stronger innate immune responses (expression of chemokines, infiltration of inflammatory cells, and enhancement of BBB permeability) than street RABV or recombinant viruses expressing the G from street RABVs. Fixed viruses induce disease via an immune-mediated pathogenic mechanism while street viruses or recombinant viruses expressing the G from street RABVs induce diseases via a mechanism other than immune-mediated pathogenesis. Therefore, RABV G is an important determinant for the induction of innate immune responses and consequently the pathogenic mechanisms.
Keywords: Rabies virus, innate immune response, inflammation, blood brain barrier
Introduction
Rabies is an ancient zoonotic disease and today still causes more than 55,000 human deaths around the globe each year (WHO, 2005). The causative agent, rabies virus (RABV), belongs to the genus Lyssavirus in the family Rhabdoviridae. Its genome is a single-strand, negative-sense RNA of approximately 12 kb in length and encodes for five proteins in the order of nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G) and RNA-dependent RNA polymerase (RdRp) or large protein (L) (Tordo et al., 1986; Wunner, 2007). RABV N plays important roles in regulating viral transcription and replication (Patton et al., 1984; Wunner, 2007) via encapsdation of the genomic RNA (Sokol et al., 1969). RABV P binds to the nucleoprotein-RNA template and is also part of the RdRp (Mebatsion, 2001; Raux et al., 1997; Takamatsu et al., 1998; Vidy et al., 2007). The P has also been reported to interact with cellular factors such as dynein, contributing to RABV spread (Raux et al., 2000) and promoting RABV transcription (Tan et al., 2007). The viral envelope is composed of the M and the transmembrane G proteins. The G is the only surface protein of the rabies virion and thus capable of inducing virus neutralizing antibodies (VNA) (Benmansour et al., 1991; Wiktor et al., 1973). The G plays important roles in rabies pathogenesis (Ito et al., 2001; Morimoto et al., 1999; Wunner, 2007) by binding to neural receptors such as acetylcholine receptor (Lentz et al., 1982; Lentz et al., 1984), neural cell adhesion molecules (NCAM) (Thoulouze et al., 1998), or low affinity neurotrophic receptor (Tuffereau et al., 1998), contributing to the exclusive neurotropism and neuroinvasiveness of RABV (Morimoto et al., 2000).
Despite the lethality of rabies, only mild inflammation and little neuronal destruction were observed in the central nervous system (CNS) of rabies patients (Miyamoto and Matsumoto, 1967; Murphy, 1977). On the other hand, fixed (laboratory-adapted) RABV induces extensive inflammation and neuronal degeneration in experimentally infected animals (Miyamoto and Matsumoto, 1967; Murphy, 1977). It was further shown that fixed RABV induces inflammation by activating the innate immune responses (type 1 interferon, chemokines and complements) in the CNS (Johnson et al., 2006; Nakamichi et al., 2004; Prehaud et al., 2005). The induction of innate immune responses and inflammation in the CNS is also associated with the enhancement of blood-brain-barrier (BBB) permeability (Fabis et al., 2008; Phares et al., 2007; Roy and Hooper, 2007; Roy et al., 2007). The BBB was more permeable in mice infected with fixed RABV than in those infected with street RABV. It is hypothesized that the induction of inflammatory responses, on one hand, can lead in the clearance of the RABV from the CNS (virus attenuation) when the virus dose is low. However, extensive inflammation in the CNS can result in diseases and death when large doses of fixed virus are used to infect mice (Sarmento et al., 2005; Wang et al., 2005). These studies led to the hypothesis that street and fixed RABV induce disease and death by different mechanisms (Kuang et al., 2009; Wang et al., 2005). Infection with fixed RABV induces diseases via immune-mediated pathogenesis while the pathogenic mechanism by which street RABV induces rabies remains to be determined.
It has been reported that virulent or street RABV expresses low level, while fixed RABV expresses high level, of the G (Morimoto et al., 1998; Yan et al., 2001). The high level of G expression by fixed RABV has been correlated with the induction of apoptosis, enhancement of the BBB permeability, and increases of innate immune responses in the CNS (Faber et al., 2002). On the other hand, street RABV expresses low level of the G, thus evading the host innate immune responses (Wang et al., 2005). In the present study attempts were made to determine if RABV G is a determinant for the induction of innate immune responses and the pathogenic mechanisms by exchanging the G between street and fixed RABVs. It was found that fixed viruses induced stronger innate immune responses and inflammation than street RABV or recombinant viruses expressing the G from street RABVs. These results confirm that fixed viruses induce disease via an immune-mediated pathogenic mechanism while street viruses or recombinant viruses expressing the G from street RABVs induce diseases via mechanism other than immune-mediated pathogenesis. Therefore, RABV G is an important determinant for the induction of innate immune responses and consequently the pathogenic mechanisms.
Materials and Methods
Viruses, Cells and Antibodies
Three parent RABVs were used in this study and they are silver-haired bat rabies virus (SHBRV), dog rabies virus (DRV), and BHK-adapted CVS virus (B2c). SHBRV is a wt RABV isolated from a human patient (Morimoto et al., 1996). DRV is a wt virus originated from a Mexican dog (Dietzschold et al., 2000). B2c is a fixed virus isolated from challenge virus standard (CVS-24) by serial passaging in BHK cells (Morimoto et al., 1998). Virus stocks were prepared as described previously (Sarmento et al., 2005; Wang et al., 2005). Briefly, one-day-old suckling mice were inoculated with 10 ! l of viral preparation by the intracerebral (IC) route. When moribund, mice were euthanized and brains removed. A 10% (w/v) suspension was prepared by homogenizing the brain in Dulbecco’s modified Eagle’s medium (DMEM). The homogenate was centrifuged to remove debris and the supernatant collected and stored at −80°C. Fluorescein isothiocyanate (FITC)-conjugated antibody against the RABV N was purchased from FujiRebio (FujiRebio Diagnostic INC, PA). Anti-RABV N monoclonal antibody (N42) was prepared as described (Jiang et al., 2010). Anti-RABV G polyclonal antibody was prepared in rabbits as described (Fu et al., 1993) and has been shown to have similar affinity to the G from street and fixed RABVs (Yan et al., 2001). Anti-CD3 polyclonal antibody was purchased from Dako (Dako North America, CA). Mouse neuroblastoma (NA) cells were maintained in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY). BSR cells, a cloned cell line derived from BHK-21 cells, were maintained in Dulbecco’s modified Eagle’s medium (Mediatech, VA) containing 10% FBS. Four to 6 weeks old female ICR mice and 6 to 8 weeks old female Balb/c mice were purchased from Harlan (Harlan, IN) and housed in temperature- and light-controlled quarters in the Animal Facility, College of Veterinary Medicine, University of Georgia. All animal experiments were carried out under Institutional Animal Care and Use Committee-approved protocols (animal welfare assurance no. A3085-01).
Sequencing the complete B2c virus genome
Total RNA was extracted from B2c-infected mouse brain tissue using TRIZOL® LS Reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer’s recommendation. Briefly, 250μl of the brain suspension was mixed with 3 volumes of TRIzol LS reagent and extracted with 240μl of chloroform. The RNA containing aqueous phase was precipitated with 600μl of isopropanol. The RNA was used to amplify the B2c sequences using reverse transcription polymerase chain reaction (RT-PCR) with 10 pairs of oligonucleotide primers (Supplemental Table 1) based on the genomic sequences of the HEP-Flury and CVS-AVO1 strains (Inoue et al., 2003; Wang et al., 2010). PCR amplification was performed for a total of 35 cycles (denaturation at 95°C for 15 sec, annealing at 55°C to 58°C for 30 sec, and extension at 68°C for 1 min per kb pair) in the presence of 10μM each of forward and reverse primers, 0.3mM of dNTPs, 20mM KCl, 30mM Tris-HCl (pH 8.8), 1mM MgSO4, 1mM DTT, and 1.5 units AccuPrime™ Pfx DNA Polymerase (Invitrogen, NY). PCR products were purified using QIAquick Gel Extraction Kit (QIAGEN, Germantown, MD) and cloned into the pCR-Blunt II Vector (Invitrogen, NY) according to the manufacturer’s instructions. Cloned DNA was sequenced using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit and ABI Prism 3730 sequencer at Tufts University Core Facility. At least three clones were sequenced from each amplified fragment. The assembly of genomic sequences was carried out with the aid of SeqMan software (DNASTAR Inc).
Construction of recombinant B2c clones
The PCR products were digested with enzyme sets SnaB I &Blp I, Blp I &Nhe I, Nhe I &Nar I and Nar I &Kpn I and then were ligated into vector pcDNA3.1 (Fig. 1A). Nhe I was added as the genomic marker to differentiate the cDNA clone from the parental B2c. The recombinant cDNA clones rB2c/DRVG and rB2c/SHBRVG were constructed by switching the coding sequence of the G with DRV/Wt and SHBRV/Wt, respectively, without altering the start/stop signals outside the ORF (Fig. 1B). The primers used for cloning rB2c are listed in Supplemental Table 2.
Fig. 1. Schematic diagram for the construction and characterization of rRABVs.

Construction of the infectious B2c clone is accomplished by amplification of the indicated fragments from RNA extracted from B2c-infected mouse brain and ligation of these fragment into the pcDNA vector (A). Restriction enzyme Nhe I was used as a genetic marker. The G gene from the street DRV or SHBRV was inserted between the Pac I and Nhe I sites of the B2c full infectious clone (B). BSR or NA cells were infected with each of the viruses and virus titers determined at indicated time points (C). FFU, fluorescent focus units.
Rescue of recombinant RABVs from cloned cDNA
Recombinant RABVs were rescued as described previously (Inoue et al., 2003). Briefly, BSR cells grown in six-well plates were transfected with 2.0 μg of the full-length clone and 0.5, 0.25, 0.15 and 0.1 μg of pcDNA-N,-P, -G and -L, respectively using 15 μl of SuperFect transfection reagent (Qiangen, Valencia, CA) in a final volume of 2 ml. The transfection was allowed to proceed for 2 to 4 hours and then the transfecting medium was removed and replaced with fresh DMEM containing 10% FBS. After incubation at 34°C for 4 days, the culture medium was removed and fresh medium with 2% FBS added to the cells. After incubation for another 3 days, the culture medium was harvested and the cells examined for the presence of rescued virus by using FITC-conjugated antibodies against the RABV N protein.
Virus propagation and titration
All rRABV strains were propagated in newborn mouse and harvested from the brain as described previously (Yan et al., 2001). For virus titration by direct fluorescent antibody assay (dFA), NA cells or BSR in 96-well plates were infected with serial 10-fold dilutions of the virus in DMEM and incubated at 37°C for 2 days (Zhao et al., 2009). The relative neurotropism for each virus was determined by division of the virus titers in NA cells by those in BSR cells.
Single step growth assays
Monolayer cultures of 3×106 NA cells and 3×106 of BSR cells in six-well plates were infected with virus at a multiplicity of infection (MOI) of 1 fluorescent focus units (FFU). Virus was adsorbed in 1ml of growth medium for 1 hour and then the cells were washed three times with PBS. Afterward, fresh medium was added into the cells and incubated at 37°C. Samples of culture medium were harvested at 12, 24, 48, 72 hours post-inoculation (p.i.). Virus titer was determined in NA cells by the dFA as described (Zhao et al., 2009).
Quantitative real-time RT-PCR (qRT-PCR)
Brain tissues were removed from infected mice after perfusion at indicated time points, and flash frozen on dry ice before being stored at −80°C. Total RNA was extracted with Trizol and used for qRT-PCR in an Mx3000P apparatus (Stratagene, La Jolla, CA) as described previously (Wang et al., 2005). Each reaction was carried out in duplicate with approximately 100ng of DNase-treated RNA and 5nM each primer pairs by using a One-Step Brilliant II SYBR green qRT-PCR master mix kit (Stratagene, CA) according to the manufacturer’s instruction. For absolute quantification of viral RNA, a standard curve was generated from using a serially diluted RNA in vitro transcribed from plasmids expressing RABV N or G and the copy numbers of viral RNA were normalized to 1μg of total RNA (Wen et al., 2011). For chemokine expression, mRNA copy numbers of a particular gene were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Levels of gene expression in a test sample are presented as the fold increase over that detected in sham-infected controls (Wen et al., 2011).
Western blotting
Western blotting was performed as described previously (Wang et al., 2005). Briefly, brain extract was resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with respective antibodies overnight at 4°C or for 1 hour at room temperature, followed by horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, UK) for 1 hour at room temperature. Proteins were detected by enhanced chemiluminescence (Amersham Biosciences, UK). Band signals corresponding to immunoreactive proteins were measured and scanned by image densitometry using Adobe Photoshop CS4 software.
Histopathology and immunohistochemistry
For histopathology, animals were anesthetized with ketamine-xylazine at a dose of 0.1ml/10 g body weight and then perfused by intracardiac injection of PBS followed by 10% neutral buffered formalin as described previously (Li et al., 2005). Brain tissues were removed and embedded with paraffin. Histopathology was performed by staining the paraffin-embedded sections with hematoxylin and eosin (H&E). For immunohistochemistry, the tissue sections were de-paraffinized by heating at 70°C for 10 min and then dipped in CitriSolv (Fisher Scientific, PA) three times for 5 min and dried until chalky white. Slides were heated in antigen unmasking solution (Vector Laboratories, CA) above 90°C for 20 min and naturally cooled down to room temperature. Anti-RABV N monoclonal antibody or anti-RABV G polyclonal antibodies were used to detect viral antigens. Avidin-biotin-peroxidase complex (Vector Laboratories, CA) was then used to localize the biotinylated antibodies. Finally, diaminobenzidine (DAB) was used as a substrate for color development.
Measurement of BBB permeability
BBB permeability was assessed using a modification of a previously described technique in which Na-fluorescein (NaF) is utilized as a tracer molecule (Phares et al., 2007). Briefly, Mice were given 100 μl of 10% NaF in PBS intravenously under anesthesia. After 10 min to allow circulation of the NaF, cardiac blood was collected and the animals were transcardially perfused with PBS. Brain tissues were homogenized in 7.5% trichloroacetic acid (TCA) and centrifuged for 10min at 10,000×g to remove insoluble precipitates. After addition of 0.25 ml 5N NaOH, the fluorescence of a 100 μl supernatant was determined using a BioTek Spectrophotometers (Bio-Tek Instruments, Inc.) with excitation at 485 nm and emission at 530 nm. NaF uptake into tissue is expressed as (μg fluorescence brain/mg tissue) / (μg fluorescence serum/ml blood) to normalize values for blood levels of the dye at the time of tissue collection.
Flow cytometry
Flow cytometery was performed as described (Zhao et al., 2010). Briefly, mouse brains were harvested and digested with 1 μg/μl DNase I (Sigma-Aldrich), 2 μg/μl collagenase D (Worthington Biochemical Corporation, Lakewood, NJ) in Hanks balanced salt solution (with Ca2+ and Mg2+) for 1 h to disperse the tissue into single-cell suspension. Viable cells were separated by discontinuous Percoll gradient (70/30%) centrifugation (650 × g, 25 min). After being washed once with Hanks balanced salt solution (without Ca2+ and Mg2+) and counted, cells were stained with Ly6G/PerCP, CD19/PE-Cy7, CD45/PE, CD3/Alexa 647, or CD11b/APC (BD Biosciences Pharmingen, San Jose, CA) for 30 min at 4°C. Flow cytometery was performed on a BD LSR-II flow cytometer and a minimum of 30,000 events were counted. Data were analyzed using FlowJo software (Tree star, Inc., San Carlos, CA).
Data analysis
All experiments were repeated at least three times. Data were analyzed by GraphPad Prism® 5 (GraphPad software, Inc.), or One-Way ANOVA. For all tests, the following notations are used to indicate significance between groups: *, p<0.05; **, p<0.01; ***, p<0.001
Results
Complete nucleotide sequences of B2c
To obtain the full genome sequence for B2c, ten pairs of primers were designed (Supplemental Table 1) based on the sequences from HEP-Flury and AVO1 strains. The sequences of the ten fragments were amplified, sequenced, and assembled into consecutive sequence. The genome of B2c has 11927 nucleotides in length. The coding sequences for B2c are as follows: 1353nt for N, 894nt for P, 609nt for M, 1575nt for G, and 6384nt for L. B2c shares 100%, 99.9%, 99.5%, 99.1% and 99.7% on the N, P, M, G and L respectively, with CVS-11 (Wang et al., 2010). The complete sequence of B2c was submitted to GenBank (GenBank accession number: HQ891318).
Construction and in vitro characterization of rRABVs
To further investigate the role of RABV G in the induction of host innate immune responses, an infectious clone of pcDNA/B2c was constructed as shown in Fig. 1A. Based on the rB2c, two more recombinant RABV infectious clones were constructed by replacing B2c G with DRV and SHBRV G to obtain rB2c/DRVG and rB2c/SHBRVG, respectively (Fig. 1B). The recombinant RABVs were rescued and propagated in newborn mouse as described previously (Zhao et al., 2009). Viruses were harvested from the brain and titrated in NA or BSR cells in comparison with the parent viruses. As shown in Table 1, all the viruses show similar titers in NA cells while the titers in BSR cells varied greatly. Fixed virus B2c and the rB2c grow to high titers (107 FFU/ml or above). SHBRV and rB2c/SHBRVG grow modestly to 106.25 and 104.08, respectively. DRV and rB2c/DRVG grow poorly in BSR cells with titers of only 104.25 and 103.75, respectively. When the neurotropism index was calculated (Table 1), B2c and rB2c had a very similar index (0.046 and 0.056) while the neurotropism indices of DRV and SHBRV were about 200-fold higher than that of B2c. On the other hand, the neurotropism indices of rB2c/DRVG and rB2c/SHBRVG were about 30-fold higher than those of DVR and SHBRV, but were more than 5,000-fold higher than that of B2c or rB2c.
Table 1.
Titers and neurotropism of rabies viruses in NA and BSR cells.
| Virus | Adaptation/selection | Titer (FFU/ml) |
Neurotropism | |
|---|---|---|---|---|
| NA | BSR | Indexa | ||
| B2c (Wt) | Mouse Brain | 106.33 | 107.67 | 0.046 |
| DRV (Wt) | Mouse Brain | 105.33 | 104.25 | 12.02 |
| SHBRV (Wt) | Mouse Brain | 107.17 | 106.25 | 8.32 |
| rB2c | Mouse Brain | 105.75 | 107 | 0.056 |
| rB2c/DRVG | Mouse Brain | 106.25 | 103.75 | 316.23 |
| rB2c/SHBRVG | Mouse Brain | 106.5 | 104.08 | 263.03 |
The neurotropism index was the logarithm of the titer in NA cells minus the logarithm of the titer in BSR cells. All titrations were carried out in quadruplicate.
The growth kinetics of the recombinant and parental viruses in BSR and NA cells were determined in the single-step growth curve assays (Fig. 1C). It was found that rB2c grew to higher titers than its parental virus B2c during the early phase of virus infection (12 hpi.) in both BSR and NA cells. Otherwise, all the viruses grew to similar titers in NA cells (105.33 -107.17 FFU/ml). In BSR cells, B2c, rB2c, and SHBRV grew to high titers (~107 FFU/ml) while DRV, rB2c/DRVG, and rB2c/SHBRVG grow poorly (103.75 - 104.25 FFU/ml).
To correlate the virus titers with the rate of viral transcription/replication, the copy number of the viral mRNA (N and G) was determined by qRT-PCR at 24 hr after infection. As shown in Fig. 2A, the copy number of the N mRNA was similar for all the viruses in NA cells except the DRV (2 logs lower) and SHBRV (1 log lower) while the copy number of the G mRNA was significantly lower (2-6 logs) in DRV, rB2c/DRVG, and rB2c/SHBRVG than those in B2c/wt, SHBRV/wt, and rB2c. In BSR cells (Fig. 2C). The copy number of the N and G mRNA was 2 and 4 logs more, respectively, for the fixed viruses (B2c and rB2c) than those for the street viruses or the rRABVs expressing the G from the street viruses. To investigate the ability of the rRABV to induce innate immune responses, NA or BSR cells were infected with each of the viruses and the cells were harvested for RNA extraction and quantification of chemokine expression (MIP-1! , RANTES, and IP-10) by RT-PCR as described (Kuang et al., 2009). As shown in Fig. 2B and 2D, the expression of MIP-1! was very low in cells infected with each of the viruses. The level of RANTES was significantly up-regulated in NA cells when infected with either B2c or rB2c than infected with DRV, SHBRV, rB2c/DRVG or rB2c/SHBRVG. The RANTES expression is minimal in BSR cells infected with each of the viruses. In both NA and BSR cells, IP-10 was highly up-regulated when infected with each of the viruses over the sham-infected controls. Furthermore, the increase was significantly more in cells infected with B2c or rB2c than in those infected with street RABV or rRABV expressing the G from street viruses.
Fig. 2. Viral gene expression and induction of innate immune gene expression in cell cultures after infected with rRABVs.

Mouse NA or BSR cells were infected with each of the viruses at MOI of 1.Total RNA was prepared from virus-infected cultures and used for qRT-PCR. Each reaction was carried out in duplicate and repeated for three times. RABV mRNA (G and N) (A, C) and the expression of chemokine mRNA (MIP-1! , RANTES, IP-10) (B, D) were analyzed by qRT-PCR. Data are given as mean values ± standard errors. Asterisks indicate significant differences between the indicated experimental groups as analyzed by One-Way ANOVA: (* p<0.05, ** p<0.01 and *** p<0.001)
Viral replication and the level of the G gene expression in mouse brain after infection with rRABV
To determine if the observation in vitro reflect the situation in vivo, mice were infected with 10 ICLD50 of each virus by the IC route and sacrificed at day 6 post infection (dpi). Mouse brains were harvested for measuring the level of N and G expression using immunohistochemistry (Fig. 3A), qRT-PCR (Fig. 3B and 3C) and Western blotting (Fig. 3D). Overall, the level of N expression was similar in mice infected with each of the viruses while more G expression was found in mice infected with fixed viruses B2c or rB2c than in mice infected with street viruses or rRABV expressing the G from the street RABV (Fig. 3A). To investigate whether the differences in antigen expression is due to viral transcription and/or translation, viral mRNA was determined in mouse brain at 6 dpi using real-time PCR. The copy numbers for the N or the G mRNA are shown in Fig. 3B and the G/N ratio in Fig. 3C. Overall, the rate of replication as shown by the copy number of N and G mRNA was higher in mice infected with B2c or rB2c than in mice with street viruses or rRABV expressing the G from the street RABV. The copy number for the N and G mRNA was similar in mice infected with B2c or rB2c while the copy number of the N mRNA was significantly higher than those of the G mRNA in mice with street viruses or rRABV expressing the G from the street RABV. The G/N ratio is about 1 in mice infected with B2c and rB2c and about 0.4-0.6 in mice infected with street viruses or rRABV expressing the G from the street RABVs. Similar results were obtained by Western blotting (Fig. 3D). The levels of N expression were slightly more in the brain of mice infected with fixed RABV than in those infected with street RABV or rRABV expressing the G from street RABVs. Furthermore, the G expression is significantly lower in mice infected with street viruses or rRABV expressing the G from street RABVs than with B2c or rB2c. Quantitation of the intensity of DAB signals by Image-pro Plus Software revealed that the G/N ratio is higher in mice infected with B2c (0.68) or rB2c (0.82) than that in mice infected with street viruses (0.34 and 0.42) or recombinant viruses expressing the G from the street RABVs (0.45 and 0.41) (Fig. 3D). These data confirms previous findings that it is the G that determines the level of G expression and the expression of street RABV G is inhibited (Kuang et al., 2009; Sarmento et al., 2005; Wang et al., 2005; Yan et al., 2001). The overall rate of viral transcription/replication is slightly higher in mice infected with B2c and rB2c than that in mice infected with street viruses or rRABV expressing the G from street viruses.
Fig. 3. Detection of viral antigens in mice at day 6 p.i. with rRABV.

RABV N and G antigens were detected by immunohistochemistry in mouse brain section (A, magnification ×20). RABV viral mRNAs were analyzed by qRT-PCR (B) and RVBV proteins (N and G) detected by Western blotting (D). The ratio between G and N was calculated with RNA copy number (C) or determined after measurement of the band density (D). As a loading control, β-tubulin was measured in the same sample preparation using anti-β-tubulin (anti-T) antibody in the Western blot assay. Significance of differences between the experimental groups was assessed by One-Way ANOVA (*, p<0.05; **, p<0.01 and *** p<0.001).
Induction of innate immune responses and infiltration of inflammatory cells in mouse brain after infection with rRABV
To investigate the ability of the rRABV to induce innate immune responses, mice were infected with 10 ICLD50 of each virus by IC and at 6 dpi, mice were sacrificed and brains harvested for measuring the expression of chemokines (MIP-1! , RANTES, and IP-10) by qRT-PCR as described (Kuang et al., 2009). As shown in Fig. 4A, the expression of MIP-1! was very low in mice infected with all the viruses although significantly higher level of MIP-1! was detected in mice infected with B2c than with DRV. The level of RANTES and IP-10 in mice infected with B2c or rB2c was significantly higher than that in mice infected with street RABV or rRABV expressing the G from street viruses. To determine if the expression of chemokines results in infiltration of inflammatory cells into the CNS, immunohistochemistry was performed by using anti-CD3 antibodies. There were significantly more infiltration of CD3+ cells in the brain of mice infected with B2c or rB2c than in mice infected with street RABV or rRABV expressing the G from street viruses (Figs. 4B and 4C).
Fig. 4. Expression of chemokines and inflammatory responses in the CNS after infection with rRABV.

Groups of 3 female ICR mice at the age of 4 to 6 weeks were infected with each of the viruses at 10ICLD50 and transcardially perfused with 10% formalin at day 6 p.i. Brains were used for preparation of RNA and chemokine expression was assessed by qTR-PCR (A). Paraffin sections were subjected to immunohistochemistry for detecting CD3-positive cells (B). CD3-postive cells were quantified (C). Significance of differences between experimental groups was assessed by One-Way ANOVA. (*, p<0.05; **, p<0.01).
To differentiate the inflammatory cells infiltrated into or activated in the CNS after infection with RABVs, leukocytes were recovered from mouse brains and analyzed by flow cytometery. The populations of activated macrophages, neutrophils, B cells and T cells were differentiated using cell surface markers CD11b, Ly6G, CD19 and CD3, respectively. CD45 was used as a maker for all the inflammatory cells. Fig. 5A shows representative flow plots for macrophages, neutrophils, CD19 B cells, and CD3+ T cells, in mice infected with B2c. The detailed results for each of the cell types were shown in Fig. 5B for mice infected with each of the viruses. At 3 dpi, the total number of inflammatory cells in the CNS was less than 18,000 in each group, but increased dramatically by 6 and 9 dpi. Overall significantly more inflammatory cells were detected in mice infected by fixed viruses than those by street RABV or rRABV expressing the G from street viruses. The exception is SHBRV that induced the activation or infiltration of more macrophages at 6 and 9 dpi as well as neutrophils and CD19+ B cells at 9 dpi. By 9 dpi, the numbers of CD3+ T cells are no longer significantly different among the different groups. These results indicate that expression of the G from fixed RABV induces high level of innate immune responses including the expression of chemokines and infiltration of inflammatory cells in the CNS while street RABV or rRABV expressing the G from street viruses induce only low level of innate immune responses and inflammation.
Fig. 5. Slow cytometry analysis of inflammatory cells in the CNS.

Groups of 3 female BALB/c mice at the age of 6 to 8 weeks were infected with each of the viruses at 10 ICLD50 and brains were collected after extensive perfusion at days 3, 6 and 9 p.i. Activated macrophages, neutrophils, CD19 B cells and CD3 positive cells were analyzed by flow cytometery. Four regions were identified: CD11b/CD45 (region R1, activated macrophages), Ly6G/CD45 (region R2, neutrophils), CD19/CD45 (region R3, CD19 B cells) and CD3/CD45 (region R4, CD3+ T cells). Total number of monocytes (per mouse brain) in regions R1, R2, R3 and R4 were shown in mice on 3, 6, or 9 dpi with B2c (A). Detailed analysis was performed for mice infected with each of the viruses and for each of the types of inflammatory cells (B). The results show the means±SE. The differences between the indicated experimental groups were analyzed by On-Way ANOVA (*, p<0.05; **, p<0.01.).
Enhancement of BBB permeability in mouse brain after infection with rRABV
To investigate whether the innate immune responses (expression of chemokines and infiltration of inflammatory cells) induced by infection with rRABV results in enhancement of BBB permeability, mice were infected with 10ICLD50 of each virus and the leakage of the NaF from circulation into the CNS tissues was measured in the cerebellum and cerebrum at 6 dpi. As shown in Fig. 6, BBB permeability was enhanced in both the cerebellum and cerebrum when infected with B2c and rB2c, but not with street RABV or rRABV expressing the G from street viruses. These data indicate that the expression of G induced innate immune responses (expression of chemokines and infiltration of inflammatory cells), which leads to the enhancement of BBB permeability.
Fig.6. BBB permeability changes in the CNS tissues of mice infected with each of viruses.
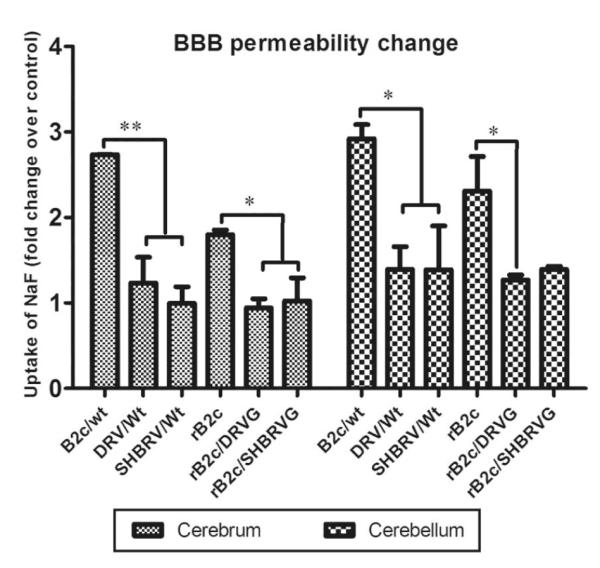
ICR mice (4-6 weeks of age) were infected with each of the viruses at 10 ICLD50, BBB permeability in the cortex and the cerebellum was assessed by measuring NaF leakage from the circulation into CNS tissues at day 6 post infection. Data are given as mean values±SE. Asterisks indicate significant differences between the indicated experimental groups: *, p<0.05; **, p<0.01.
Other pathological changes in mouse brain after infection with rRABV
To investigate if other damage in the CNS is associated with infection of rRABV, pathological changes were observed in the cortex, cerebellum, and hippocampus as shown in Fig. 7A and the quantification is shown in Fig. 7B. Overall neural degeneration is detected more in the cortex and hippocampus of mice infected with fixed viruses than in mice infected with street RABV or rRABV expressing the G from street viruses except in mice infected with rB2c/DRVG. In the cerebellum, significant difference was observed between mice infected with B2c and DRV. Significantly more infiltration of inflammatory cells was observed in the hippocampus of mice infected with fixed viruses than in mice infected with street RABV or rRABV expressing the G from street viruses. These studies overall indicate that infection with fixed RABV induces more pathological changes in the CNS than street RABV or rRABV expressing the G from street viruses.
Fig. 7. Histopathological changes in mice after infection with rRABVs.

Groups of 3 female ICR mice at the age of 4 to 6 weeks were infected with each of the viruses at 10ICLD50 and were transcardially perfused with 10% formalin at 6 dpi. Pathological changes were observed in paraffin sections after H&E staining (A, magnification x20). Neurodegeneration/cell death and infiltration score were determined from 8 regions of three slides for each group (B). Significance of differences between different groups was assessed by One-Way ANOVA. *, p<0.05; **, p<0.01.
Discussion
B2c is a virus derived from CVS-24 by passaging in BHK cells (Morimoto et al., 1998). Previous studies have shown that infection of mice with B2c activates strong innate immune responses in the CNS by inducing the expression of IFN and chemokines, infiltration of inflammatory cells and enhancement of BBB permeability (Kuang et al., 2009; Wang et al., 2005). Here in the present study, the complete sequence of B2c was obtained and its genome has 11927 nucleotides in length, the same as another CVS strain, CVS-11, and many other fixed RABV (Inoue et al., 2003; Wang et al., 2010; Wirblich and Schnell, 2011). Overall B2c shares more than 99% identity with CVS-11. Based on the entire sequence, an infectious clone was constructed and infectious virus rescued. The rB2c has the characteristics of B2c in the rate of virus replication, expression of viral proteins, and induction of innate immune responses (expression of chemokines, infiltration of inflammatory cells, and enhancement of BBB permeability).
Previous studies have demonstrated that fixed RABV induces innate immune responses in the CNS while street RABV evades such activation (Wang et al., 2005). The induction of innate immune responses correlates with the expression of the G protein. To determine if RABV G (or the level of G expression) is responsible for the induction of innate immunity, rRABV based on the B2c backbone was constructed by replacing its G with the G derived from street viruses SHBRV and DRV. These viruses were compared with the parent viruses from which the G is derived in the rate of virus replication, the level of G and N expression, the induction of innate immune responses (expression of chemokines) in vitro and in vivo. Furthermore, the infiltration of inflammatory cells and the enhancement of BBB permeability were also compared in the mouse model for these viruses. These studies demonstrate that RABV G protein determines the rate of viral replication, the level of the G expression and the induction of innate immunity. Recombinant B2c, like the parent B2c, replicated to high level, expressed high level of the G, and stimulated strong innate immune responses (expression of chemokines, infiltration of inflammatory cells, and enhancement of BBB permeability (Kuang et al., 2009; Wang et al., 2005). On the other hand, replacing B2c G with the G from street RABVs (SHBRV and DRV) resulted in the reduction of viral replication, the G expression, and the innate immune responses (expression of chemokines, infiltration of inflammatory cells and the enhancement of BBB permeability). The biological properties of the rRABV are similar to the parent viruses from which the G is derived despite the fact that rRABV derived all the other viral genes from B2c. All these data confirm previous findings that it is the G that determines the level of G expression and consequently the induction of innate immune responses (Dietzschold et al., 2005; Sarmento et al., 2005).
In addition to the differentiation in the G expression, the overall rate of virus replication is higher in cells or animals infected with fixed viruses than with street RABV or rRABV expressing the G from street RABV. This can be reflected by virus titer, particularly in non-neuronal cells, and the copy numbers of viral RNA in animals. It is not known if the higher rate of fixed virus replication is at least partially responsible for the higher level of its G expression. It has been reported previously that pathogenic RABV suppresses its rate of replication to stay pathogenic (Dietzschold et al., 2004; Pulmanausahakul et al., 2008). The only exception is the SHBRV that grows well in non-neuronal BSR cells as has been reported previously (Morimoto et al., 1996). It also induced more infiltration of inflammatory cells by 9 dpi. It is not known if the high rate of replication and induction of moderate innate immune responses is associated with the perceived low virulence associated with RABV isolated from silver-haired bats (Roy et al., 2007; Wang et al., 2005).
It has been known for a long time that only mild inflammation and little neuronal destruction were observed in the CNS of rabies patients (Miyamoto and Matsumoto, 1967; Murphy, 1977). Yet, fixed RABV induces extensive inflammation and neuronal degeneration in experimentally infected animals (Miyamoto and Matsumoto, 1967; Murphy, 1977). These observations raise the question as to how the street and fixed RABV differ in terms of pathogenic mechanisms. Results from previous studies demonstrate that fixed RABV induces extensive inflammation, apoptosis, and neuronal degeneration in the CNS while street RABV causes little or no neuronal damage in the mouse model (Kuang et al., 2009; Sarmento et al., 2005; Wang et al., 2005; Yan et al., 2001). These studies also revealed that the induction of innate immunity, apoptosis, and inflammation correlate with the expression of the G protein. Using the backbone of SN-10 (SAD B19) (Conzelmann et al., 1990; Morimoto et al., 1998; Morimoto et al., 1996; Schnell et al., 1994; Yan et al., 2002), it has been demonstrated that it is the G that determines the level of G expression and the induction of apoptosis (Sarmento et al., 2005). In this present study, we used B2c as the backbone and replaced the G from those of street RABV DRV and SHBRV. Again, it is the G that determines the level of the G expression and the induction of innate immune responses. Most importantly our present study demonstrates that it is the G that determines the pathogenic mechanisms for the fixed and street RABVs. Direct IC infection of mice with fixed RABV (large doses, 10 ICLD50) resulted in the induction of innate immunity and the infiltration of inflammatory cells in the CNS. It is thus possible that infected mice develop disease and death via immune-mediated pathogenesis. On the other hand, street RABV or rRABV expressing the G from street viruses induce only limited innate immune responses, mild inflammation or other pathological changes in the CNS. Despite the fact that the level of RABV N is similar for all the viruses in the brain of mice, the level of G expression is higher in mice infected with fixed viruses than in mice infected with street RABV or rRABV expressing the G from street viruses. It is thus the G that determines the level of G expression, the induction of the innate immunity, and consequently the pathogenic mechanisms. Although the exact mechanism by which street RABV induces rabies remains to be determined, hypothesis has been proposed such as neuronal dysfunction (Tsiang, 1982). Further studies are warranted to decipher the mechanism(s) by which street RABV induces rabies.
Supplementary Material
Acknowledgements
This work was supported partially by Public Health Service grant AI-051560 from the National Institute of Allergy and Infectious Diseases (Z.F.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benmansour A, Leblois H, Coulon P, Tuffereau C, Gaudin Y, Flamand A, Lafay F. Antigenicity of rabies virus glycoprotein. Journal of virology. 1991;65:4198–4203. doi: 10.1128/jvi.65.8.4198-4203.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann KK, Cox JH, Schneider LG, Thiel HJ. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology. 1990;175:485–499. doi: 10.1016/0042-6822(90)90433-r. [DOI] [PubMed] [Google Scholar]
- Dietzschold B, Morimoto K, Hooper DC, Smith JS, Rupprecht CE, Koprowski H. Genotypic and phenotypic diversity of rabies virus variants involved in human rabies: implications for postexposure prophylaxis. Journal of human virology. 2000;3:50–57. [PubMed] [Google Scholar]
- Dietzschold B, Schnell M, Koprowski H. Pathogenesis of rabies. Current topics in microbiology and immunology. 2005;292:45–56. doi: 10.1007/3-540-27485-5_3. [DOI] [PubMed] [Google Scholar]
- Dietzschold ML, Faber M, Mattis JA, Pak KY, Schnell MJ, Dietzschold B. In vitro growth and stability of recombinant rabies viruses designed for vaccination of wildlife. Vaccine. 2004;23:518–524. doi: 10.1016/j.vaccine.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Faber M, Pulmanausahakul R, Hodawadekar SS, Spitsin S, McGettigan JP, Schnell MJ, Dietzschold B. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response. Journal of virology. 2002;76:3374–3381. doi: 10.1128/JVI.76.7.3374-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabis MJ, Phares TW, Kean RB, Koprowski H, Hooper DC. Blood-brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc Natl Acad Sci U S A. 2008;105:15511–15516. doi: 10.1073/pnas.0807656105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZF, Rupprecht CE, Dietzschold B, Saikumar P, Niu HS, Babka I, Wunner WH, Koprowski H. Oral vaccination of racoons (Procyon lotor) with baculovirus-expressed rabies virus glycoprotein. Vaccine. 1993;11:925–928. doi: 10.1016/0264-410x(93)90379-c. [DOI] [PubMed] [Google Scholar]
- Inoue K, Shoji Y, Kurane I, Iijima T, Sakai T, Morimoto K. An improved method for recovering rabies virus from cloned cDNA. Journal of virological methods. 2003;107:229–236. doi: 10.1016/s0166-0934(02)00249-5. [DOI] [PubMed] [Google Scholar]
- Ito N, Takayama M, Yamada K, Sugiyama M, Minamoto N. Rescue of rabies virus from cloned cDNA and identification of the pathogenicity-related gene: glycoprotein gene is associated with virulence for adult mice. Journal of virology. 2001;75:9121–9128. doi: 10.1128/JVI.75.19.9121-9128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Luo Y, Michel F, Hogan RJ, He Y, Fu ZF. Characterization of conformation-specific monoclonal antibodies against rabies virus nucleoprotein. Archives of virology. 2010;155:1187–1192. doi: 10.1007/s00705-010-0709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, McKimmie CS, Mansfield KL, Wakeley PR, Brookes SM, Fazakerley JK, Fooks AR. Lyssavirus infection activates interferon gene expression in the brain. The Journal of general virology. 2006;87:2663–2667. doi: 10.1099/vir.0.82024-0. [DOI] [PubMed] [Google Scholar]
- Kuang Y, Lackay SN, Zhao L, Fu ZF. Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after rabies virus infection. Virus research. 2009;144:18–26. doi: 10.1016/j.virusres.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetylcholine receptor a rabies virus receptor? Science. 1982;215:182–184. doi: 10.1126/science.7053569. [DOI] [PubMed] [Google Scholar]
- Lentz TL, Wilson PT, Hawrot E, Speicher DW. Amino acid sequence similarity between rabies virus glycoprotein and snake venom curaremimetic neurotoxins. Science. 1984;226:847–848. doi: 10.1126/science.6494916. [DOI] [PubMed] [Google Scholar]
- Li XQ, Sarmento L, Fu ZF. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. Journal of virology. 2005;79:10063–10068. doi: 10.1128/JVI.79.15.10063-10068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. Journal of virology. 2001;75:11496–11502. doi: 10.1128/JVI.75.23.11496-11502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K, Matsumoto S. Comparative studies between pathogenesis of street and fixed rabies infection. The Journal of experimental medicine. 1967;125:447–456. doi: 10.1084/jem.125.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Foley HD, McGettigan JP, Schnell MJ, Dietzschold B. Reinvestigation of the role of the rabies virus glycoprotein in viral pathogenesis using a reverse genetics approach. Journal of neurovirology. 2000;6:373–381. doi: 10.3109/13550280009018301. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Hooper DC, Carbaugh H, Fu ZF, Koprowski H, Dietzschold B. Rabies virus quasispecies: implications for pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3152–3156. doi: 10.1073/pnas.95.6.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. Journal of virology. 1999;73:510–518. doi: 10.1128/jvi.73.1.510-518.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Patel M, Corisdeo S, Hooper DC, Fu ZF, Rupprecht CE, Koprowski H, Dietzschold B. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5653–5658. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FA. Rabies pathogenesis. Archives of virology. 1977;54:279–297. doi: 10.1007/BF01314774. [DOI] [PubMed] [Google Scholar]
- Nakamichi K, Inoue S, Takasaki T, Morimoto K, Kurane I. Rabies virus stimulates nitric oxide production and CXC chemokine ligand 10 expression in macrophages through activation of extracellular signal-regulated kinases 1 and 2. Journal of virology. 2004;78:9376–9388. doi: 10.1128/JVI.78.17.9376-9388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JT, Davis NL, Wertz GW. N protein alone satisfies the requirement for protein synthesis during RNA replication of vesicular stomatitis virus. Journal of virology. 1984;49:303–309. doi: 10.1128/jvi.49.2.303-309.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares TW, Fabis MJ, Brimer CM, Kean RB, Hooper DC. A peroxynitrite-dependent pathway is responsible for blood-brain barrier permeability changes during a central nervous system inflammatory response: TNF-alpha is neither necessary nor sufficient. J Immunol. 2007;178:7334–7343. doi: 10.4049/jimmunol.178.11.7334. [DOI] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. Journal of virology. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulmanausahakul R, Li J, Schnell MJ, Dietzschold B. The glycoprotein and the matrix protein of rabies virus affect pathogenicity by regulating viral replication and facilitating cell-to-cell spread. Journal of virology. 2008;82:2330–2338. doi: 10.1128/JVI.02327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raux H, Flamand A, Blondel D. Interaction of the rabies virus P protein with the LC8 dynein light chain. Journal of virology. 2000;74:10212–10216. doi: 10.1128/jvi.74.21.10212-10216.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raux H, Iseni F, Lafay F, Blondel D. Mapping of monoclonal antibody epitopes of the rabies virus P protein. The Journal of general virology. 1997;78(Pt 1):119–124. doi: 10.1099/0022-1317-78-1-119. [DOI] [PubMed] [Google Scholar]
- Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. Journal of virology. 2007;81:7993–7998. doi: 10.1128/JVI.00710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Phares TW, Koprowski H, Hooper DC. Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. Journal of virology. 2007;81:1110–1118. doi: 10.1128/JVI.01964-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento L, Li XQ, Howerth E, Jackson AC, Fu ZF. Glycoprotein-mediated induction of apoptosis limits the spread of attenuated rabies viruses in the central nervous system of mice. Journal of neurovirology. 2005;11:571–581. doi: 10.1080/13550280500385310. [DOI] [PubMed] [Google Scholar]
- Schnell MJ, Mebatsion T, Conzelmann KK. Infectious rabies viruses from cloned cDNA. The EMBO journal. 1994;13:4195–4203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol F, Schlumberger HD, Wiktor TJ, Koprowski H. Biochemical and biophysical studies on the nucleocapsid and on the RNA of rabies virus. Virology. 1969;38:651–665. doi: 10.1016/0042-6822(69)90184-6. [DOI] [PubMed] [Google Scholar]
- Takamatsu F, Asakawa N, Morimoto K, Takeuchi K, Eriguchi Y, Toriumi H, Kawai A. Studies on the rabies virus RNA polymerase: 2. Possible relationships between the two forms of the non-catalytic subunit (P protein) Microbiology and immunology. 1998;42:761–771. doi: 10.1111/j.1348-0421.1998.tb02350.x. [DOI] [PubMed] [Google Scholar]
- Tan GS, Preuss MA, Williams JC, Schnell MJ. The dynein light chain 8 binding motif of rabies virus phosphoprotein promotes efficient viral transcription. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7229–7234. doi: 10.1073/pnas.0701397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoulouze MI, Lafage M, Schachner M, Hartmann U, Cremer H, Lafon M. The neural cell adhesion molecule is a receptor for rabies virus. Journal of virology. 1998;72:7181–7190. doi: 10.1128/jvi.72.9.7181-7190.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordo N, Poch O, Ermine A, Keith G, Rougeon F. Walking along the rabies genome: is the large G-L intergenic region a remnant gene? Proceedings of the National Academy of Sciences of the United States of America. 1986;83:3914–3918. doi: 10.1073/pnas.83.11.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang H. Neuronal function impairment in rabies-infected rat brain. The Journal of general virology. 1982;61(Pt 2):277–281. doi: 10.1099/0022-1317-61-2-277. [DOI] [PubMed] [Google Scholar]
- Tuffereau C, Benejean J, Blondel D, Kieffer B, Flamand A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. The EMBO journal. 1998;17:7250–7259. doi: 10.1093/emboj/17.24.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidy A, El Bougrini J, Chelbi-Alix MK, Blondel D. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. Journal of virology. 2007;81:4255–4263. doi: 10.1128/JVI.01930-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cao S, Du J, Dong G, Tang Q, Tang J, Shi L, Li J, Wu X. Sequencing of Complete Genome of Rabies Virus CVS-11 Strain. Zhongguo Sheng Wu Zhi Pin Xue Za Zhi (in Chinese) 2010;23:455–459. [Google Scholar]
- Wang ZW, Sarmento L, Wang Y, Li XQ, Dhingra V, Tseggai T, Jiang B, Fu ZF. Attenuated rabies virus activates, while pathogenic rabies virus evades, the host innate immune responses in the central nervous system. Journal of virology. 2005;79:12554–12565. doi: 10.1128/JVI.79.19.12554-12565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Wang H, Wu H, Yang F, Tripp RA, Hogan RJ, Fu ZF. Rabies virus expressing dendritic cell-activating molecules enhances the innate and adaptive immune response to vaccination. Journal of virology. 2011;85:1634–1644. doi: 10.1128/JVI.01552-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . WHO Technical Report Series. No 931. World Health Organization; Geneva: 2005. Expert consultation on rabies, first report. [PubMed] [Google Scholar]
- Wiktor TJ, Gyorgy E, Schlumberger D, Sokol F, Koprowski H. Antigenic properties of rabies virus components. J Immunol. 1973;110:269–276. [PubMed] [Google Scholar]
- Wirblich C, Schnell MJ. Rabies virus (RV) glycoprotein expression levels are not critical for pathogenicity of RV. Journal of virology. 2011;85:697–704. doi: 10.1128/JVI.01309-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunner WH. Rabies virus. In: Jackson AC, Wunner WH, editors. Rabies. 2nd edn Elsevier/Academic Press; London: 2007. pp. 23–68. [Google Scholar]
- Yan X, Mohankumar PS, Dietzschold B, Schnell MJ, Fu ZF. The rabies virus glycoprotein determines the distribution of different rabies virus strains in the brain. Journal of neurovirology. 2002;8:345–352. doi: 10.1080/13550280290100707. [DOI] [PubMed] [Google Scholar]
- Yan X, Prosniak M, Curtis MT, Weiss ML, Faber M, Dietzschold B, Fu ZF. Silver-haired bat rabies virus variant does not induce apoptosis in the brain of experimentally infected mice. Journal of neurovirology. 2001;7:518–527. doi: 10.1080/135502801753248105. [DOI] [PubMed] [Google Scholar]
- Zhao L, Toriumi H, Kuang Y, Chen H, Fu ZF. The roles of chemokines in rabies virus infection: overexpression may not always be beneficial. Journal of virology. 2009;83:11808–11818. doi: 10.1128/JVI.01346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Toriumi H, Wang H, Kuang Y, Guo X, Morimoto K, Fu ZF. Expression of MIP-1alpha (CCL3) by a recombinant rabies virus enhances its immunogenicity by inducing innate immunity and recruiting dendritic cells and B cells. Journal of virology. 2010;84:9642–9648. doi: 10.1128/JVI.00326-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.