

Abstract
Despite numerous approaches for the development of l-threonine-producing strains, strain development is still hampered by the intrinsic inefficiency of metabolic reactions caused by simple diffusion and random collisions of enzymes and metabolites. A scaffold system, which can promote the proximity of metabolic enzymes and increase the local concentration of intermediates, was reported to be one of the most promising solutions. Here, we report an improvement in l-threonine production in Escherichia coli using a DNA scaffold system, in which a zinc finger protein serves as an adapter for the site-specific binding of each enzyme involved in l-threonine production to a precisely ordered location on a DNA double helix to increase the proximity of enzymes and the local concentration of metabolites to maximize production. The optimized DNA scaffold system for l-threonine production significantly increased the efficiency of the threonine biosynthetic pathway in E. coli, substantially reducing the production time for l-threonine (by over 50%). In addition, this DNA scaffold system enhanced the growth rate of the host strain by reducing the intracellular concentration of toxic intermediates, such as homoserine. Our DNA scaffold system can be used as a platform technology for the construction and optimization of artificial metabolic pathways as well as for the production of many useful biomaterials.
INTRODUCTION
As the building blocks of life, amino acids have long played an important role in both human and animal nutrition and health maintenance. On account of its functionality and the special features arising from chirality, this class of compounds is extremely important and of great interest for the chemical industry (1). Of the 20 standard amino acids, the 9 essential amino acids (histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine) occupy a key position, in that they are not synthesized in animals and humans but must be ingested with feed or food (2).
Production methods for these essential amino acids are broadly classified into three types: extraction, chemical synthesis, and microbial fermentation. Among these methods, the microbial fermentation method is being widely applied to the industrial production of most essential amino acids, except for a few kinds of l-amino acids for which high production yields have not been achieved by fermentation (1). The advances in the industrial production of amino acids are closely connected with screening or selection of suitable putative production hosts and subsequent improvement of production strains. Previous attempts at strain improvement have relied on classical mutagenesis and screening procedures, which focused on deleting competing pathways and eliminating feedback regulations in the biosynthetic pathway (1, 3–7). The recent trend of whole-genome analysis and systems biology has begun to exert a profound effect on the strategy of strain development. The barrage of information has led to a better understanding of the architecture of cellular metabolism. These studies allow a novel approach of assembling only beneficial genetic modifications in a production host to be taken. This genomics-based strain construction is now being carried out successfully (8–11).
Despite all these efforts, strain improvements are still hampered by intrinsic drawbacks of metabolic reactions. The actual metabolic reactions of amino acid biosynthetic pathways in microorganisms are mediated by simple diffusion and random collisions of metabolites and enzymes, which cause metabolic reactions to be inefficient by lowering the local metabolite concentration around the enzymes (12). Furthermore, the accumulation of toxic intermediates, such as a homocysteine in the methionine biosynthesis pathway, within a host cell inhibits the activity of many cellular functions by lowering cytoplasmic pH (13). These intrinsic drawbacks of metabolic reactions have prompted researchers to develop novel biological systems that make enzymatic reactions more efficient (14). Among many suggested solutions, a scaffold system, which organizes enzymes spatially and temporally and increases the local concentration of intermediates (enforced proximity or substrate channeling), turns out to be one of the most promising approaches (14–25). Recently, Dueber et al. (18) expressed scaffolds built from the interaction domains of metazoan signaling proteins to assemble metabolic enzymes and reported significant increases in the production of mevalonate and, separately, glucaric acid. Delebecque et al. (26) created RNA aptamer-based scaffolds to control the spatial organization of two metabolic enzymes involved in biological hydrogen production, and Conrado et al. (27) reported a DNA-based scaffold system to promote improved catalytic efficiency: application of the DNA scaffold to produce resveratrol, 1,2-propandiol, and mevalonate significantly enhanced the titer of these metabolites. These reports demonstrate the potential of scaffold systems as a powerful tool for metabolic engineering.
Here we describe a DNA scaffold system designed for the improved metabolic efficiency in the l-threonine biosynthetic pathway of Escherichia coli (Fig. 1). Application of the DNA scaffold system to l-threonine production significantly increased the efficiency of the l-threonine biosynthetic pathway in E. coli, substantially reducing the production time for l-threonine (by over 50%). In addition, the DNA scaffold system significantly enhanced the growth rate of the l-threonine-producing strain by reducing the intracellular concentration of toxic intermediates such as homoserine. Therefore, our results confirm that DNA scaffold systems should be useful as a platform technology for the construction and optimization of numerous artificial metabolic pathways.
Fig 1.

General scheme of a DNA scaffold system. Enzymes of a desired metabolic pathway are fused with artificial DNA binding domains consisting of 4 ZFPs. The fused enzymes are then bound to their specific target sites on the DNA scaffold to form an artificial enzymatic cascade. The number of enzymes bound to the DNA scaffold, order of enzymes, relative angles of enzymes to each other, and distance between the binding sites for each enzyme can be controlled in a designable manner by simply changing the sequence of binding sites on the DNA scaffold.
MATERIALS AND METHODS
Bacterial strains, plasmids, enzymes, and chemicals.
The bacterial strains and plasmids used in this work are listed in Table 1. E. coli strain K-12 MG1655 (28) was used as a cloning host, E. coli strain BL21(DE3) (Invitrogen, Carlsbad, CA) was used for the expression of enzymes, and E. coli strain MG-105 (30) was used as an l-threonine production host. All of the oligonucleotides used in this work (Table 2) were synthesized by Genotech (Daejeon, South Korea). All enzymes were purchased from New England BioLabs (Beverly, MA), except Taq polymerase, which was from TaKaRa Bio Inc. (Shiga, Japan). All antibiotics and chemicals were from Sigma-Aldrich (St. Louis, MO). Ampicillin, chloramphenicol, and kanamycin were used at concentrations of 50, 17, and 25 μg/ml, respectively.
Table 1.
Strains used in this study
| E. coli strain | Description | Reference or source |
|---|---|---|
| K-12 MG1655 | λ− F− ilvG rfb-50 rph-1 | Blattner et al. (28) |
| ATCC 21277 | K-12 supE relA+ Kmr-Ptac-thrA*BC ilvA422 | Shiio et al. (29) |
| BL21(DE3) | F− ompT hsdSB(rB−, mB−) gal dcm(DE3) | Invitrogen |
| MG-105 | K-12 MG1655 Ptac-thrA*BC ΔlacI Δtdh ΔtdcC::rhtA23 ΔsstT::rhtA23 | Lee et al. (30) |
| MG-105E | MG-105(pET16tac-thrABC) | This study |
| MG-105ES | MG-105(pET16tac-thrABC)(pSC-4) | This study |
Table 2.
Oligonucleotides used in this study
| Name | Sequence (5′ → 3′) |
|---|---|
| A-for | GAG CCA TAT GAT GCG AGT GTT GAA GTT CGG CG |
| A-rev | CCC GGG GAT CCC GCT ACC TCC GCC ACC GCT ACC TCC GCC ACC ACT CCT AAC TTC CAT GAG AGG GT |
| B-for | GAC CCA TAT GGT TAA AGT TTA TGC CCC GGC TT |
| B-rev | CCC GGG GAT CCC GCT ACC TCC GCC ACC GCT ACC TCC GCC ACC GTT TTC CAG TAC TCG TGC GCC CGC C |
| C-for | GAG CCA TAT GAA ACT CTA CAA TCT GAA AGA TC |
| C-rev | CCC GGG GAT CCC GCT ACC TCC GCC ACC GCT ACC TCC GCC ACC CTG ATG ATT CAT CAT CAA TTT ACG CAA CG |
Combinatorial assembly of ZFPs for construction of artificial DNA binding domains.
All of the plasmids containing DNA fragments encoding single zinc finger proteins (ZFPs), which were used for the construction of artificial DNA binding domains (ADBs), were obtained from Lee et al. (31). To minimize the undesired binding of ADBs to the E. coli genomic DNA, which might change the expression of downstream genes, the three ADBs used in this study (ADB1, ADB2, and ADB3) were designed such that their recognition sites were not present in the E. coli genome. pADB1, which encodes ADB1 (RSNR-RDHT-VSTR-QSNI), a binding domain that recognizes the 12-bp DNA sequence 5′-CAAGCTAGGGAG-3′, was constructed as follows. The DNA fragment encoding the second ZFP, RDHT, was obtained by digesting pRDHT with XmaI and EcoRI, and then the fragment was ligated into the AgeI/EcoRI site of pRSNR to generate pRSNR-RDHT. The DNA fragment encoding the third ZFP, VSTR, was obtained by digesting pVSTR with XmaI and EcoRI, and then the fragment was ligated into the AgeI/EcoRI site of pRSNR-RDHT to generate pRSNR-RDHT-VSTR. Subsequently, the DNA fragment encoding the fourth ZFP, QSNI, was obtained by digesting pQSNI with XmaI and EcoRI, and then the fragment was ligated into the AgeI/EcoRI site of pRSNR-RDHT-VSTR to generate pADB1. pADB2 and pADB3, encoding ADB2 (VSSR-RSHR-RSNR-CSNR) and ADB3 (QSSR-RSHR-RSHR-QTHQ), respectively, which recognize the 12-bp DNA sequences 5′-GACGAGGGGGTG-3′ and 5′-GAAGGGGGGGTA-3′, respectively, were constructed in the same manner as pADB1, except for using plasmids that encoded different ZFPs (pVSSR, pRSHR, pRSNR, and pCSNR for the construction of pADB2 and pQSSR, pRSHR, and pQTHQ for the construction of pADB3).
Construction of plasmids encoding fusions between threonine biosynthesis enzymes and artificial DNA binding domains.
The enzymes involved in l-threonine biosynthesis, homoserine dehydrogenase (HDH), homoserine kinase (HK), and threonine synthase (TS) (Fig. 2), were fused with artificial DNA binding domains as follows. The thrA*, thrB, and thrC genes, encoding feedback-inhibition-resistant HDH, HK, and TS, respectively, were amplified from E. coli K-12 ATCC 21277 genomic DNA (29) using the forward and reverse primers described in Table 2. The amplified DNA fragments for the thrA, thrB, and thrC genes, after digestion with NdeI and BamHI, were cloned into the NdeI/BamHI site of pET16b to produce pET16b-thrA, pET16b-thrB, and pET16b-thrC, respectively. Then, the DNA fragments containing a T7 promoter and each gene, obtained by digesting pET16b-thrA, pET16b-thrB, and pET16b-thrC with BglII and BamHI, were cloned into the BglII/BamHI site of pET21c to generate pET21c-thrA, pET21c-thrB, and pET21c-thrC, respectively. Next, DNA fragments encoding ADB1, ADB2, and ADB3 were obtained by digesting pADB1, pADB2, and pADB3, respectively, with BamHI and EcoRI and then ligated into the BamHI/EcoRI site of pET21c-thrA, pET21c-thrB, and pET21c-thrC, respectively, producing pET21c-thrA-ADB1, pET21c-thrB-ADB2, and pET21c-thrC-ADB3, respectively.
Fig 2.
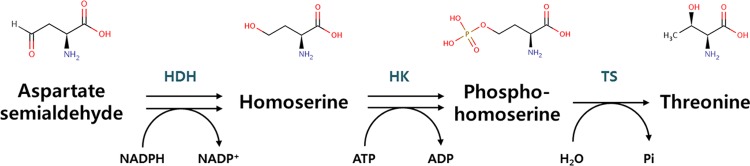
l-Threonine biosynthesis from aspartate semialdehyde in E. coli. The three steps for the l-threonine synthesis pathway are catalyzed by homoserine dehydrogenase (HDH), homoserine kinase (HK), and threonine synthase (TS).
To express the three enzyme fusions as an artificial operon, pETtac-thrABC was constructed as follows. First, a DNA fragment containing a T7 promoter and the thrA-ADB1 gene fusion, obtained by digesting pET21c-thrA-ADB1 with BglII and NotI, was ligated into the BglII/XbaI site of pET21c-thrB-ADB2, in which the XbaI site was blunted by treating with mung bean nuclease, to generate pET21c-thrAB. Then, a DNA fragment containing a T7 promoter and the thrA-ADB1 and thrB-ADB2 gene fusions, obtained by digesting pET21c-thrAB with BglII and NotI, was ligated into the BglII/XbaI site of pET21c-thrC-ADB3, in which the XbaI site was blunted by treating with mung bean nuclease, producing pET21c-thrABC. A DNA fragment containing the thrA-ADB1, thrB-ADB2, and thrC-ADB3 gene fusions, obtained by digesting pET21c-thrABC with NcoI and EcoRI, was ligated into the NcoI/EcoRI site of plasmid pETtac (31), to generate pETtac-thrABC.
All of the scaffold plasmids used in this study (Fig. 3) were constructed by the ligation of binding site sequences synthesized by Genotech (Daejeon, South Korea) into the pCR2.1-TOPO plasmid using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Fig 3.
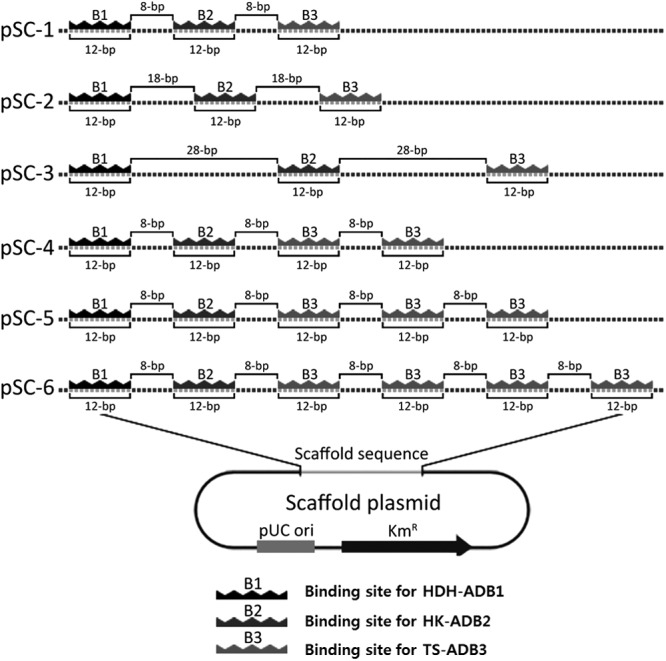
Design and construction of scaffold plasmids for the optimization of l-threonine production. To optimize the design of a DNA scaffold system for the production of l-threonine, six different scaffold plasmids were constructed (i) with different distances between the binding sites for artificial DNA binding domain-fused enzymes (HDH-ADB1, HK-ADB2, and TS-ADB3) or (ii) with different stoichiometric ratios of three enzymes (HDH-ADB1/HK-ADB2/TS-ADB3). Scaffold plasmids pSC-1, pSC-2, and pSC-3 were constructed with spacer DNA sequences of different lengths (8, 18, and 28 bp, respectively) between the 12-bp binding sites for ADBs (ADB1, ADB2, and ADB3) to locate an ADB-fused enzyme every 20, 30, and 40 bp, respectively. Scaffold plasmids pSC-4, pSC-5, and pSC-6 were constructed to bind different stoichiometric ratios (1:1:2, 1:1:3, and 1:1:4, respectively) of three enzymes, with 8-bp spacer DNA sequences between the sites.
In vitro production of l-threonine with a DNA scaffold system.
For the purification of enzymes involved in the l-threonine biosynthesis pathway (HDH, HK, TS, HDH-ADB1, HK-ADB2, and TS-ADB3), plasmids for the expression of each enzyme (pET21c-thrA, pET21c-thrB, pET21c-thrC, pET21c-thrA-ADB1, pET21c-thrB-ADB2, and pET21c-thrC-ADB3, respectively) were transformed into E. coli BL21(DE3) competent cells. Each transformed clone was grown in 100 ml LB supplemented with 50 μg ampicillin to early log phase (optical density at 600 nm [OD600], 0.4) at 30°C with constant shaking at 200 rpm. Enzyme expression was induced by adding 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to the culture medium. After 4 h incubation, cultured cells were harvested by centrifuging at 1,000 × g for 5 min at 4°C. The pellet was suspended in 20 ml extraction buffer II (100 mM Tris-HCl [pH 8.0], 25 mM imidazole). The cells in suspension were lysed by three freeze-thaw cycles, and the clear supernatant, obtained after centrifugation at 10,000 × g for 10 min at 4°C, was processed for protein purification on a Ni-nitrilotriacetic acid agarose affinity column. The affinity resin was equilibrated with extraction buffer II, the supernatant was loaded onto the matrix, and fractions were collected at a flow rate of 3 ml/min. The unbound proteins were washed away with extraction buffer II, and the bound protein was eluted with a step gradient of imidazole (50, 100, 200, and 300 mM) in elution buffer containing 100 mM Tris-HCl (pH 8.0). The eluted protein was dialyzed against 20 mM Tris-HCl (pH 8.0) containing 20% (vol/vol) glycerol. Protein concentrations were determined using a commercial bicinchoninic acid assay (Pierce, Rockford, IL) with bovine serum albumin (0.05 to 2 mg/ml) as the standard.
All enzymatic activities were determined as described by Szczesiul and Wampler (32) with the following modifications. HDH activity was measured by following NADPH oxidation at 340 nm for the forward reaction and NADP+ reduction at 340 nm for the reverse reaction (33). The forward reaction was performed in 1 ml of assay buffer {23 mM TES [N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid] buffer, pH 7.5, 114 mM KCl, 6 mM MgCl2} that contained 0.3 mM NADPH, 1 mM aspartate semialdehyde, and 40 μg of purified enzyme. The reverse reaction was carried out in 1 ml of assay buffer that contained 0.8 mM NADP+, 10 mM homoserine, and 100 μg of purified HDH enzyme.
HK activity was measured in a coupled assay with pyruvate kinase and lactate dehydrogenase, as described by Wampler and Westhead (34) by following NADH oxidation at 340 nm. The reaction was performed in 1 ml of assay buffer containing 2 mM homoserine, 4 mM ATP, 1.5 mM phosphoenolpyruvate, 0.3 mM NADH, 2 units of pyruvate kinase, 2.25 units of lactate dehydrogenase, and 100 μg of purified HK enzyme.
TS activity was determined by measuring the amount of inorganic phosphate released from phosphohomoserine. The reaction was started by adding 2 mM phosphohomoserine as a substrate in 3 ml of assay buffer containing 0.1 mM pyridoxal phosphate and 200 μg of purified TS enzyme. Then, 400 μl of the reaction mixture was removed and mixed with 40 μl of 40% (wt/vol) perchloric acid; the solution was neutralized with 2 M KOH–0.2 M MOPS (morpholinepropanesulfonic acid) and then centrifuged for 15 min at 4°C and 10,000 × g. The inorganic phosphate concentration was measured as described by Kyaw et al. (35). Triton X-100 (150 μl) and ammonium molybdate reagent [150 μl; 2.5 g (NH4)6Mo7O24 · 4H2O dissolved in 30 ml of 10 N sulfuric acid, after which the solution was diluted to 100 ml with water] were added to 200 μl of centrifuged solution containing inorganic phosphate, shaking after each addition. After standing for 10 min at 25°C, the absorbance of the solution was read at 375 nm against a blank control. The difference between the original and the final inorganic phosphate concentrations gave the rate of the reaction.
To test the effect of our DNA scaffold system on the in vitro production of l-threonine, artificial enzymatic cascades were formed by the addition of 50, 100, or 200 ng of the scaffold plasmids to 100 μl of the enzyme mixture containing 200 μg of each enzyme (HDH-ADB1, HK-ADB2, and TS-ADB3). The activities of the artificial enzymatic cascades were determined by measuring the inorganic phosphate released from phosphohomoserine as described above, with the following modifications. The reaction was started by adding 4 mM aspartate semialdehyde (ASA) to 3 ml of assay buffer containing 4 mM ATP, 0.3 mM NADPH, 0.1 mM pyridoxal phosphate, and 100 μl of artificial enzymatic cascade.
In vivo production of l-threonine with a DNA scaffold.
The threonine-producing strains (Table 1) were grown overnight on LB plates and then transferred to a 250-ml flask containing 50 ml of seed medium [32.5 g glucose, 24.35 g K2HPO4, 9.5 g KH2PO4, 15 g yeast extract, 5 g (NH4)2SO4, 1 g MgSO4 · 7H2O per liter at pH 7.0]. After growth for 24 h at 37°C, an aliquot (5 ml) of the seed culture was transferred to 500 ml of fermentation medium 1 [100 g glucose, 10 g (NH4)2SO4, 2 g KH2PO4, 0.5 g MgSO4 · 7 H2O, 5 mg FeSO4 · 7H2O, 5 mg MnSO4 · 4H2O, and 3 g yeast extract per liter at pH 7.5] in a 1-liter jar fermentor supplemented with 0.1 mM IPTG. During batch-phase fermentation, the pH was maintained at 7.5 with NH4OH, the temperature was maintained at 37°C, the aeration rate was maintained at 1 vvm (air volume · working volume−1 · min−1), and the agitation speed was maintained at 800 rpm. During the fermentation, the concentrations of the metabolites l-threonine, glucose, and homoserine were determined by high-pressure liquid chromatography (HPLC) using precolumn derivation with o-phthaldehyde-thiol (OPA) developed by Joseph and Marsden (36) with the following modifications. Metabolites were analyzed on a Micra NPS ODS-1 1.5-μm column (33 mm by 4.6 mm; Eichrom Technologies, IL) in reversed phase with a concentration gradient of sodium acetate buffer. This gradient was formed from two buffers, 100 mM sodium acetate, pH 5.9 (adjusted with 1 M HCl; buffer A) and pure methanol (buffer B), with a flow rate of 0.5 ml/min. The time course of the gradient was as follows: at the starting point, buffer A/buffer B at 98:2 (vol/vol); at 1 min, buffer A/buffer B at 85:15 (vol/vol); at 5 min, buffer A/buffer B at 50:50 (vol/vol); at 10 min, buffer A/buffer B at 30:70 (vol/vol); and at 18 min, buffer A/buffer B at 2:98 (vol/vol). The retention times and response factors of the metabolites were evaluated by injecting known amounts of standard molecules.
RESULTS
An artificial enzymatic cascade assembled on a DNA scaffold.
The general scheme of our DNA scaffold system is depicted in Fig. 1. ADBs consisting of 4 ZFPs, which serve as ligands for the precise localization of enzymes on a DNA scaffold, were constructed by combinatorial assembly of 40 selected ZFPs (31, 37, 38). This process theoretically provides 2.56 × 106 (= 40 × 40 × 40 × 40) different ADBs, which recognize and bind to specific 12-bp target DNA sequences. These ADBs were then fused with the enzymes involved in the desired metabolic pathway via three tandem repeats of a flexible G4S linker (39). In the presence of Zn2+, enzymes fused with the ADBs precisely bind to their target DNA sequences on a DNA scaffold, forming an artificial enzymatic cascade. The number of enzymes bound to the DNA scaffold and order of enzymes can be controlled in a designable manner by simply changing the sequence of binding sites on the DNA scaffold, and the relative position of enzymes to each other and the distance between the binding sites for each enzyme might be also controllable to some extent. Through the artificial enzymatic cascade on a DNA scaffold, the metabolic efficiency of a desired pathway should be significantly improved because of the increased local concentrations of substrates around enzymes and reduced loss of intermediates to competing pathways (14, 27).
Effect of a DNA scaffold on the production of l-threonine in vitro.
The effect of the DNA scaffold on the l-threonine biosynthesis pathway in E. coli was examined. l-Threonine is synthesized from ASA in E. coli by the sequential action of three enzymes (HDH, HK, and TS) (40, 41) (Fig. 2). An extensive analysis of enzymatic kinetics revealed that the final step, the conversion of phosphohomoserine to l-threonine by TS, is slower than the other preceding reactions. Furthermore, it has been reported that the intracellular accumulation of homoserine or phosphohomoserine, both of which are toxic reaction intermediates, might inhibit host cell growth during fermentation (32, 33, 42, 43). Therefore, to improve the metabolic efficiency and relieve the toxic effect of accumulated intermediates, rapid channeling of the intermediates to l-threonine was explored using an artificial enzymatic cascade assembled on a DNA scaffold.
To construct an artificial enzymatic cascade for l-threonine production, first, HDH, HK, and TS enzymes were fused to the N termini of ADBs, termed ADB1, ADB2, and ADB3, respectively. The expression of HDH-ADB1, HK-ADB2, or TS-ADB3 fusions did not show any noticeable changes in the growth of E. coli BL21(DE3) (see Fig. S1 in the supplemental material). Likewise, the kinetic parameters of the ADB-fused enzymes (HDH-ADB1, HK-ADB2, and TS-ADB3) are comparable to those of the wild-type enzymes (Table 3). In addition, the binding of the ADB-fused enzymes to their target DNA sequences was confirmed by a gel shift assay (see Fig. S2 in the supplemental material). The ability of this artificial enzymatic cascade to produce l-threonine was confirmed by an in vitro enzymatic assay. The rate of l-threonine production was significantly increased by the addition of the scaffold plasmid pSC-1, which contains one binding site for each enzyme (HDH-ADB1, HK-ADB2, and TS-ADB3) separated by 8-bp spacers (Fig. 3 and 4a). Without pSC-1, it took about 30 min to convert 80% of the substrate ASA to l-threonine under the tested conditions. Notably, it took less than 10 min to produce the same amount of l-threonine in the presence of pSC-1. The increase in the reaction rate was dependent on the concentration of pSC-1 added (Fig. 4a). Thus, the reaction rate of the l-threonine biosynthesis pathway could be significantly improved by the formation of an artificial, DNA scaffold-based enzymatic cascade.
Table 3.
Kinetic parameters for the enzymes used in this studya
| Enzyme | Km (mM) | kcat (1/s) |
Ki (mM) |
|
|---|---|---|---|---|
| Threonine | Homoserine | |||
| HDH | 0.238 ± 0.04 | 27.9 ± 0.45 | 0.1 | |
| HDH-ADB1 | 0.233 ± 0.08 | 28.3 ± 0.21 | 0.09 | |
| HK | 0.112 ± 0.007 | 17.8 ± 0.38 | 1.2 | 4.6 |
| HK-ADB2 | 0.109 ± 0.02 | 17.1 ± 0.32 | 1.08 | 4.4 |
| TS | 0.321 ± 0.03 | 7.48 ± 0.19 | ||
| TS-ADB3 | 0.313 ± 0.05 | 7.23 ± 0.25 | ||
The data shown are the means and standard deviations for three independent experiments.
Fig 4.

Optimization of the DNA scaffold for the production of l-threonine in vitro. Rates of threonine production from the different scaffold plasmids were measured by following the in vitro enzymatic conversion of aspartate semialdehyde to threonine (refer to Materials and Methods). The increase in the l-threonine production rate was dependent on the concentrations of the scaffold plasmid used (50, 100, or 200 ng) (a), the distances between the binding sites of artificial DNA binding domains (20, 30, or 40 bp) (b), and the numbers of TS-ADB3 enzymes bound to the scaffold plasmid (2, 3, or 4 enzymes) (c). The data shown are the means for three independent experiments.
Optimization of a DNA scaffold in a designable manner.
To study the relation between the spatial arrangement of enzymes on a DNA scaffold and the reaction efficiency of the enzymes, a series of scaffold plasmids, pSC-1, pSC-2, and pSC-3, with spacer DNA sequences of different lengths (8, 18, and 28 bp, respectively) between the binding sites for ADBs was designed to locate ADB-fused enzymes (HDH-ADB1, HK-ADB2, and TS-ADB3) every 20, 30, and 40 bp, respectively (Fig. 3). Analysis of l-threonine production by these scaffold plasmids (pSC-1, pSC-2, and pSC-3) showed that pSC-1, which contained the 8-bp spacer sequence among the tested scaffold plasmids, was associated with the most efficient l-threonine production (Fig. 4b). In addition, analysis of the l-threonine production rate using pSC-1a and pSC-1b with 3- and 13-bp spacer DNA sequences, respectively, between the binding sites for ADBs to locate HDH-ADB1, HK-ADB2, and TS-ADB3 every 15 and 25 bp, respectively, showed a decreased production rate compared to that of pSC-1 (see Fig. S3 in the supplemental material).
We next tested how the number of TS enzymes on the DNA scaffold affected the l-threonine production rate. In addition to single binding sites for HDH-ADB1 and HK-ADB2, scaffold plasmids pSC-4, pSC-5, and pSC-6 have 2, 3, and 4 binding sites for TS-ADB3, respectively, with 8-bp spacers between the sites, because the TS enzyme is responsible for the rate-limiting step (Fig. 3). An increased number of TS-ADB3 enzyme binding sites on the DNA scaffold resulted in an increased l-threonine production rate: the scaffold plasmid pSC-4, with 2 copies of the TS-ADB3 binding site, caused the highest production rate (>3-fold higher than that of pSC-1) (Fig. 4c). The scaffold plasmids with over 2 copies of the TS-ADB3 binding site showed a similar production rate (Fig. 4c). Thus, the reaction efficiency of a whole metabolic pathway might be controlled by varying the distance between the enzymes assembled on the DNA scaffold and the number of enzymes bound to the DNA scaffold.
Enhancing in vivo production of l-threonine with a DNA scaffold.
We next tested the effect of the DNA scaffold-based artificial enzymatic cascade on the in vivo production of l-threonine. Scaffold plasmid pSC-4, which caused the highest production rate in vitro, and the pET16tac-thrABC plasmid, which expressed three ADB-fused enzymes (HDH-ADB1, HK-ADB2, and TS-ADB3) under the control of a tac promoter, were transformed into the l-threonine-producing strain MG-105, to create strain MG-105ES (Table 1). Interestingly, time course analysis of l-threonine production during fermentation revealed that MG-105ES had already produced the maximum titer within 24 h of fermentation and that almost all glucose in the medium was consumed (Fig. 5), whereas for the strain without the DNA scaffold system, MG-105, it took about 48 h to produce the same maximum titer of l-threonine. Production of l-threonine by the MG-105E strain, which contained only the pET16tac-thrABC plasmid and not the scaffold plasmid pSC-4 (Table 1), was similar to that by MG-105. This result clearly shows that the increase in l-threonine production efficiency was due to the presence of the DNA scaffold in l-threonine-producing strain MG-105ES. In addition, the growth rate of the host strain was significantly improved by the introduction of the DNA scaffold system (Fig. 5). An analysis of the intracellular concentrations of the homoserine showed that the homoserine concentration in strain MG-105ES (318.3 ± 11.46 μM [mean ± standard deviation]) was 15-fold lower than that in strain MG-105 (4,730.7 ± 28.11 μM) after 24 h of fermentation. The homoserine concentration in strain MG-105E was 4,288.4 ± 42.85 μM. This result implies that our DNA scaffold system reduced the accumulation of the intermediate homoserine, which might inhibit the growth of the host cell (44), as well as improved the reaction efficiency by increasing the local concentration of intermediates.
Fig 5.

Improved production of l-threonine by using the DNA scaffold system in vivo. l-Threonine-producing strains (Table 1) were cultivated in a 1-liter fermentor containing 500 ml of fermentation medium 1 at 37°C (refer to Materials and Methods). During the 48-h fermentation, the concentrations of l-threonine and glucose in the culture broth were measured by HPLC (refer to Materials and Methods). The data shown are the means for three independent experiments.
DISCUSSION
For decades, the development of industrial host strains for l-threonine production has relied on the traditional metabolic engineering and systems biology approach, which focused on increases in metabolic flux through the l-threonine biosynthetic pathway by deleting the competing pathways, elimination of feedback inhibition in the biosynthetic pathway, amplification of the rate-limiting enzyme, and export of the l-threonine produced (1, 3, 4, 9, 11, 30, 45–49). The engineered host strains were able to produce l-threonine with a high yield close to a theoretical maximum (8, 50). The remaining engineering issues to be solved for the cost-effective production of l-threonine include imbalanced metabolic flux, formation of by-products, and accumulation of toxic intermediates (14).
Inspired by natural enzymatic complexes (e.g., fatty acid biosynthases, polyketide synthases, cellulosome complex, and signaling scaffolds) and engineered scaffolds (e.g., protein-, RNA-, and DNA-base scaffolds), which increase the production of target molecules or signal cascades by the enforced proximity and substrate channeling through assembly lines of proteins on a scaffold (14, 18, 25–27, 51–60), we devised a DNA scaffold strategy that facilitates the design and optimization of artificial enzymatic cascades. This approach enables us to increase the overall metabolic efficiency of the l-threonine biosynthetic pathway by colocalization of enzymes on a DNA scaffold in a programmable manner using an interaction between well-characterized ZFPs and their target DNA sequences. Compared with the protein/protein interactions used in natural enzymatic complexes or synthetic protein scaffolds, which are limited by the number of characterized domains and their ligands, the ADBs used as ligands in our DNA scaffold system bypass these limitations, because they are provided by the combinatorial modular assembly of ZFPs (31). Moreover, the affinity of the ADB for a DNA sequence can be controlled by the number of ZFPs used in its assembly (61–63).
Our DNA scaffold system has been successfully applied to the production of l-threonine in E. coli, reducing the production time by more than 50%. Additionally, it seems that our DNA scaffold system does not interfere with the structural characteristics of enzymes involved in the l-threonine production in E. coli. It is expected that because the homoserine dehydrogenase (HDH) and homoserine kinase (HK) enzymes used in this study consist of a homotetramer of HDH and homodimer of HK, respectively (33, 34, 43, 64), the fusion of ADBs to these enzymes may restrict the activities of artificial enzymatic cascades on the DNA scaffold. However, our in vitro assay clearly demonstrates that there was no significant change in the kinetic parameters of enzymes fused with the ADBs (Table 3).
The increased metabolic efficiency in l-threonine production in vivo as well as in vitro (Fig. 4 and 5) can be explained by the enforced proximity and substrate channeling effect facilitated by our DNA scaffold system through the following specific effects: (i) increasing the local concentration of intermediates around the enzymes on the DNA scaffold, (ii) preventing the loss of intermediates to diffusion or by competing reactions, and (iii) circumventing feedback inhibition on other pathways due to the rapid conversion of feedback inhibitors (14, 18, 27). Therefore, the production of l-threonine through multiple enzymatic reactions could be achieved as a single-step reaction by assembly lines on a DNA scaffold.
In our DNA scaffold system, the design of the DNA binding site sequences in the scaffold plasmid dictates the number of enzymes bound, the distance between enzymes, and the 3-dimensional distribution of enzymes. The helical structure of DNA repeats every 10 bp, turning 360° and covering 3.4 nm over that interval. Therefore, the addition of a 1-bp spacer between the ADB binding sites adds a 0.34-nm distance in a side view and a 36° turn in an axial view. We constructed our scaffold plasmids to position the ADB-enzyme fusions (HDH-ADB1, HK-ADB2, and TS-ADB3) every 20, 30, or 40 bp (Fig. 3). This design, based on a consideration of DNA structure, was selected so that all of the scaffold-bound enzymes would be heading in the same direction in 3-dimensional space and the distance between the enzymes would be proportional to the length of the spacer sequences between the ADB binding sites. As shown in our results, the distance between the binding sites for the enzymes that causes changes in the relative position of ADB-fused enzymes on the scaffold DNA, as well as the number of enzymes in the complex, is an important factor for the optimization of this metabolic pathway (Fig. 4; see Fig. S3 in the supplemental material).
In addition, our DNA scaffold system may prevent the undesired binding of ADBs to the host genome, because the ADBs, which recognize orthogonal 12-bp DNA sequences and have no specific binding sites in the host genome, were selected from more than 2 × 106 different possible combinations of ZFPs (31).
The in vivo assembly of enzymes on the scaffold plasmid into functional artificial enzymatic cascades may be affected by many factors, such as the scaffold plasmid copy number and supercoiling, the number of molecules and the functionality of the expressed enzymes, or the density and viscosity of the cytoplasmic space, which affect the diffusion rate. Thus, it is not realistic to assume that 100% of the scaffold plasmid inside the cells will be assembled with the desired enzymes. In vivo and in vitro binding of ZFP to a supercoiled plasmid DNA was previously reported by Woodgate et al. (65) and Desjarlais and Berg (66). In addition, Conrado et al. (27) proved that the ZFP domains bind specifically to their corresponding DNA target sites in vivo and thus are ideally suited for directing diverse cellular enzymes to specific sites on plasmid DNA. These reports suggest that artificial enzymatic cascades might be assembled on a supercoiled plasmid DNA in vivo to some extent, and previous studies showed that the actual assembly of functional protein scaffolds is only 10% (67, 68). Theoretical analysis revealed that assembly of only 10% of expressed enzymes into scaffolds still dramatically increases the reaction efficiency by enforced proximity (25, 69). Therefore, further controls on the expression levels of each enzyme and the copy number of the scaffold plasmid might increase the proportion of the scaffold that is fully assembled, which would result in a lower metabolic burden for the host cell by avoiding any unnecessary overexpression of enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Intelligent Synthetic Biology Center of Global Frontier Project (2011-0031955), funded by the Ministry of Education, Science and Technology, Republic of Korea, and a grant from the Next-Generation BioGreen 21 Program (SSAC; grant PJ008110), Rural Development Administration, Republic of Korea.
Footnotes
Published ahead of print 16 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02578-12.
REFERENCES
- 1. Ikeda M. 2003. Amino acid production processes. Adv. Biochem. Eng. Biotechnol. 79:1–35 [DOI] [PubMed] [Google Scholar]
- 2. Reeds PJ. 2000. Dispensable and indispensable amino acids for humans. J. Nutr. 130:1835S–1840S [DOI] [PubMed] [Google Scholar]
- 3. Guillouet S, Rodal AA, An G, Lessard PA, Sinskey AJ. 1999. Expression of the Escherichia coli catabolic threonine dehydratase in Corynebacterium glutamicum and its effect on isoleucine production. Appl. Environ. Microbiol. 65:3100–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guillouet S, Rodal AA, An GH, Gorret N, Lessard PA, Sinskey AJ. 2001. Metabolic redirection of carbon flow toward isoleucine by expressing a catabolic threonine dehydratase in a threonine-overproducing Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 57:667–673 [DOI] [PubMed] [Google Scholar]
- 5. Ikeda M, Katsumata R. 1992. Metabolic engineering to produce tyrosine or phenylalanine in a tryptophan-producing Corynebacterium glutamicum strain. Appl. Environ. Microbiol. 58:781–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikeda M, Nakanishi K, Kino K, Katsumata R. 1994. Fermentative production of tryptophan by a stable recombinant strain of Corynebacterium glutamicum with a modified serine-biosynthetic pathway. Biosci. Biotechnol. Biochem. 58:674–678 [DOI] [PubMed] [Google Scholar]
- 7. Ikeda M, Ozaki A, Katsumata R. 1993. Phenylalanine production by metabolically engineered Corynebacterium glutamicum with the pheA gene of Escherichia coli. Appl. Microbiol. Biotechnol. 39:318–323 [DOI] [PubMed] [Google Scholar]
- 8. Lee KH, Park JH, Kim TY, Kim HU, Lee SY. 2007. Systems metabolic engineering of Escherichia coli for l-threonine production. Mol. Syst. Biol. 3:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee SY, Park JH. 2010. Integration of systems biology with bioprocess engineering: l-threonine production by systems metabolic engineering of Escherichia coli. Adv. Biochem. Eng. Biotechnol. 120:1–19 [DOI] [PubMed] [Google Scholar]
- 10. Park JH, Jang YS, Lee JW, Lee SY. 2011. Escherichia coli W as a new platform strain for the enhanced production of l-valine by systems metabolic engineering. Biotechnol. Bioeng. 108:1140–1147 [DOI] [PubMed] [Google Scholar]
- 11. Park JH, Lee SY. 2010. Metabolic pathways and fermentative production of l-aspartate family amino acids. Biotechnol. J. 5:560–577 [DOI] [PubMed] [Google Scholar]
- 12. Tinoco I. 2002. Physical chemistry: principles and applications in biological sciences, 4th ed Prentice Hall, Upper Saddle River, NJ [Google Scholar]
- 13. Roe AJ, O'Byrne C, McLaggan D, Booth IR. 2002. Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiology 148:2215–2222 [DOI] [PubMed] [Google Scholar]
- 14. Conrado RJ, Varner JD, DeLisa MP. 2008. Engineering the spatial organization of metabolic enzymes: mimicking nature's synergy. Curr. Opin. Biotechnol. 19:492–499 [DOI] [PubMed] [Google Scholar]
- 15. Bashor CJ, Horwitz AA, Peisajovich SG, Lim WA. 2010. Rewiring cells: synthetic biology as a tool to interrogate the organizational principles of living systems. Annu. Rev. Biophys. 39:515–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blouzard JC, Coutinho PM, Fierobe HP, Henrissat B, Lignon S, Tardif C, Pages S, de Philip P. 2010. Modulation of cellulosome composition in Clostridium cellulolyticum: adaptation to the polysaccharide environment revealed by proteomic and carbohydrate-active enzyme analyses. Proteomics 10:541–554 [DOI] [PubMed] [Google Scholar]
- 17. Bomble YJ, Beckham GT, Matthews JF, Nimlos MR, Himmel ME, Crowley MF. 2011. Modeling the self-assembly of the cellulosome enzyme complex. J. Biol. Chem. 286:5614–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD. 2009. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 27:753–759 [DOI] [PubMed] [Google Scholar]
- 19. Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lim WA. 2010. Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 11:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitsuzawa S, Kagawa H, Li Y, Chan SL, Paavola CD, Trent JD. 2009. The rosettazyme: a synthetic cellulosome. J. Biotechnol. 143:139–144 [DOI] [PubMed] [Google Scholar]
- 22. Nordon RE, Craig SJ, Foong FC. 2009. Molecular engineering of the cellulosome complex for affinity and bioenergy applications. Biotechnol. Lett. 31:465–476 [DOI] [PubMed] [Google Scholar]
- 23. Peisajovich SG, Garbarino JE, Wei P, Lim WA. 2010. Rapid diversification of cell signaling phenotypes by modular domain recombination. Science 328:368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yaniv O, Shimon LJ, Bayer EA, Lamed R, Frolow F. 2011. Scaffoldin-borne family 3b carbohydrate-binding module from the cellulosome of Bacteroides cellulosolvens: structural diversity and significance of calcium for carbohydrate binding. Acta Crystallogr. D Biol. Crystallogr. 67:506–515 [DOI] [PubMed] [Google Scholar]
- 25. Zeke A, Lukacs M, Lim WA, Remenyi A. 2009. Scaffolds: interaction platforms for cellular signalling circuits. Trends Cell Biol. 19:364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. 2011. Organization of intracellular reactions with rationally designed RNA assemblies. Science 333:470–474 [DOI] [PubMed] [Google Scholar]
- 27. Conrado RJ, Wu GC, Boock JT, Xu H, Chen SY, Lebar T, Turnsek J, Tomsic N, Avbelj M, Gaber R, Koprivnjak T, Mori J, Glavnik V, Vovk I, Bencina M, Hodnik V, Anderluh G, Dueber JE, Jerala R, Delisa MP. 2012. DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res. 40:1879–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462 [DOI] [PubMed] [Google Scholar]
- 29. Shiio I, Nakamori S, Sano K. May 1971. Fermentation production of l-threonine. US patent 3,580,810
- 30. Lee JH, Sung BH, Kim MS, Blattner FR, Yoon BH, Kim JH, Kim SC. 2009. Metabolic engineering of a reduced-genome strain of Escherichia coli for l-threonine production. Microb. Cell Fact. 8:2 doi:10.1186/1475-2859-8-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JY, Sung BH, Yu BJ, Lee JH, Lee SH, Kim MS, Koob MD, Kim SC. 2008. Phenotypic engineering by reprogramming gene transcription using novel artificial transcription factors in Escherichia coli. Nucleic Acids Res. 36:e102 doi:10.1093/nar/gkn449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szczesiul M, Wampler DE. 1976. Regulation of a metabolic system in vitro: synthesis of threonine from aspartic acid. Biochemistry 15:2236–2244 [DOI] [PubMed] [Google Scholar]
- 33. Chassagnole C, Rais B, Quentin E, Fell DA, Mazat JP. 2001. An integrated study of threonine-pathway enzyme kinetics in Escherichia coli. Biochem. J. 356:415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wampler DE, Westhead EW. 1968. Two aspartokinases from Escherichia coli. Nature of the inhibition and molecular changes accompanying reversible inactivation. Biochemistry 7:1661–1671 [DOI] [PubMed] [Google Scholar]
- 35. Kyaw A, Maung UK, Toe T. 1985. Determination of inorganic phosphate with molybdate and Triton X-100 without reduction. Anal. Biochem. 145:230–234 [DOI] [PubMed] [Google Scholar]
- 36. Joseph MH, Marsden CA. 1986. Amino acids and small peptides, p 13–28 In Lim CK. (ed), HPLC of small molecules, a practical approach. IRL Press, Oxford, United Kingdom [Google Scholar]
- 37. Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, Yang HY, Lee DK, Lee Y, Park J, Kwon HS, Kim HW, Yeh BI, Lee HW, Sohn SH, Yoon J, Seol W, Kim JS. 2003. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat. Biotechnol. 21:275–280 [DOI] [PubMed] [Google Scholar]
- 38. Park KS, Lee DK, Lee H, Lee Y, Jang YS, Kim YH, Yang HY, Lee SI, Seol W, Kim JS. 2003. Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat. Biotechnol. 21:1208–1214 [DOI] [PubMed] [Google Scholar]
- 39. Dall'Acqua W, Simon AL, Mulkerrin MG, Carter P. 1998. Contribution of domain interface residues to the stability of antibody CH3 domain homodimers. Biochemistry 37:9266–9273 [DOI] [PubMed] [Google Scholar]
- 40. Neidhardt FC, Curtiss R. 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed ASM Press, Washington, DC [Google Scholar]
- 41. Shames SL, Ash DE, Wedler FC, Villafranca JJ. 1984. Interaction of aspartate and aspartate-derived antimetabolites with the enzymes of the threonine biosynthetic pathway of Escherichia coli. J. Biol. Chem. 259:15331–15339 [PubMed] [Google Scholar]
- 42. Shames SL, Wedler FC. 1984. Homoserine kinase of Escherichia coli: kinetic mechanism and inhibition by l-aspartate semialdehyde. Arch. Biochem. Biophys. 235:359–370 [DOI] [PubMed] [Google Scholar]
- 43. Wedler FC, Ley BW. 1993. Kinetic and regulatory mechanisms for (Escherichia coli) homoserine dehydrogenase-I. Equilibrium isotope exchange kinetics. J. Biol. Chem. 268:4880–4888 [PubMed] [Google Scholar]
- 44. Kotre AM, Sullivan SJ, Savageau MA. 1973. Metabolic regulation by homoserine in Escherichia coli B-r. J. Bacteriol. 116:663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishida M, Kawashima H, Sato K, Hashiguchi K, Ito H, Enei H, Nakamori S. 1994. Factors improving l-threonine production by a three l-threonine biosynthetic genes-amplified recombinant strain of Brevibacterium lactofermentum. Biosci. Biotechnol. Biochem. 58:768–770 [DOI] [PubMed] [Google Scholar]
- 46. Ishida M, Sato K, Hashiguchi K, Ito H, Enei H, Nakamori S. 1993. High fermentative production of l-threonine from acetate by a Brevibacterium flavum stabilized strain transformed with a recombinant plasmid carrying the Escherichia coli thr operon. Biosci. Biotechnol. Biochem. 57:1755–1756 [DOI] [PubMed] [Google Scholar]
- 47. Kruse D, Kramer R, Eggeling L, Rieping M, Pfefferle W, Tchieu JH, Chung YJ, Jr, Saier MH, Burkovski A. 2002. Influence of threonine exporters on threonine production in Escherichia coli. Appl. Microbiol. Biotechnol. 59:205–210 [DOI] [PubMed] [Google Scholar]
- 48. Park JH, Lee SY. 2010. Fermentative production of branched chain amino acids: a focus on metabolic engineering. Appl. Microbiol. Biotechnol. 85:491–506 [DOI] [PubMed] [Google Scholar]
- 49. Park JH, Lee SY. 2008. Towards systems metabolic engineering of microorganisms for amino acid production. Curr. Opin. Biotechnol. 19:454–460 [DOI] [PubMed] [Google Scholar]
- 50. Hong SH, Kim JS, Lee SY, In YH, Choi SS, Rih JK, Kim CH, Jeong H, Hur CG, Kim JJ. 2004. The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nat. Biotechnol. 22:1275–1281 [DOI] [PubMed] [Google Scholar]
- 51. Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. 2006. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75:655–680 [DOI] [PubMed] [Google Scholar]
- 52. Dueber JE, Yeh BJ, Chak K, Lim WA. 2003. Reprogramming control of an allosteric signaling switch through modular recombination. Science 301:1904–1908 [DOI] [PubMed] [Google Scholar]
- 53. Ferrell JE, Jr, Cimprich KA. 2003. Enforced proximity in the function of a famous scaffold. Mol. Cell 11:289–291 [DOI] [PubMed] [Google Scholar]
- 54. Fontes CM, Gilbert HJ. 2010. Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 79:655–681 [DOI] [PubMed] [Google Scholar]
- 55. Gilbert HJ. 2007. Cellulosomes: microbial nanomachines that display plasticity in quaternary structure. Mol. Microbiol. 63:1568–1576 [DOI] [PubMed] [Google Scholar]
- 56. Gulder TA, Freeman MF, Piel J. 1 March 2011. The catalytic diversity of multimodular polyketide synthases: natural product biosynthesis beyond textbook assembly rules. Top. Curr. Chem. [Epub ahead of print.] doi:10.1007/128_2010_113 [DOI] [PubMed] [Google Scholar]
- 57. Kwan DH, Schulz F. 2011. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules 16:6092–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lim WA. 2002. The modular logic of signaling proteins: building allosteric switches from simple binding domains. Curr. Opin. Struct. Biol. 12:61–68 [DOI] [PubMed] [Google Scholar]
- 59. Maier T, Leibundgut M, Boehringer D, Ban N. 2010. Structure and function of eukaryotic fatty acid synthases. Q. Rev. Biophys. 43:373–422 [DOI] [PubMed] [Google Scholar]
- 60. Peer A, Smith SP, Bayer EA, Lamed R, Borovok I. 2009. Noncellulosomal cohesin- and dockerin-like modules in the three domains of life. FEMS Microbiol. Lett. 291:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bhakta MS, Segal DJ. 2010. The generation of zinc finger proteins by modular assembly. Methods Mol. Biol. 649:3–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Imanishi M, Nakamura A, Morisaki T, Futaki S. 2009. Positive and negative cooperativity of modularly assembled zinc fingers. Biochem. Biophys. Res. Commun. 387:440–443 [DOI] [PubMed] [Google Scholar]
- 63. Jantz D, Berg JM. 2010. Probing the DNA-binding affinity and specificity of designed zinc finger proteins. Biophys. J. 98:852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Theze J, Kleidman L, St Girons I. 1974. Homoserine kinase from Escherichia coli K-12: properties, inhibition by l-threonine, and regulation of biosynthesis. J. Bacteriol. 118:577–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Woodgate J, Palfrey D, Nagel DA, Hine AV, Slater NK. 2002. Protein-mediated isolation of plasmid DNA by a zinc finger-glutathione S-transferase affinity linker. Biotechnol. Bioeng. 79:450–456 [DOI] [PubMed] [Google Scholar]
- 66. Desjarlais JR, Berg JM. 1993. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc. Natl. Acad. Sci. U. S. A. 90:2256–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PI, Knop M. 2007. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat. Cell Biol. 9:1319–1326 [DOI] [PubMed] [Google Scholar]
- 68. Slaughter BD, Schwartz JW, Li R. 2007. Mapping dynamic protein interactions in MAP kinase signaling using live-cell fluorescence fluctuation spectroscopy and imaging. Proc. Natl. Acad. Sci. U. S. A. 104:20320–20325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fischer H, Polikarpov I, Craievich AF. 2004. Average protein density is a molecular-weight-dependent function. Protein Sci. 13:2825–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.