

Abstract
We developed a high-throughput method based on terminal restriction fragment length polymorphisms (T-RFLP) to identify ospC genotypes from field-collected samples of Borrelia burgdorferi. We first validated the method by analyzing B. burgdorferi ospC previously identified by sequencing. We then analyzed and compared ospC genotypes detected from ear biopsy tissue from natural populations of the white-footed mouse, a major B. burgdorferi reservoir host species in the eastern United States, and larval ticks feeding on those individual mice. The T-RFLP method enabled us to distinguish all 17 ospC genotypes tested, as well as mixed samples containing more than one genotype. Analysis costs compare favorably to those of alternative ospC identification methods. The T-RFLP method will facilitate large-scale field studies to advance our understanding of genotype-specific transmission patterns.
INTRODUCTION
The causative agent of Lyme disease, Borrelia burgdorferi, is a spirochete that is transmitted among vertebrate hosts by the black-legged tick, Ixodes scapularis, and other ticks within the Ixodes ricinus species complex. Based on variation in the B. burgdorferi outer surface protein C (ospC) gene, 17 genotypes, or major groups, have been identified from the northeastern United States, designated A through O, T, and U (1), with a few additional genotypes occurring outside this region (2, 3). ospC is involved in establishing initial infection in mammalian hosts during tick bites (4, 5) and appears to have a role in protecting B. burgdorferi from the host's innate immune response (6). Although it is one of the most variable genes known for B. burgdorferi (7), within-genotype sequences are highly conserved throughout their distributions across the United States (2) and in Europe (3), with less than 2% sequence divergence within and more than 8% sequence divergence among genotypes (1). Some ospC genotypes show phenotypic differences in infecting host tissues, including human hosts (8–10), and ospC has been proposed to play a role similar to that of a “speciation gene” in the evolution of the bacterium (11).
Studies of this polymorphism rely on a set of molecular methods to identify ospC genotypes from field-sampled ticks and hosts. Previous studies have used any of three methods: single-strand conformation polymorphism (SSCP) (1, 12, 13), traditional Sanger sequencing (1, 3, 13, 14), and reverse line blotting (15, 16). SSCP precludes direct identification to the genotype level, because there are only 13 defined mobility classes for 17 genotypes. Traditional Sanger sequencing requires generating a clone library for resolving and identifying multiple genotypes present within a single sample, a common situation, as hosts and ticks are often multiply infected (14, 15). Reverse line blotting is capable of identifying multiple ospC genotypes within a sample but requires skilled technical experience to achieve consistent results, and the blotting apparatus is limited to running approximately 45 samples per day, due to both space and time constraints. Studies of the distribution of B. burgdorferi genotypes will benefit from the development of a relatively inexpensive and high-throughput identification method capable of characterizing mixed infections.
Here, we propose an alternative method to identify ospC genotypes of B. burgdorferi from field-collected samples. Terminal restriction fragment length polymorphism (T-RFLP) analysis has been used successfully to profile bacterial communities, typically analyzing 16S rRNA genes (17), but we adapted this method to the ospC gene instead of 16S rRNA genes. Because ospC sequences are well characterized and highly conserved within genotypes (1–3), they can be reliably distinguished from one another using this method. All steps can be performed in 96-well plates, permitting high-throughput processing. We also compare the costs of performing T-RFLP analysis to those of existing methods.
Ecological studies of B. burgdorferi that involve host sampling have sampled either from host ear tissue (12, 13, 18) or from ticks feeding on the hosts (14, 15). We use T-RFLP to analyze both of these sample types to demonstrate the applicability of this method for future studies. Peromyscus leucopus, the white-footed mouse, is a major reservoir host for B. burgdorferi in the northeastern United States (19). Individuals are often simultaneously infected with multiple ospC genotypes (12, 14, 15) and host numerous larval ticks (20, 21), which are uninfected prior to their first blood meal (22). P. leucopus is thus an ideal study subject for examining B. burgdorferi genotypes in both host tissues and ticks.
MATERIALS AND METHODS
T-RFLP begins with PCR amplification of ospC, using fluorescently labeled primers. The labeled PCR product is then digested with selected restriction enzymes, yielding fluorescent fragments in sizes unique to each genotype (Fig. 1). Fragment analysis is performed on a capillary sequencer, and the fluor color and fragment size (±0.1 bp) identify the ospC genotype(s) present in the sample. Because a single enzyme may cut at the same site for multiple genotypes, more than one enzyme is needed to identify certain genotypes. Multiple digestions are performed with different enzymes, each digesting the original-length PCR product to give a set of fragment lengths that characterize the genotype. In the interest of efficiency, we labeled the PCR products to be digested by different enzymes with different fluor colors. We combined all the fragments into a single sample for the fragment analysis.
Fig 1.
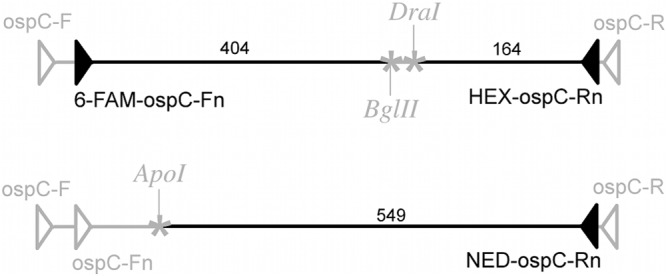
Schematic diagram of ospC PCR product digestion of ospC genotype A. Filled triangles, fluor-labeled primers; open triangles, unlabeled primers; asterisks, restriction enzyme cut sites; numbers, lengths (bp) of labeled fragments.
In silico enzyme selection.
From a reference collection of strains with known ospC genotypes and sequences, we analyzed B. burgdorferi ospC sequences in silico for genotypes occurring in the northeastern United States. We selected two of the most divergent strains within each genotype, based on the sequences of eight housekeeping loci (3). Sequences were input into an online interface similar to that at RestrictionMapper (http://www.restrictionmapper.org/) and virtually digested with all the enzymes. The resulting fragment lengths for each enzyme digest were compared to find the most variable restriction site locations among ospC genotypes. Three enzymes that produced combinations of fragments unique to, but consistent within, each genotype were chosen: BglII, DraI, and ApoI.
Because some fragment sizes are shared among genotypes with similar sequences, certain mixtures of multiple genotypes may create combinations in which the presence of additional genotypes is ambiguous (see Appendix). The in silico digests were examined to find an additional pair of enzymes that could resolve these ambiguities: ApeKI and Cac8I. Digesting ambiguous samples with these enzymes confirms the presence of any masked genotypes.
Amplification and digestion.
B. burgdorferi ospC was amplified using nested PCR in 25-μl volumes as described in a previous study (23), with the following thermocycling conditions: 95°C for 15 min; 35 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 30 s; and 72°C for 5 min (Fig. 1). The second round used the PCR products from the first round as a template and amplified them under the same conditions but with 40 cycles (23). The first PCR round used unlabeled outer primers, and the second round used inner primers labeled with fluors that enable detection by the automated sequencer (Table 1). Synthesized primers containing the fluors 6-carboxyfluorescein (6-FAM) and hexachlorofluorescein (HEX) are widely available from several reagent suppliers, and NED is a proprietary fluor available in primers from Applied Biosystems. The resulting fluorescent PCR products were digested with restriction enzymes as follows. Five microliters of PCR product labeled with 6-FAM and HEX was added to 2 μl of 10× NEBuffer 2, 10 units of DraI, 10 units of BglII, and water for a total volume of 20 μl (all digestion reagents were from New England BioLabs). Samples were incubated at 37°C for 1 h, followed by 65°C for 20 min to heat inactivate DraI. Five microliters of PCR product labeled with NED (Applied Biosystems) was added to 2 μl of 10× NEBuffer 3, 4 μl bovine serum albumin (BSA), 5 units of ApoI, and water for a total volume of 20 μl. Samples were incubated at 50°C for 1 h, followed by 80°C for 20 min to heat inactivate ApoI. For the ApeKI-Cac8I digestion, 5 μl of 6-FAM/HEX-labeled PCR product was added to 2 μl of 10× NEBuffer 3, 2 units of Cac8I, 2 units of ApeKI, and water for a total volume of 20 μl. Samples were incubated at 37°C for 1 h, followed by 75°C for 1 h. The digested products were stored at −20°C until they were ready for fragment analysis.
Table 1.
Primers used in ospC amplification and enzymes used to digest the resulting PCR products
| Primera | Fluor sequence (5′ to 3′) | Enzyme to digest PCR productb |
|---|---|---|
| First round | ||
| Outer-forward | ATGAAAAAGAATACATTAAGTGC | NA |
| Outer-reverse | ATTAATCTTATAATATTGATTTTAATTAAGG | |
| Second (nested) round | ||
| Inner-forward (6-FAM) | [6-FAM]-TATTAATGACTTTATTTTTATTTATATCT | DraI + BglII (Cac8I + ApeKI) |
| Inner-reverse (HEX) | [HEX]-TTGATTTTAATTAAGGTTTTTTTGG | |
| Inner-forward | TATTAATGACTTTATTTTTATTTATATCT | ApoI |
| Inner-reverse (NED) | [NED]-TTGATTTTAATTAAGGTTTTTTTGG | |
Second-round PCRs use primers containing the fluor 6-FAM, HEX, or NED, enabling detection by the automated sequencer.
Enzymes Cac8I and ApeKI are used only if samples contain ambiguous combinations (see Appendix). NA, not applicable.
Fragment analysis.
Both DraI-BglII and ApoI digests (0.5 μl each) were combined with 13.5 μl of Hi-Di formamide (Applied Biosystems). The DraI-BglII digests were added to the formamide first to deactivate BglII. ROX-labeled MapMarker size standard (0.5 μl) containing 50-, 100-, 150-, 200-, 250-, 300-, 350-, 400-, 450-, 500-, 550-, and 600-bp fragments (BioVentures) was added for a total volume of 15 μl, and the mixture was denatured for 5 min at 95°C. Fragment analysis was performed on an ABI 3730 capillary sequencer (Applied Biosystems). Peaks were analyzed using freely available PeakScanner software (Applied Biosystems) (Fig. 2). Code was written in the R statistical programming environment to make preliminary identifications of genotypes based on the fragment analysis data (available for download at http://www.kimtsao.com) (Fig. 2).
Fig 2.

Fragment analysis chromatograms. (Top) Mouse host tissue with genotypes A and G, with peaks at 163 (green), 401 and 407 (blue), and 557 (black) bp, respectively. (Bottom) Larval tick from the same mouse, with genotypes A and D, with peaks at 163, 407 and 455, and 557 and 560 bp. Fragments smaller than 100 bp were disregarded as primer peaks.
Validation of known samples and mixed samples.
We analyzed samples from our reference collection of known ospC sequences (Table 2) according to this protocol and compared the observed fragments to those predicted from the in silico analysis.
Table 2.
Fragment sizes from in silico and experimental analyses
| ospC genotype | IGSa | Sequence type(s) (MLST)b | Fragment sizec |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DraI + BglII, 6-FAM (5′) |
DraI + BglII, HEX (3′) |
ApoI, NED (3′) |
ApeKI + Cac8I, 6-FAM (5′) |
ApeKI + Cac8I, HEX (3′) |
||||||||
| Obs | Exp | Obs | Exp | Obs | Exp | Obs | Exp | Obs | Exp | |||
| A | 1A | 1 | 406.1 | 404 | 163.4 | 164 | 557.2 | 549 | 194.0 | 193 | 66.4 | 70 |
| B | 3A | 7, 59 | 333.7 | 334 | 166.6 | 167 | 560.3 | 552 | 189.9 | 191 | 67.3 | 70 |
| C | 5 | 11 | 332.9 | 334 | 163.7 | 164 | 246.1 | 242 | 481.2 | 484 | 84.7 | 86 |
| D | 5 | 38, 226, 268 | 456.4 | 457 | 163.6 | 164 | 560.3 | 552 | 518.3 | 522 | 83.3 | 86 |
| E | 9 | 19, 266 | 449.0 | 451 | 109.3 | 112 | 246.4 | 242 | 480.4 | 481 | 84.7 | 86 |
| F | 4A | 8 | 449.9 | 451 | 119.2 | 125 | 299.7 | 294 | 413.9 | 415 | 83.4 | 86 |
| G | 6B, 5 | 34, 14, 50 | 401.2 | 398 | 163.7 | 164 | 557.7 | 549 | 346.2 | 349 | 75.0 | 77 |
| H | 2D | 4 | 451.4 | 454 | 119.0 | 125 | 557.5 | 549 | 170.7 | 173 | 240.3 | 237 |
| I | 7A | 16 | 406.3 | 404 | 163.8 | 164 | 246.3 | 242 | 189.0 | 191 | 83.2 | 86 |
| J | 1, 5 | Unknown, 34 | 455.9 | 457 | 163.2 | 164 | 247.8 | 242 | 170.8 | 173 | 355.6 | 352 |
| K | 2A | 3, 269 | 508.1 | 509 | 109.6 | 112 | 298.3 | 294 | 170.8 | 173 | 67.5 | 70 |
| L | 2D, D | 29, 267 | 452.2 | 454 | 163.6 | 164 | 246.9 | 242 | 189.8 | 191 | 67.3 | 70 |
| M | 6A | 12 | 230.3 | 230 | 163.1 | 164 | 305.0 | 300 | 170.8 | 173 | 84.7 | 86 |
| N | 4A | 36 | 509.4 | 509 | 110.6 | 112 | 304.9 | 300 | 189.2 | 191 | 84.8 | 86 |
| O | 6C | 54 | 449.9 | 451 | 163.9 | 164 | 555.0 | 546 | 189.3 | 191 | 97.3 | 99 |
| T | 8C | 37 | 461.3 | 463 | 163.9 | 164 | 567.4 | 558 | 170.9 | 173 | 84.5 | 85 |
| U | 8A | 18 | 461.0 | 463 | 109.6 | 112 | 246.9 | 242 | 170.4 | 173 | 65.4 | 70 |
IGS, intergenic spacer.
MLST, multilocus sequence typing.
Fragment sizes (bp) observed (Obs) and expected (Exp) with the indicated enzymes and fluors.
Because host and tick samples often contain multiple strains with different ospC genotypes, we mixed digested samples in various quantities to see if all the genotypes present would be detectable. Mixtures were selected so that multiple genotypes would produce the same bands, potentially producing peaks larger than those produced by a single genotype. The genotype combinations tested (for DraI, BglII, and ApoI digests only) were as follows: A, B, C, D, and E with and without G; F, G, H, and I with and without L; U, N, and F with and without K and E; G, I, and T with and without A; H, J, and M with and without L; J, K, L, and M with and without N; N, O, T, and U with and without E; and F, J, and L. Quantitative PCR was performed on each of the reference samples to look for correlations between DNA quantity and peak height, width, or area.
Cost comparison of methods.
We compared the analysis costs of processing 3,000 samples using sequencing, reverse line blotting, and T-RFLP and also varied the number of samples between 1 and 10,000. Because SSCP cannot identify all 17 unique genotypes, it was not included in the cost comparison analysis. We examined only postextraction processing steps, because DNA extraction and identification of positive samples by PCR are the same among these analyses.
PCR.
Nested PCRs are performed as described above for all methods, except that T-RFLP uses fluorescent primers for the second, nested, round, for which additional costs are reflected. Other reactions include the following: for reverse line blotting, equipment and reagents for membrane preparation and processing of X-ray images; for T-RFLP, materials for enzyme digestion and fragment analysis. Run costs reflect equipment costs incurred in running the analysis. For sequencing and T-RFLP, estimates were taken from the Yale DNA Sequencing Facility's price list for sequencing and fragment analysis, respectively. For reverse line blotting, the equipment purchase cost was divided over the total number of samples run. Pricing quotes are for customers based in the United States. Labor for all methods accounts for the amount of time required for post-PCR processing and was estimated at $15 an hour. In an 8-h day, we estimated that three 96-well plates could be processed for sequencing and two plates for T-RFLP. Reverse line blotting is limited to approximately 45 samples per day due to time and space constraints on the blotting equipment.
Tissue and tick sample comparison.
We trapped P. leucopus mice from natural populations in northwestern Connecticut from June to October 2008. Each captured mouse was identified with a uniquely numbered ear tag. At initial capture and subsequent recaptures of a mouse, an ear punch biopsy specimen was taken (ca. 25 mg), all visible attached ticks were collected, and the animal was released. We analyzed only individual mice captured in four or more trapping sessions and their attached larval-stage ticks in order to have infection profiles as complete as possible for those individuals. Each tissue sample and tick was analyzed individually. Whole genomic DNA was extracted from ear tissue and larval ticks using NucleoSpin tissue extraction kits (Macherey-Nagel Inc., Bethlehem, PA). T-RFLP analysis was performed as described above.
RESULTS
T-RFLP validation.
Our in silico analyses predicted that each ospC type would produce a unique combination of fluorescently labeled fragments of different sizes, and these predictions were supported by the fragment analysis we conducted on strains with known sequences (Table 2). The differences between the observed fragments from known sequences and the expected sizes are consistent with previous demonstrations that fragment migration rates, and thus the sizes determined by fragment analysis, vary according to the sequence content and fluor (24). Fragments of the same sizes labeled with the same fluors are consistent within 1 bp, and fragments from different samples of the same genotype (and therefore containing very similar sequences) are consistent within 0.1 bp. Thus, these called sizes can be used as the reference sizes and do not compromise our ability to identify ospC samples to the genotype level.
Individual ospC genotypes are identifiable from mixtures of multiple genotypes using this method. While all of the expected bands in each mixed infection were present, quantifiable measures of peak signal strength (area, height, or width) were not reliable indicators of copy numbers (due to multiple genotypes producing the same size fragment), volume of the sample, or quantity of DNA. R2 values for these correlations ranged from 0.01 to 0.05, with P values from 0.08 to 0.4. While mixed samples are identifiable by this method, if the sample contains potentially ambiguous mixtures (see Appendix), the ApeKI and Cac8I enzyme combination is needed to definitively identify the genotypes in the mixture.
Cost analysis.
Overall per-sample analysis costs came to $8.50 for sequencing, $7.00 for reverse line blotting, and $6.00 for T-RFLP (Table 3) at the level of 3,000 samples. Cost differences are largely attributable to reagent and equipment costs for sequencing and equipment and labor for reverse line blotting. However, because reverse line blotting requires an initial equipment investment for the blotter, UV cross-linker, and X-ray processor, the first 2,000 samples analyzed by this method are more expensive than either sequencing or T-RFLP would be (Fig. 3). As the number of samples processed increases, the reverse line blotting cost per sample decreases. Reverse line blotting per-sample costs fall below those of T-RFLP if more than 5,500 samples are processed. Despite their higher cost, the lengths of time required to perform T-RFLP and sequencing are shorter, facilitated by automated sequencing machines.
Table 3.
Per-sample cost comparison of ospC identification methods for processing 3,000 samples
| Step or item | Cost/sample (U.S. dollars) |
Source or details | ||
|---|---|---|---|---|
| Sequencing | RLBa | T-RFLP | ||
| PCR | ||||
| 1st round | 1.06 | 1.06 | 1.06 | Qiagen HotStart MasterMix |
| 2nd (nested) round | 0.95 | 0.95 | 1.28 | Bioline Immomix, Applied Biosystems FAM/HEX primers |
| 2nd round (additional primers) | 1.21 | Bioline Immomix, Applied Biosystems NED primer | ||
| Other | ||||
| Oligonucleotidess to bind (19) | <0.01 | Invitrogen | ||
| 20× SSCb | 0.01 | Promega | ||
| SDS 10% | 0.01 | Promega | ||
| 1 M Tris HCl | 0.01 | Promega | ||
| 0.5 M EDTA | <0.01 | Promega | ||
| Maleic acid buffer | 0.01 | Sigma Aldrich | ||
| DIG luminescence detection kit | 0.29 | Roche | ||
| 96-well plates | 0.08 | 0.23 | USA Scientific | |
| Enzymes, buffers | 0.70 | New England Biolabs | ||
| Size standard | 0.23 | BioVentures | ||
| Formamide | 0.02 | Applied Biosystems | ||
| Run costs | ||||
| PCR purification, cycle seq | 6.00 | DNA Analysis Facility at Yale University (external price) | ||
| MiniSlot30 | 0.36 | Immunetics | ||
| UV cross-linker | 0.53 | VWR Scientific | ||
| X-ray processor | 1.13 | Konica Minolta | ||
| Fragment analysis | 0.75 | DNA Analysis Facility at Yale University (external price) | ||
| Labor | 0.42 | 2.67 | 0.63 | |
| Total cost/sample | 8.51 | 7.02 | 6.10 | |
RLB, reverse line blotting.
SSC, 1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate.
Fig 3.
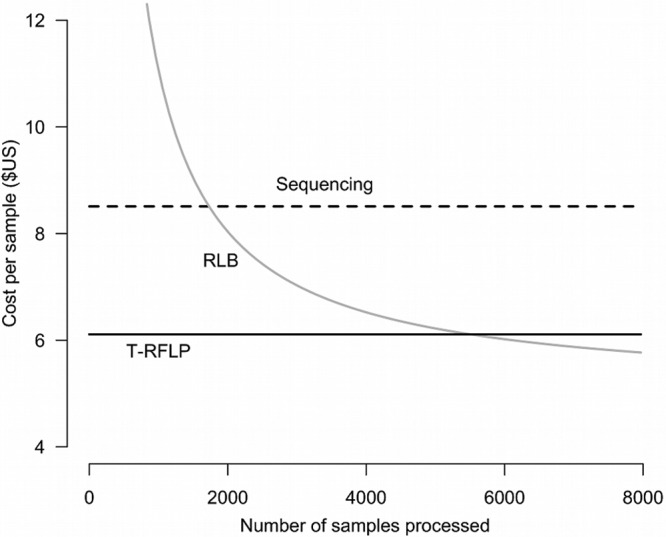
Cost comparison of ospC identification using sequencing, reverse line blotting, and T-RFLP. The per-sample costs for reverse line blotting are equal to those of sequencing at 1,700 samples and match those of T-RFLP at 5,500 samples.
Sample type comparison.
We analyzed ear tissues from 26 individual mice and 615 larval ticks collected from the mice. We amplified B. burgdorferi ospC from 28 (36%) of 77 mouse ear tissue samples and from 180 (29%) of the 615 larval ticks and were able to identify 200 of these 208 ospC-positive samples to the genotype level. The remaining eight samples (all ticks) were unidentifiable due to weak or noisy signals. Five tick samples required further resolution with the Cac8I-ApeKI enzyme combination. Ten of the 26 mice did not produce any positive ticks or ear tissues.
Analyzing only one sample type or the other gave different perspectives of the B. burgdorferi community. Considering only ear biopsy samples, the average infected mouse harbored 1.8 ospC genotypes over the course of the transmission season (standard deviation, 1.6). Considering only tick data from these mice increases the average to 2.7 genotypes per mouse (standard deviation, 2.0). Combining these data, using both sample types to determine the genotypes present in each mouse, yields an average of 3 genotypes per mouse (standard deviation, 2.0). Part of these detection differences may be attributable to specific genotypes; we did not detect ospC genotype D in any mouse ear tissue but detected it in 22 larval ticks removed from four individual mice.
DISCUSSION
T-RFLP correctly identified B. burgdorferi samples with known ospC sequences and is a viable method for identifying ospC genotypes present within unknown samples. Furthermore, like reverse line blotting, this method can identify multiple genotypes within a single sample, usually without requiring additional steps. Certain combinations of genotypes may be ambiguous in that their combinations of fragments may mask the presence of other genotypes. However, these cases were in the minority in our field-collected samples and were resolved simply by reanalyzing the PCR products with another pair of enzymes. T-RFLP combines the benefits of identifying multiple genotypes within a sample with the speed and processing ease of automated 96-well plate handling. Unlike reverse line blotting, no investment in specialized equipment is required, and compared to sequencing, data on fragment sizes are faster and less expensive to generate than nucleotide identities.
While the costs of reverse line blotting are estimated to drop below those of T-RFLP if more than 5,500 samples are processed, this assumes the equipment life span is effectively indefinite. Furthermore, while the per-hour cost was considered in the cost analysis, the total time required to complete sample processing was not. At 45 samples per day compared to 288 (samples from three 96-well plates), it would take six times as long to process the same number of samples with reverse line blotting as with T-RFLP. This time lag increases with sample numbers. Although the actual costs vary by geographic region, equipment and reagent suppliers, quantity discounts, and technical expertise, these estimates are intended to provide a starting point for calculations, and the overall relative costs among methods will likely be similar to what we have described.
Quantifying peak data did not provide additional information about relative genotype abundance within samples. This could be due to amplification bias by primer sets, inconsistent enzymatic activity, or pipetting error, although repeated efforts yielded a similar lack of correlations. Our test samples are a “worst case scenario,” deliberately constructed to yield ambiguous peaks. In real-world samples with small numbers of genotypes, it may be possible to achieve more quantitative results with this method. Other methods should also be explored to estimate quantities of genotypes relative to one another to provide additional information about within-host and vector genotype interaction and transmission.
The ospC genotypes identified in our mouse and tick samples are generally consistent in identity and relative prevalence with previous findings for this geographic region (14, 15). Previous studies also reported slightly lower detection of B. burgdorferi from ear biopsy tissue than from feeding larval ticks (25). Our findings suggest these differences may be partly attributable to genotype identity. A previous study did report a few (3/127) genotype D-positive ear biopsy tissue samples (13), yet genotype D is fairly common among feeding larval ticks.
These findings were surprising; contrary to hosts acting as a filter for the transmission of particular genotypes, we detected more genotypes transmitted to feeding ticks than in host tissue. Determining the mechanism for this outcome is unfortunately beyond the scope of the data presented here, and many more details of genotype- and species-specific transmission of B. burgdorferi are yet to be described. However, T-RFLP will make it easier to perform larger-scale studies capable of answering these and many other questions.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation under grant no. 1110472.
We are grateful to L. Rollend for laboratory assistance and A. D. Paltiel for comments on the cost comparison analysis. S. Hogan and C. Tackett provided assistance in the field and laboratory. L. Doss, L. Fortin, S. Heth, S. Childs, and J. Bronson of the Great Mountain Forest Corporation and the Audubon Center at Bent of the River graciously provided access to field sites.
APPENDIX
Table A1 provides a list of potentially ambiguous genotype combinations using only DraI, BglII, and ApoI enzymes. Samples containing these combinations can be resolved using enzymes Cac8I and ApeKI.
Table A1.
Potentially ambiguous genotype combinations using only DraI, BglII, and ApoI enzymes
| Genotypes contained in sample | Genotype(s) possibly concealed |
|---|---|
| AC | I |
| AE | I |
| AJ | I |
| AL | I |
| AU | I |
| BI | C |
| BJ | CD |
| BL | C |
| CD | J |
| CH | L |
| DE | J |
| DI | J |
| DL | J |
| DU | J |
| ET | U |
| FN | K |
| FU | E |
| GI | A |
| HI | AL |
| HJ | L |
| KM | N |
| OU | E |
| ABE | C |
| ABU | C |
| AEH | L |
| AFL | H |
| AHU | L |
| BDE | C |
| BDU | C |
| BEG | C |
| BEM | C |
| BEO | C |
| BET | C |
| BGU | C |
| BMU | C |
| BOU | C |
| BTU | C |
| CFK | E |
| CFN | E |
| CKO | E |
| CKT | U |
| CNO | E |
| CNT | U |
| DEH | L |
| DHU | L |
| EGH | L |
| EHK | F |
| EHM | L |
| EHO | L |
| EHT | L |
| FGL | H |
| FIK | E |
| FIN | E |
| FJK | E |
| FJN | E |
| FKL | E |
| FLN | E |
| GHU | L |
| HKO | F |
| HMU | L |
| HOU | L |
| HTU | L |
| IKO | E |
| IKT | U |
| INO | E |
| INT | U |
| JKO | E |
| JKT | U |
| JNO | E |
| JNT | U |
| KLO | E |
| KLT | U |
| LNO | E |
| LNT | U |
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. Wang IN, Dykhuizen DE, Qiu W, Dunn JJ, Bosler EM, Luft BJ. 1999. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics 151:15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barbour AG, Travinsky B. 2010. Evolution and distribution of the ospC gene, a transferable serotype determinant of Borrelia burgdorferi. mBio 1:e00153-10 doi:10.1128/mBio.00153-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Margos G, Gatewood AG, Aanensen DM, Hanincová K, Terekhova D, Vollmer SA, Cornet M, Piesman J, Donaghy M, Bormane A. 2008. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A. 105:8730–8735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilbert MA, Morton EA, Bundle SF, Samuels DS. 2007. Artificial regulation of ospC expression in Borrelia burgdorferi. Mol. Microbiol. 63:1259–1273 [DOI] [PubMed] [Google Scholar]
- 5. Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, VanRaden MJ, Stewart P. 2006. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect. Immun. 74:3554–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tilly K, Bestor A, Jewett MW, Rosa P. 2007. Rapid clearance of Lyme disease spirochetes lacking OspC from skin. Infect. Immun. 75:1517–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qiu WG, Schutzer SE, Bruno JF, Attie O, Xu Y, Dunn JJ, Fraser CM, Casjens SR, Luft BJ. 2004. Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proc. Natl. Acad. Sci. U. S. A. 101:14150–14155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dykhuizen DE, Brisson D, Sandigursky S, Wormser GP, Nowakowski J, Nadelman RB, Schwartz I. 2008. The propensity of different Borrelia burgdorferi sensu stricto genotypes to cause disseminated infections in humans. Am. J. Trop. Med. Hyg. 78:806–810 [PMC free article] [PubMed] [Google Scholar]
- 9. Seinost G, Dykhuizen DE, Dattwyler RJ, Golde WT, Dunn JJ, Wang IN, Wormser GP, Schriefer ME, Luft BJ. 1999. Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect. Immun. 67:3518–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wormser GP, Brisson D, Liveris D, Hanincová K, Sandigursky S, Nowakowski J, Nadelman RB, Ludin S, Schwartz I. 2008. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J. Infect. Dis. 198:1358–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Attie O, Bruno JF, Xu Y, Qiu D, Luft BJ, Qiu WG. 2007. Co-evolution of the outer surface protein C gene (ospC) and intraspecific lineages of Borrelia burgdorferi sensu stricto in the northeastern United States. Infect. Genet. Evol. 7:1–12 [DOI] [PubMed] [Google Scholar]
- 12. Swanson KI, Norris DE. 2008. Presence of multiple variants of Borrelia burgdorferi in the natural reservoir Peromyscus leucopus throughout a transmission season. Vector Borne Zoonotic Dis. 8:397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anderson JM, Norris DE. 2006. Genetic diversity of Borrelia burgdorferi sensu stricto in Peromyscus leucopus, the primary reservoir of Lyme disease in a region of endemicity in southern Maryland. Appl. Environ. Microbiol. 72:5331–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanincova K, Kurtenbach K, Diuk-Wasser M, Brei B, Fish D. 2006. Epidemic spread of Lyme borreliosis, northeastern United States. Emerg. Infect. Dis. 12:604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brisson D, Dykhuizen DE. 2004. ospC Diversity in Borrelia burgdorferi: different hosts are different niches. Genetics 168:713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qiu WG, Dykhuizen DE, Acosta MS, Luft BJ. 2002. Geographic uniformity of the Lyme disease spirochete (Borrelia burgdorferi) and its shared history with tick vector (Ixodes scapularis) in the northeastern United States. Genetics 160:833–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu WT, Marsh TL, Cheng H, Forney LJ. 1997. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hofmeister EK, Ellis BA, Glass GE, Childs JE. 1999. Longitudinal study of infection with Borrelia burgdorferi in a population of Peromyscus leucopus at a Lyme disease-enzootic site in Maryland. Am. J. Trop. Med. Hyg. 60:598–609 [DOI] [PubMed] [Google Scholar]
- 19. Tsao JI, Wootton JT, Bunikis J, Luna MG, Fish D, Barbour AG. 2004. An ecological approach to preventing human infection: vaccinating wild mouse reservoirs intervenes in the Lyme disease cycle. Proc. Natl. Acad. Sci. U. S. A. 101:18159–18164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brunner JL, Ostfeld RS. 2008. Multiple causes of variable tick burdens on small-mammal hosts. Ecology 89:2259–2272 [DOI] [PubMed] [Google Scholar]
- 21. Goodwin BJ, Ostfeld RS, Schauber EM. 2001. Spatiotemporal variation in a lyme disease host and vector: black-legged ticks on white-footed mice. Vector Borne Zoonotic Dis. 1:129–138 [DOI] [PubMed] [Google Scholar]
- 22. Patrican LA. 1997. Absence of Lyme disease spirochetes in larval progeny of naturally infected Ixodes scapularis (Acari: Ixodidae) fed on dogs. J. Med. Entomol. 34:52–55 [DOI] [PubMed] [Google Scholar]
- 23. Bunikis J, Garpmo U, Tsao J, Berglund J, Fish D, Barbour AG. 2004. Sequence typing reveals extensive strain diversity of the Lyme borreliosis agents Borrelia burgdorferi in North America and Borrelia afzelii in Europe. Microbiology 150:1741–1755 [DOI] [PubMed] [Google Scholar]
- 24. Kaplan CW, Kitts CL. 2003. Variation between observed and true terminal restriction fragment length is dependent on true TRF length and purine content. J. Microbiol. Methods 54:121–125 [DOI] [PubMed] [Google Scholar]
- 25. Sinsky RJ, Piesman J. 1989. Ear punch biopsy method for detection and isolation of Borrelia burgdorferi from rodents. J. Clin. Microbiol. 27:1723–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]