

Abstract
A natural Spodoptera exigua multiple nucleopolyhedrovirus (SeMNPV) isolate from Florida shares a strikingly similar genotypic composition to that of a natural Spodoptera frugiperda MNPV (SfMNPV) isolate from Nicaragua. Both isolates comprise a high proportion of large-deletion genotypes that lack genes that are essential for viral replication or transmission. To determine the likely origins of such genotypically similar population structures, we performed genomic and functional analyses of these genotypes. The homology of nucleotides in the deleted regions was as high as 79%, similar to those of other colinear genomic regions, although some SfMNPV genes were not present in SeMNPV. In addition, no potential consensus sequences were shared between the deletion flanking sequences. These results indicate an evolutionary mechanism that independently generates and sustains deletion mutants within each virus population. Functional analyses using different proportions of complete and deletion genotypes were performed with the two viruses in mixtures of occlusion bodies (OBs) or co-occluded virions. Ratios greater than 3:1 of complete/deletion genotypes resulted in reduced pathogenicity (expressed as median lethal dose), but there were no significant changes in the speed of kill. In contrast, OB yields increased only in the 1:1 mixture. The three phenotypic traits analyzed provide a broader picture of the functional significance of the most extensively deleted SeMNPV genotype and contribute toward the elucidation of the role of such mutants in baculovirus populations.
INTRODUCTION
The fall armyworm, Spodoptera frugiperda (J. E. Smith) (Lepidoptera: Noctuidae), and the beet armyworm, Spodoptera exigua (Hübner) (Lepidoptera: Noctuidae), are polyphagous pests of agricultural crops. S. frugiperda is native to the tropical and subtropical regions of the Americas, from the United States to Argentina (1). S. frugiperda adults disperse over long distances during the summer months. S. exigua originated in the Middle East, but it is now found in tropical and subtropical areas worldwide, including Europe and the Americas (2). The geographical distributions of S. exigua and S. frugiperda have overlapped in North America for more than a century (3).
The multiple nucleopolyhedroviruses (genus Alphabaculovirus, family Baculoviridae) isolated from S. exigua multiple nucleopolyhedrovirus (SeMNPV) and S. frugiperda multiple nucleopolyhedrovirus (SfMNPV) are important species-specific mortality factors, particularly in the high-density populations of their respective hosts. Alphabaculoviruses, including SeMNPV, have proven to be effective for use as a base in biological insecticides (4). A detailed understanding of the genetic and phenotypic diversity within and between baculovirus populations can facilitate the selection of highly insecticidal strains for development as a base of commercial products (5).
The diversity of SfMNPV and SeMNPV has been described both between (6–8) and within (9–13) populations. The genomes of SfMNPV and SeMNPV are strikingly similar (14, 15), with 72 to 85% nucleotide homology in colinear regions covering 30% of the genome, and with >85% of genes being shared; this has led researchers to suggest that these two viruses are closely related (16, 17). The large-deletion genotypes present in a Floridan SeMNPV isolate, SeMNPV-US2 (SeUS2), were observed to share parallel genetic and phenotypic characteristics with an SfMNPV isolate from Nicaragua, SfMNPV-NIC (SfNIC) (18). SeMNPV and SfMNPV represent different virus species (19). The genotypic structure was detected later in other geographically distant populations of these viruses. Specifically, a Californian SeMNPV isolate (SeUS1) includes three genotypes with deletions in similar locations and of comparable lengths to those of SeUS2, namely, SeUS1-B (K. Zimmermann, D. Muñoz, and P. Caballero, unpublished data), SeUS1-XD1 (9), and a 25-kb deletion genotype (20) that we designated here as SeUS1-JH (Fig. 1). Five samples from Japan and Thailand show restriction endonuclease profiles (10) with signs of deletion genotypes, characterized by submolar PstI-C and PstI-D fragments. Finally, in SfMNPV isolates from the United States (17) and Colombia (G. Barrera, O. Simón, and P. Caballero, unpublished data), the presence of genotypes with sizable genomic deletions in the same genomic region has been reported recently. The host ranges of these viruses differ. SeMNPV is not pathogenic for S. frugiperda or any other heterologous host, whereas SfMNPV, although it is lethal to S. exigua, produces atypical pathogenesis, lack of integument rupture, and no progeny virus in this (S. exigua) host. The existence of such similar population structures in distinct viruses that infect different host species raises questions concerning the origins and functions of these genotypes.
Fig 1.

Gene content of SeMNPV and SfMNPV deletion genotypes. (A) Schematic representation of the SeMNPV-US1A (SeUS1-A) genome (above). The PstI restriction sites and fragments are represented right below the genome line. The shaded rectangles right above the genome line correspond to origins of replication within homologous regions (hr) or not (non-hr), and numbers correspond to nucleotide position in kb. ORFs (below) are represented as arrowed bars, where size and arrowhead direction indicate their respective length and transcription direction; the colors are described in the key in panel B. The numbers below each ORF indicate its number. (B) Linear maps (thick horizontal lines) and corresponding ORFs (colored arrowed bars) of the SeMNPV (Se, above) and SfMNPV (Sf, below) auxiliary gene-rich (agr) regions. The horizontal dashed arrows represent the areas of deletion in SeUS2-C, SeUS2-E, SeUS2-F, SeUS1-JH, SeUS1-XD1, and the SfNIC deletion variants. The lengths of the SeUS2-C, SeUS2-E, and SeUS2-F deletions are indicated in bp. The approximate deletion points of the SeUS1 genotypes SeUS1-B (left breakpoint) and SeUS1-JH have also been included. The SeUS2-D insertion point is also represented as a vertical dotted arrow and the insertion as a solid line. The homolog ORFs between SeMNPV and SfMNPV are connected by dotted lines.
In this study, we analyzed the gene content of SeUS2 genotypic variants and compared the genotypic structures of SeUS2, SeUS1, and SfNIC in an attempt to determine the origins of such genotypically similar population structures that would provide clues as to the ecological or evolutionary factors that determine the population structures in these viruses. In doing so, we also identify a series of characteristics that can be employed as genotype selection criteria for the development of biological insecticides that are based on these viruses.
MATERIALS AND METHODS
Insects, cell lines, and viruses.
S. exigua larvae were obtained from a laboratory colony, maintained under constant environmental conditions (25 ± 1°C, 50% ± 5% relative humidity, and a photoperiod of 16 h light and 8 h dark), and reared on a wheat germ-based semisynthetic diet (21). This colony was known to be free from latent or covert nucleopolyhedrovirus infections (22). S. exigua Se301 cells, which were kindly provided by S. Herrero (Universidad de Valencia, Spain), were maintained at 28 ± 0.5°C in Grace's insect cell culture medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Scotland, United Kingdom). The SeMNPV isolate from Florida, SeUS2-wild type (WT), which was used in this study, is the active ingredient of the bioinsecticide Spod-X and was kindly provided by DuPont Ibérica S.A, Barcelona, Spain. The virus was amplified by infecting S. exigua fourth instars from the laboratory colony using the droplet feeding method (23).
Initially, the SeUS2 genotypic variants SeUS2-A, SeUS2-C, SeUS2-D, SeUS2-E, and SeUS2-F were obtained by in vivo cloning in S. exigua larvae (12). SeUS2-E could not be fully isolated, but several samples contained enriched mixtures of this genotype with SeUS2-A as a contaminant, and so these were used to construct the physical map of SeUS2-E (12). Viral occlusion bodies (OBs) were produced by feeding healthy fourth instars with a virus-contaminated diet. OBs were extracted from dead diseased larvae using homogenization in water, and they were purified by filtration and differential centrifugation (12).
Viral DNA purification, restriction endonuclease digestion, cloning, and PCR amplification.
Viral DNA was extracted from ∼109 OBs in 300 μl of water by dissolving the polyhedrin matrix with the addition of 100 μl of 0.5 M Na2CO3 and 50 μl of 10% (wt/vol) SDS at 60°C for 10 min. Undissolved OBs and other particulates were pelleted by low-speed centrifugation (2,700 × g for 5 min). Supernatant containing occlusion-derived virions (ODVs) was recovered and incubated with 500 μg/ml proteinase K at 50°C for 2.5 h. Viral genomic DNA was extracted with phenol and chloroform, precipitated by the addition of 0.1 volume 3 M sodium acetate (pH 5.2) and 2.5 volumes 96% ethanol, washed with 70% ethanol, and dissolved in 0.1× Tris-EDTA (TE) buffer (10 mM Tris-acetate and 1 mM EDTA [pH 8.0]). For restriction endonuclease digestion, ∼2 μg of viral genomic DNA was treated with 10 U of PstI (TaKaRa, Shiga, Japan); on occasion, it was treated with BamHI (Amersham) following the manufacturer's recommendation, which in the case of BamHI resulted in cloneable polymorphic fragments for SeUS2-C and SeUS2-D. Electrophoresis was performed in horizontal 1% agarose gels in Tris-acetate-EDTA (TAE) (0.04 M Tris-acetate, 0.001 M EDTA [pH 8.0]), and the DNA fragments were visualized by staining with ethidium bromide. The cloneable SeUS2-C PstI-J fragment, which is characteristic of this genotype and results from the shortening and fusion of SeUS2-A PstI-C and PstI-D fragments (12), was gel-extracted, purified through GFX PCR columns (Amersham Biosciences, Little Chalfont, United Kingdom), and cloned in pUC19 plasmid (Promega Biotech Ibérica, Madrid, Spain) using a DNA ligation kit (LigaFast, Promega Biotech Ibérica). The same procedure was followed using a cloneable 3.1-kb product of BamHI digestion of the characteristic SeUS2-D PstI-C fragment, containing the 2.5-kb insertion. PCR amplifications were performed in a total reaction mixture volume of 50 μl containing 250 μM each deoxynucleoside triphosphate, 56 pmol of each primer, polymerase buffer, and Accuzyme DNA polymerase (Bioline Supply, Segovia, Spain). The following cycling conditions were used: 3 min of initial denaturation at 94°C, 25 amplification cycles (30 s denaturation at 94°C, 1 min of annealing at 58°C, and 2 min of extension at 72°C) with a final extension step at 72°C for 10 min.
Nucleotide sequencing.
Sequencing was performed in an ABI Prism BigDye Terminator ready reaction cycle sequencing kit on a 9600 PE model thermocycler. The reaction products were loaded into an automated DNA sequencer ABI Prism (Sistemas Genómicos, Valencia, Spain).
Determination of the deletion/insertion breakpoints for each variant.
For SeUS2-C, the characteristic SeUS2-C PstI-J fragment was purified from an agarose gel, cloned into pUC19, and sequenced using a universal reverse primer. The deletion in SeUS2-E was mapped using the SeUS1-based primers prE-F01 and prE-R01 (Table 1), designed to target a 2.7-kb fragment around the deletion breaking point. The SeUS2-F deletion was mapped by cloning a 4.8-kb restriction fragment of the SeUS2-F genome obtained with PstI and BamHI and sequencing with a universal forward primer. To map the insertion point on SeUS2-D, the SeUS2-D PstI-C fragment containing the insertion was digested with BamHI, cloned into pUC19, and sequenced with the primers universal forward, prD13-F02, and prD13-F03 (Table 1). The last two primers were designed from the sequences obtained with the universal forward and prD13-F02 primers, respectively.
Table 1.
Name and sequence of the primers used to map the deletion breakpoint for SeUS2-E and for the quantification of the SeUS2-C and SeUS2-A genotypes
| Primer | Sequence | Genotype | Use |
|---|---|---|---|
| prE-F01 | 5′-TGTTACTTGCGTGGCTATCG-3′ | SeUS2-E | Mapping |
| prE-R01 | 5′-GTGACACGCTTCGATCTTGA-3′ | SeUS2-E | Mapping |
| prD13-F02 | 5′-AGTGTGGTACCCAGGCAAAG-3′ | SeUS2-D | Mapping |
| prD13-F03 | 5′-CATGACCCTTGCAGACTTCC-3′ | SeUS2-D | Mapping |
| P1 | 5′-CACGTAGCGCAACAAATCCTC-3′ | SeUS2-C | Quantification |
| P2 | 5′-CGAGGACGAGTAGAGTGTTG-3′ | SeUS2-C and SeUS2-A | Quantification |
| P3 | 5′-CAGTCACCTTCGCCCACAGC-3′ | SeUS2-A | Quantification |
Sequence computer analysis.
DNA and protein comparisons with entries in the updated GenBank/EMBL databases were performed with BLASTn, FASTA format, and BLASTp (24, 25). A pairwise alignment of sequences was made using the Needleman-Wunsch global alignment algorithm in Emboss software (26).
In vitro virus cloning.
For complementation analyses, SeUS2-A and SeUS2-C were cloned in vitro. Fifth instars of S. exigua from the laboratory colony were inoculated per os with SeUS2-WT OBs at a 90% lethal concentration (LC90) (2.3 × 105 OBs/ml). The hemolymphs of these insects were extracted at 48 h postinfection (h.p.i.), added to 500 μl Grace's culture medium supplemented with 10% fetal bovine serum, filtered throughout a 0.45-μm filter, and used for 10-fold serial dilutions (10−1 to 10−6). A 100-μl volume of this mixture was added to Se301 cells at a density of 8 × 105 cells/ml in 96-well tissue culture plates. At 5 days postinfection, the virion-containing supernatants from wells with only one infection plaque were collected and inoculated onto Se301 cells, which were plated at a density of 2 × 106 cells/ml in 6-well tissue culture plates and incubated in 2 ml Grace's insect cell culture medium supplemented with 10% FBS and 2% penicillin-streptomycin (Gibco, Scotland, United Kingdom). At 10 days postinfection, the medium and the cells were collected and centrifuged at 2,300 × g for 5 min to separate OB-containing cells from the budded virus (BV)-containing supernatant. The individual genotypes purified by this procedure were stored at 4°C. For the production of OBs, fourth-instar S. exigua insects from the laboratory colony were injected with 8 μl of each of the BV suspensions produced in vitro. Inoculated larvae were reared individually on a semisynthetic diet until their death. The dead larvae were stored at −20°C. SeUS2-A and SeUS2-C were identified by using restriction endonuclease analysis of viral DNA as described above.
Production of occlusion body mixtures and co-occlusion of genotypes.
Two types of virus populations were constructed: (i) mixtures of OBs of two genotypes, Se-US2A and Se-US2C, and (ii) co-occlusion of SeUS2-A and SeUS2C in ODVs within the same OBs, following methods developed previously (27). For the OB mixtures, suspensions of SeUS2-A OBs and SeUS2-C OBs that had been grown in vitro were quantified by titration in an improved Neubauer hemocytometer, diluted in distilled water to a concentration of 1 × 108 OBs/ml, and mixed in the following proportions: 90% SeUS2-A and 10% SeUS2-C (9A + 1C), 75% SeUS2-A and 25% SeUS2-C (3A + 1C), 50% SeUS2-A and 50% SeUS2-C (1A + 1C), 25% SeUS2-A and 75% SeUS2-C (1A + 3C), and 10% SeUS2-A and 90% SeUS2-C (1A + 9C). These mixtures were then used in bioassays. To produce co-occluded genotypes, 100-μl samples of each of the previous OBs mixtures (1 × 108 OBs/ml) were incubated with 1 volume 1 M Na2CO3 at 50°C for 60 min to release ODVs. Eight-microliter volumes of each ODV suspension were injected into S. exigua fourth instars that were reared individually on a semisynthetic diet until their death. OBs were extracted from dead larvae, purified as described above, resuspended in 50 μl distilled water, and used in bioassays.
The relative proportions of each genotype in the OB mixtures and the co-occluded preparations were estimated by using semiquantitative PCR. The primers P1, P2, and P3 (Table 1) were designed to differentiate between SeUS2-A and SeUS2-C, which produced the amplicons 691 and 791 bp, respectively. Amplifications were performed using genomic DNA extracted from OBs. The reactions were stopped at the mid-logarithmic phase of amplification (17 cycles), which was before the rate of amplification began to decrease. The relative intensities of the two amplicons were compared using the Scion Image PC program (Scion Corporation, MD) as described previously (28).
Bioassays.
Bioassays with OB mixtures and with co-occluded genotypes were carried out using the droplet feeding method (23). Second-instar S. exigua larvae were starved for 12 h and then were allowed to drink from an aqueous suspension containing 10% sucrose, 0.001% Fluorella blue, and OB mixtures or co-occluded genotypes at five different concentrations (2.45 × 105, 8.1 × 104, 2.7 × 104, 9 × 103, and 3 × 103 OBs/ml). Control larvae were fed a solution of sucrose and Fluorella blue without OBs. Larvae that ingested the suspension within 10 min were transferred individually to 24-well tissue culture plates and given a semisynthetic diet. Groups of 24 larvae were treated with each concentration. Each assay was performed three times. The insects were incubated at 25°C, and mortality was recorded daily until larvae died or pupated. Virus-induced mortality was subjected to probit analysis using the Polo-PC program (29).
Mean time to death (MTD) and OB production were calculated for SeUS2-WT, SeUS2-A, and the co-occluded genotype mixtures 9A + 1C, 3A + 1C, and 1A + 1C. For this, groups of 24 S. exigua second instars were inoculated with an LC90 of each inoculum (1.50 × 105 OBs/ml for SeUS2-WT, 1.64 × 105 OBs/ml for SeUS2-A, 2.14 × 105 OBs/ml for 9A + 1C, 1.26 × 105 OBs/ml for 3A + 1C, and 2.07 × 105 OBs/ml for 1A + 1C), which were estimated in the previous bioassay. Inoculated larvae were reared individually at 25°C, and mortality was recorded at 8-h intervals until death or pupation. Cadavers were collected individually and homogenized in 100 μl distilled water. The whole experiment was performed three times. Time mortality data were subjected to Weibull survival analysis using GLIM (generalized linear interactive modeling) software (30). OB yields per larva were estimated by counting the OBs in a Neubauer hemocytometer. Two OB counts were performed for each larva, and the whole experiment was performed three times with 24 larvae per replicate. The results were normalized by square root transformation and were subjected to analysis of variance (ANOVA) in SPSS v15 (IBM, NY).
RESULTS
Population structure of SeUS2.
The large genomic deletions in the SeUS2 genotypic variants SeUS2-C and SeUS2-E were mapped between nucleotides (nt) 16437 and 39757 of SeMNPV isolate SeUS1-A (14). These deletions encompassed all or part of the PstI-L, PstID, and PstIC fragments (Fig. 1). Additionally, a 2,535-bp insertion and a 2,639-bp deletion were detected in the SeUS2 genotypic variants SeUS2-D and SeUS2-F, respectively (Fig. 1b). Other SeUS2 genotypes, such as SeUS2-B and SeUS2-H, show small deletions in the homologous region hr1 (data not shown). The genomic regions of 19,517 to 39,757 bp in SeUS2-A and of 18,753 to 35,122 bp in SfNIC-B, which encompass the large deletions present in SeUS2-C and SfNIC-C, shared 79% nucleotide sequence identity.
Gene content of SeMNPV deletion genotypes.
SeUS2-C has a large deletion of 20,241 bp, representing 14.9% of the genome in relation to that of SeUS1-A, between nt 19517 and 39757. Compared with SeUS1-A, SeUS2-C lacked open reading frames (ORFs) 17 to 39, which include the chitinase, gp37, ptp-2, egt, pkip, arif-1, per os infection factor 2 (pif-2), pif-1, and fgf genes. The left side of the deletion results in a 48% loss in the 5′ coding region of cathepsin. The right side of the deletion is 48 bp upstream of the ORF 40 start codon, and this eliminates the promoter elements of this early-transcribed gene (14). No alternative consensus sequences were found in the 100-bp region located to the left of the deletion point. None of the genes in this region are necessary for viral replication, but pif-1 and pif-2 are essential for horizontal transmission, specifically during per os infection in the insect midgut (31, 32).
SeUS2-E has a large deletion of 12,794 bp (between nt 16437 and 29230) representing 9.4% of the genome with respect to SeUS1-A. This deletion completely eliminates ORFs 13 to 27, including genes such as 38.7kd, late expression factor 1 (lef-1), cathepsin, chitinase, gp37, ptp-2, and egt. The left side of this deletion eliminates 258 of the 629 nt that constitute the 3′ end of lef-2. Both lef-1 and lef-2 are essential genes for viral DNA replication (33). The right side of this deletion removes most of ORF 28, including the whole promoter region and 510 of the 572 encoding nucleotides. The remaining parts of lef-2 (372 bp from the 5′ end) and ORF 28 (63 bp from the 3′ end) fuse in frame and constitute a sequence encoding a chimeric protein of 107 amino acids (aa) that has the carboxyl terminus of ORF 28 and the amino terminus of lef-2.
SeUS2-F had a deletion of 2,639 bp (between nt 21836 and nt 24474), representing 1.9% of the genome in relation to SeUS1-A. ORFs 20 to 22, and 1,005 bp at the 3′ end of ORF 19 (chitinase) in the left side of the deletion, are absent in the SeUS2-F genome. The right side of the deletion starts 119 bp upstream of the ORF 23 starting codon, but the consensus baculovirus late promoter (GTAAG, at nt 24579) of this putative gene is conserved.
Alignment of the deletion flanking regions between the SeMNPV and SfMNPV genotypic variants.
To reveal any potential recombination events between SeMNPV and SfMNPV, sequences of ca. 50 bp flanking the deletion breakpoints of the SeMNPV deletion genotypes SeUS2-C, SeUS2-E, and SeUS2-F were aligned with seven of the genotypes present in the SfNIC population (Table 2). A deletion genotype present in the SeMNPV isolate from California, SeUS1-XD1 (9), was also included in the analysis. No significant homologies were detected between the SeUS2 genotypes and those of SfNIC or SeUS1-XD1, even for the genotypic variants with breakpoints on homologous genes, as was the case for the left breakpoints of SeUS2-F and SfNIC-F and of SeUS2C and SfNIC-E and for the right breakpoints of SeUS1-XD1 and SfNIC-F and of SeUS2E and SfNIC-I. Only seven alignments showed homologies of >50%; the greatest observed homology was 63.3% in the SfNIC-F and SeUS2-E right flanking sequences. However, all variants were aligned with no less than 7.5% gaps. The flanking sequences of the deletions in SeUS2-C, SeUS2-E, and SeUS2-F were analyzed for potential consensus nucleotide features, but none were found.
Table 2.
Percentages of nucleotide identity and gaps existing between the left and right flanking sequences of the deletions in the SeMNPV and SfMNPV deletion genotypes
| Nucleotide identity (gaps) (%) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SfNIC-A |
SfNIC-C |
SfNIC-E |
SfNIC-F |
SfNIC-G |
SfNIC-H |
SfNIC-I |
||||||||
| Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | |
| SeUS2-C | ||||||||||||||
| Left | 48.3 (13.8) | 38.5 (27.7) | 34.2 (49.3) | 46.2 (27.7) | 38.8 (37.3) | 45.9 (27.9) | 35.7 (41.4) | 46.7 (23.3) | 41.9 (17.7) | 44.3 (23.0) | 36.2 (41.4) | 42.6 (23.0) | 43.1 (8.6) | 43.1 (35.4) |
| Right | 37.0 (7.4) | 35.3 (41.2) | 34.4 (34.4) | 35.9 (31.2) | 34.8 (40.9) | 44.8 (25.9) | 39.1 (40.6) | 42.2 (40.6) | 35.1 (52.7) | 35.9 (37.5) | 31.6 (45.6) | 41.9 (32.3) | 44.4 (30.2) | 36.1 (31.1) |
| SeUS2-E | ||||||||||||||
| Left | 38.7 (27.4) | 40.6 (26.6) | 35.1 (58.4) | 38.6 (41.4) | 41.9 (25.8) | 39.7 (34.9) | 36.6 (46.5) | 44.1 (22.0) | 39.1 (37.7) | 44.1 (18.6) | 31.6 (40.4) | 39.0 (18.6) | 35.4 (30.8) | 31.2 (34.4) |
| Right | 53.6 (19.6) | 42.9 (33.3) | 42.9 (36.5) | 46.7 (25.0) | 51.9 (11.1) | 38.9 (18.5) | 43.5 (32.3) | 63.0 (16.7) | 33.9 (10.7) | 42.1 (22.8) | 33.9 (48.2) | 53.7 (13.0) | 36.5 (34.9) | 32.3 (38.7) |
| SeUS2-F | ||||||||||||||
| Left | 34.4 (32.0) | 35.2 (45.1) | 41.0 (23.0) | 40.6 (40.6) | 37.1 (27.4) | 59.3 (25.4) | 37.9 (33.3) | 42.6 (29.5) | 34.3 (41.4) | 32.8 (34.4) | 31.0 (44.8) | 46.8 (29.0) | 32.8 (21.3) | 36.8 (15.8) |
| Right | 35.5 (29.0) | 54.1 (18.0) | 46.8 (24.2) | 42.2 (26.6) | 38.1 (28.6) | 34.4 (37.5) | 37.1 (27.4) | 37.5 (35.9) | 34.7 (44.4) | 41.5 (35.4) | 35.7 (37.5) | 33.3 (37.9) | 33.3 (16.7) | 38.6 (14.0) |
| SeUS1-XD1 | ||||||||||||||
| Left | 42.4 (43.9) | 50.0 (21.7) | 45.9 (27.9) | 38.3 (21.7) | 51.6 (32.3) | 35.6 (30.5) | 35.9 (34.4) | 44.8 (25.9) | 43.5 (25.8) | 50.9 (12.7) | 40.7 (38.9) | 36.1 (31.1) | 41.7 (23.3) | 38.7 (35.5) |
| Right | 30.3 (40.9) | 49.1 (8.8) | 42.1 (12.3) | 45.0 (18.3) | 47.5 (26.2) | 47.3 (14.5) | 45.2 (25.8) | 43.9 (19.3) | 44.3 (42.9) | 41.3 (33.3) | 39.3 (41.1) | 39.3 (27.9) | 29.0 (43.5) | 37.1 (32.3) |
Bold text indicates nucleotide identities of >50%
Relative proportion of SeUS2-A and SeUS2-C in the wild-type population and in tissue culture plaques.
In the natural SeUS2-WT population, the full-length genome variant, SeUS2A, and the deleted genome variant, SeUS2-C, were present in relative proportions of ∼3:1, respectively (Fig. 2). The opposite occurred in tissue culture plaques. SeUS2-C was present in 81% of the 97 plaques obtained following inoculation with hemolymph from SeUS2-WT-infected larvae, based on PstI restriction profiles. The remaining 19% of plaques comprised SeUS2-A. The genotypes SeUS2-E and SeUS2-F were not identified in any of the plaques.
Fig 2.

Relative proportions of SeUS2-A and SeUS2-C in mixtures of OBs and occluded virions. Semiquantitative densitometric analysis of PCR products specific for SeUS2-A (691 bp within the variable region) and SeUS2-C (791 bp flanking the SeUS2-C deletion) in the OB mixtures (A) and co-occlusions (B). For amplification, the total DNA of the wild-type, pure genotypes SeUS2-A and SeUS2-C, and mixtures of OBs and co-occluded genotypes were used. The figures next to amplicons indicate the relative proportion of each product as estimated by densitometric analysis using the Scion Image program. The molecular marker (MM) used was a 1-kb ladder from Stratagene.
Pathogenicity of SeUS2-A and SeUS2-C OB mixtures.
The 50% lethal concentrations (LC50s) of SeUS2-WT (1.69 × 104 OBs/ml) and SeUS2-A (1.31 × 104 OBs/ml) were statistically similar (Table 3). The relative proportions of SeUS2-C in the mixtures were assessed using semiquantitative PCR and were similar to those predicted from the ratio of OBs in the inoculum: 27% SeUS2-C in the mixture 9A + 1C, 38% SeUS2-C in 3A + 1C, 59% SeUS2-C in 1A + 1C, 67% SeUS2-C in 1A + 3C, and 84% SeUS2-C in 1A + 9C (Fig. 2A).
Table 3.
LC50s for S. exigua second instars treated with (i) SeUS2-A OBs or SeUS2-WT OBs, (ii) mixtures of SeUS2-A OBs and SeUS2-C OBs, and (iii) OBs comprising co-occluded mixtures of SeUS2-A and SeUS2-C
| Virus | LC50 (OBs/ml) |
|||
|---|---|---|---|---|
| Mean | Relative potencya | 95% confidence limitsb |
||
| Lower | Upper | |||
| (i) SeUS2-A | 1.31 × 104 | 1 | – | – |
| SeUS2-WT | 1.69 × 104 | 0.77 | 0.23 | 1.30 |
| (ii) Mixtures of OBs | ||||
| 9A + 1C | 1.54 × 104 | 0.84 | 0.56 | 1.22 |
| 3A + 1C | 2.00 × 104 | 0.65 | 0.43 | 0.98 |
| 1A + 1C | 2.85 × 104 | 0.46 | 0.30 | 0.69 |
| 1A + 3C | 5.80 × 104 | 0.22 | 0.13 | 0.37 |
| 1A + 9C | 1.97 × 105 | 0.06 | 0.02 | 0.15 |
| (iii) Co-occluded genotypesc | ||||
| 9A + 1C | 1.60 × 104 | 0.81 | 0.52 | 1.26 |
| 3A + 1C | 1.50 × 104 | 0.86 | 0.57 | 1.29 |
| 1A + 1C | 9.30 × 104 | 0.14 | 0.08 | 0.23 |
Regressions could not be fitted with a common slope; a test for nonparallelism was significant (χ2 = 40.71, df = 9, P < 0.001). Relative potency was therefore calculated as the ratio of LC50 values of each inoculum and SeUS2-A. As such, the relative potencies indicate the relative pathogenicity of each inoculum compared with that of SeUS2-A.
The confidence limits refer to relative potency values.
Co-occluded mixtures of 1A + 3C and 1A + 9C OBs could not be produced, as the proportion of SeUS2-C was markedly lower than that of the ODV inoculum used to inject larvae.
Only the 9A + 1C OB mixture was as pathogenic as were SeUS2-A OBs (Table 3). However, in all other OB mixtures with SeUS2-C OBs in proportions of ≥25%, LC50s were significantly higher than those of SeUS2-A OBs alone. The relative potencies of the OB mixtures reflected the abundance of SeUS2-A OBs in the inoculum. Rapid, efficient, and nonradioactive (REN) and PCR analyses of the OB samples extracted from larval cadavers confirmed the presence of SeUS2-A alone; SeUS2-C was not detected in the larvae, reflecting the lack of per os activity of these OBs (data not shown).
Pathogenicity, virulence, and OB yield of co-occluded SeUS2-A and SeUS2C mixtures.
Densitometric analyses of genotype-specific PCR products indicated that the frequency of SeUS2-C in co-occluded mixed-genotype OBs closely reflected the frequency of this genotype in the ODV inocula used to inject the larvae (Fig. 2B). Co-occluded mixtures for 1A + 3C and 1A + 9C could not be obtained because the proportion of SeUS2-C in progeny OBs was always markedly lower than those of the ODV mixtures that were used to inject larvae.
The LC50s of the two co-occluded mixtures, in which SeUS2-A was the predominant genotype (9A + 1C and 3A + 1C), were statistically similar to that of SeUS2-A alone (Table 3). However, the LC50 of OBs comprising a mixture of 1A + 1C was 7.1-fold higher than that of SeUS2-A alone.
The MTD of the insects infected by SeUS2-A did not differ significantly from those obtained with the co-occluded mixtures involving 9A + 1C, 3A + 1C, or 1A + 1C. In contrast, the larvae infected with SeUS2-WT died significantly more slowly than did insects infected by SeUS2-A alone or by any of the co-occluded mixtures (Fig. 3A).
Fig 3.
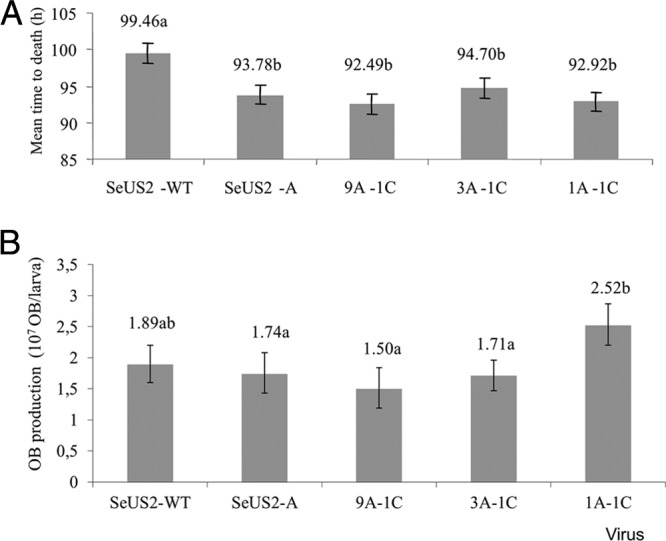
Influence of SeUS2-C on speed of kill and virus yield of occluded genotype mixtures. (A) Mean times to death (MTD) of SeUS2-WT, SeUS2-A, and co-occluded mixtures with 90% SeUS2-A and 10% SeUS2-C (9A + 1C), 75% SeUS2-A and 25% SeUS2-C (3A + 1C), and 50% SeUS2-A and 50% SeUS2-C (1A + 1C). Bars that are labeled with the same letter (a or b) were significantly similar. (B) OB yields of SeUS2-WT, SeUS2-A, and co-occlusions 9A + 1C, 3A + 1C, and 1A + 1C. The values above the bars indicate median values and those followed by the same letter (a or b) did not differ significantly for pairwise comparisons (Mann-Whitney test, P > 0.05).
The distribution of OB yield values was normalized by transformation and subjected to ANOVA (Fig. 3B). The OB yield values of SeUS2-WT, SeUS2-A, and the co-occluded mixtures 9A + 1C and 3A + 1C were statistically similar, with a mean OB production between 1.89 × 107 and 1.50 × 107 OBs/larva, whereas the co-occluded mixture 1A + 1C produced 2.52 × 107 OBs/larva and was significantly more productive (in terms of OBs) than were the other inocula tested.
DISCUSSION
Two hypotheses can explain the origin of such similar population structures in SeUS2 and SfNIC: (i) SfNIC genotypes were derived from recombination events occurring between SeMNPV deletion genotypes and SfMNPV, or vice versa; or (ii) selection has independently favored the formation and maintenance of deletion genotypes in each population.
The high colinearity and sequence homology between the genomes of SeMNPV and SfMNPV suggest a recent common ancestor for these viruses. Interestingly, both viruses are capable of replication in S. exigua when inoculated singly, and although SeMNPV cannot replicate alone in S. frugiperda, SfMNPV can assist SeMNPV replication in S. frugiperda larvae, although the molecular basis for this helper function is unknown (34).
It seems highly unlikely that the deletion genotypes present in the American SeMNPV and SfMNPV populations could have originated from Asian SeMNPV populations following the introduction and spread of Asian S. exigua biotypes in the New World (3). Specifically, two pieces of evidence suggest that this is not the case. First, none of the SfNIC deletion genotypes share homologous flanking sequences around the deletion breakpoints of SeUS2-C, SeUS2-E, SeUS2-F, or SeUS1-XD1. Second, SfNIC deletion genotypes could not have originated from SeUS2-C or SeUS2-E because the SeUS2 variants lack genes that are present in SfNIC. Theoretically, based on genetic content alone, SfNIC-F and SfNIC-I could have been generated by recombination with SeUS2-F, and SfNIC-H by recombination with SeUS1-XD1, but the flanking sequences around each deleted region could only have been generated if they had undergone additional recombination events involving identical crossovers; however, this is extremely unlikely. Similarly, the possibility that New World SeMNPV deletion genotypes were derived from SfMNPV populations is inconsistent with the presence of similar SeMNPV genotypes in isolates from Asia (10), a region where SfMNPV is not found.
This leaves the independent generation of similar population structures in SeMNPV and SfMNPV as the only viable hypothesis. Genetic and functional analyses suggest that deletion variants may have arisen by selection due to their important roles in the survival and/or transmissibility of the viral population.
Genetically, SeMNPV and SfMNPV deletion regions are auxiliary gene-rich regions that contain genes which are not essential for virus replication but which reduce dependence on the host cell machinery or increase viral fitness in other ways (35). Deletion affected two essential genes for viral replication, both of which are late expression factors (lef-1 and lef-2), only in the genotype SeUS2-E. Variants lacking genes that are essential for replication or transmission are maintained in the population through complementation with complete genotypes in multiply infected cells (36). This is possible in baculoviruses because, on average, each cell is infected by four budded virions, each carrying a single genome (37, 38). Complementation was observed in SfMNPV genotypes lacking per os infection factor (pif-1, pif-2) genes, which are required for ODV entry into midgut epithelial cells. As such, SfNIC-C and SfNIC-D are not infectious when administered per os, and the same situation is observed in SeUS2-C, which has a peroral noninfectious phenotype.
In tissue culture cells, SeUS2-C appeared to have a replicative advantage over the complete genotype, as indicated by the 1:3 SeUS2-A/SeUS2-C ratio of plaques resulting postinoculation with the SeUS2-WT population, which comprised the opposite proportions of each of these two genotypes.
The ability of a virus population to produce co-occluded genotypes was dependent on the prevalence of both deletion and complete genotypes in the SeMNPV population. When the ratio of complete/deletion genotypes in the injected inoculum was 1:3 or 1:9, the replication rate of the deletion genotype was too low to produce OBs with a frequency similar to those of co-occluded genotypes; the complete genotype invariably dominated. This differed from the situation in SfNIC genotypes, in which OBs comprising ratios up to 1:9 could be produced by injection of the appropriate inoculum (39), suggesting species-specific differences in the interactions among genotypes in their respective insect hosts.
In SfMNPV, co-occluded mixtures of complete and deletion variants at a ratio of 3:1 enhanced the pathogenicity of the mixed-genotype OBs with respect to that of the complete genotype alone, seemingly due to a dilution effect of the deletion genotype on the concentration of PIF-1 in the ODV envelope (40).
No such interaction was observed between SeUS2-A and SeUS2-C. The SeUS2-C genotype was classified previously, in a study performed prior to the development of co-occlusion techniques, as a defective parasitic genotype because it reduced the pathogenicity of OB mixtures comprising SeUS2-A OBs and ≥80% of SeUS2-C OBs (12). In the present study, the presence of SeUS2-C did not significantly affect OB pathogenicity when co-occluded mixtures were tested at ratios of 9:1 or 3:1 of SeUS2-A/SeUS2-C (Table 3), whereas the pathogenicity of OBs comprising a 1:1 ratio was severely compromised.
SeUS2-C and SfNIC-C both appear to play important roles in determining the pathogenicity of the Spodoptera virus population. Intriguingly, the prevalence of deletion variants in natural populations is maintained at a remarkably similar and highly stable frequency, representing approximately 25% of the population genotypes in SeUS2-WT (41) and 25% of the genotypes in SfNIC (38, 39). Heterogeneous host susceptibility has been demonstrated recently both to increase the prevalence of mixed-genotype baculovirus infections (42) and to mediate the selection of viral genotypes (43). Host ecology has also been observed to determine the relative fitness levels of genotypes in mixed-genotype infections (44).
The presence of genotypic diversity within ODVs and the occlusion of multiple genotypes into each OB is a natural phenomenon in multicapsid nucleopolyhedroviruses that favors the transmission of virus diversity but also allows deletion variants to survive in the population (27).
The functional complementation of co-occluded viral mixtures allowed us to examine the consequences of interactions between SeUS2-A and SeUS2-C on two additional correlates of virus transmissibility. Speed of kill (virulence) did not vary among co-occluded genotype mixtures, but it was lower for SeUS2-WT, indicating that additional genotypes present in the natural population may be capable of modulating this phenotype. Indeed, genotype SeUS2-D, which has an insertion in the auxiliary gene-rich region, was identified as a slow-killing variant in the population (45). As for OB production, all co-occluded mixtures showed OB yields/larva similar to that in the wild-type population, except for the mixture comprising 50% SeUS2-C (1A − 1C). Larvae infected by this mixture produced a significantly higher number of OBs, which suggests that SeUS2-C may have a role in promoting virus transmissibility via increased OB production. The genetic basis for this is presently unclear, but it may be related to the absence of one or more of the genes of unknown function that are present in the SeUS2-C deleted region. One such gene is Se28, the homolog of which is partially deleted in SfMNPV (sf27) in a fast-killing variant of this virus (SfMNPV-3AP2), although its influence on OB production has not been determined (17).
Although SfNIC and SeUS2 populations are clearly similar in terms of genotypic structure and diversity, the functional importance of their deletion variants appears to encompass different aspects of virus transmissibility in each virus population, ranging from OB pathogenicity effects (39) and speed-of-kill characteristics (17) to the quantities of OBs produced in each infected insect (as in this study). The three phenotypic traits analyzed here provide a broader picture of the functional significance of the defective genotype SeUS2-C. Previous studies demonstrated that the deletion variants SeUS2-C and SeUS2-E could invade a deletion variant-free SeMNPV population and become as abundant as in their original population after only four serial passages in larvae (41). Studies on SeUS2-E were hindered because this genotype lacks the essential lef-1 and lef-2 genes, making its purification impossible in cell culture. Future cotransfections with plasmids containing these factors may allow us to clone this genotype. Although the function of SeUS2-E remains veiled, it is clear that defective genotypes tend to persist in baculovirus populations because of their functional importance in key processes, particularly in transmission.
In conclusion, genomic and functional analyses of deletion genotypes present in SeMNPV and SfMNPV isolates indicate that the existence of remarkably similar population structures in distinct baculovirus species from different hosts clearly points to a shared evolutionary mechanism that generates and sustains deletion mutants within each virus population independently, rather than using a recent exchange of deletion variant genotypes between these viruses.
ACKNOWLEDGMENTS
We thank N. Gorría and I. Ibáñez for technical assistance with insect rearing.
This study received financial support from the Spanish Ministry for Science and Technology (AGL2008-05456-C03-01).
A.S. received a predoctoral fellowship from the Spanish Ministry of Education and Culture.
Footnotes
Published ahead of print 30 November 2012
REFERENCES
- 1. Sparks AN. 1979. A review of the biology of the Fall armyworm. Fla. Entomol. 62:82–87 [Google Scholar]
- 2. Zheng XL, Wang P, Cheng WJ, Wang XP, Lei CL. 2012. Projecting overwintering regions of the beet armyworm, Spodoptera exigua in China using the CLIMEX model. J. Insect. Sci. 12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capinera JL. 1999. Beet armyworm, Spodoptera exigua (Hübner) (Insecta: Lepidoptera: Noctuidae). http://edis.ifas.ufl.edu/in262 Accessed September 2012
- 4. Moscardi F. 1999. Assessment of the application of baculoviruses for control of Lepidoptera. Annu. Rev. Entomol. 44:257–289 [DOI] [PubMed] [Google Scholar]
- 5. Erlandson MA. 1990. Biological and biochemical comparison of Mamestra configurata and Mamestra brassicae nuclear polyhedrosis virus isolates pathogenic for the bertha armyworm, Mamestra configurata (Lepidoptera, Noctuidae). J. Invertebr. Pathol. 56:47–56 [Google Scholar]
- 6. Caballero P, Zuidema D, Santiago-Alvarez C, Vlak JM. 1992. Biochemical and biological characterization of four isolates of Spodoptera exigua nuclear polyhedrosis virus. Biocontrol. Sci. Tech. 2:145–157 [Google Scholar]
- 7. Escribano A, Williams T, Goulson D, Cave RD, Chapman JW, Caballero P. 1999. Selection of a nucleopolyhedrovirus for control of Spodoptera frugiperda (Lepidoptera: Noctuidae): structural, genetic, and biological comparison of four isolates from the Americas. J. Econ. Entomol. 92:1079–1085 [DOI] [PubMed] [Google Scholar]
- 8. Murillo R, Muñoz D, Lipa JJ, Caballero P. 2001. Biochemical characterization of three nucleopolyhedrovirus isolates of Spodoptera exigua and Mamestra brassicae. J. Appl. Entomol. 125:267–270 [Google Scholar]
- 9. Dai XJ, Hajos JP, Joosten NN, van Oers MM, Ijkel WFJ, Zuidema D, Pang Y, Vlak JM. 2000. Isolation of a Spodoptera exigua baculovirus recombinant with a 10.6 kbp genome deletion that retains biological activity. J. Gen. Virol. 81:2545–2554 [DOI] [PubMed] [Google Scholar]
- 10. Hara K, Funakoshi M, Kawarabata T. 1995. In vivo and in vitro characterization of several isolates of Spodoptera exigua nuclear polyhedrosis virus. Acta Virol. 39:215–222 [PubMed] [Google Scholar]
- 11. Maruniak JE, Brown SE, Knudson DL. 1984. Physical maps of SfMNPV baculovirus DNA and its genomic variants. Virology 136:221–234 [DOI] [PubMed] [Google Scholar]
- 12. Muñoz D, Castillejo JI, Caballero P. 1998. Naturally occurring deletion mutants are parasitic genotypes in a wild-type nucleopolyhedrovirus population of Spodoptera exigua. Appl. Environ. Microbiol. 64:4372–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simón O, Williams T, López-Ferber M, Caballero P. 2004. Genetic structure of a Spodoptera frugiperda nucleopolyhedrovirus population: high prevalence of deletion genotypes. Appl. Environ. Microbiol. 70:5579–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ijkel WFJ, van Strien EA, Heldens JG, Broer R, Zuidema D, Goldbach RW, Vlak JM. 1999. Sequence and organization of the Spodoptera exigua multicapsid nucleopolyhedrovirus genome. J. Gen. Virol. 80:3289–3304 [DOI] [PubMed] [Google Scholar]
- 15. Tumilasci VF, Leal E, Zanotto PM, Luque T, Wolff JL. 2003. Sequence analysis of a 5.1 kbp region of the Spodoptera frugiperda multicapsid nucleopolyhedrovirus genome that comprises a functional ecdysteroid UDP-glucosyltransferase (egt) gene. Virus Genes 27:137–144 [DOI] [PubMed] [Google Scholar]
- 16. Wolff JL, Valicente F, Martins R, Oliveira JV, Zanotto PM. 2008. Analysis of the genome of Spodoptera frugiperda nucleopolyhedrovirus (SfMNPV-19) and of the high genomic heterogeneity in group II nucleopolyhedroviruses. J. Gen. Virol. 89:1202–1211 [DOI] [PubMed] [Google Scholar]
- 17. Harrison RL, Puttler B, Popham HJ. 2008. Genomic sequence analysis of a fast-killing isolate of Spodoptera frugiperda multiple nucleopolyhedrovirus. J. Gen. Virol. 89:775–790 [DOI] [PubMed] [Google Scholar]
- 18. Simón O, Chevenet F, Williams T, Caballero P, López-Ferber M. 2005. Physical and partial genetic map of Spodoptera frugiperda nucleopolyhedrovirus (SfMNPV) genome. Virus Genes 30:403–417 [DOI] [PubMed] [Google Scholar]
- 19. Herniou E, Theilmann DA, Blissard GW, Becnel JJ, Arif BM, Harrison RL, Bonning BC, Vlak JM, Jehle JA. 2011. Baculoviridae. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy, 9th report. Elsevier Academic Press, San Diego, CA [Google Scholar]
- 20. Heldens JG, van Strien EA, Feldmann AM, Kulcsár P, Muñoz D, Leisy DJ, Zuidema D, Goldbach RW, Vlak JM. 1996. Spodoptera exigua multicapsid nucleopolyhedrovirus deletion mutants generated in cell culture lack virulence in vivo. J. Gen. Virol. 77:3127–3134 [DOI] [PubMed] [Google Scholar]
- 21. Greene GL, Leppla NC, Dickerson WA. 1976. Velvetbean caterpillar (Lepidoptera: Noctuidae): a rearing procedure and artificial medium. J. Econ. Entomol. 69:487–488 [Google Scholar]
- 22. Cabodevilla O, Ibañez I, Simón O, Murillo R, Caballero P, Williams T. 2011. Occlusion body pathogenicity, virulence and productivity traits vary with transmission strategy in a nucleopolyhedrovirus. Biol. Control 56:184–192 [Google Scholar]
- 23. Hughes PR, van Beek NAM, Wood HA. 1986. A modified droplet feeding method for rapid assay of Bacillus thuringiensis and baculoviruses in noctuid larvae. J. Invertebr. Pathol. 48:187–192 [Google Scholar]
- 24. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 25. Pearson WR, Lipman DJ. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U. S. A. 85:2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rice P, Longden I, Bleasby A. 2000. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16:276–277 [DOI] [PubMed] [Google Scholar]
- 27. Clavijo G, Williams T, Muñoz D, Caballero P, López-Ferber M. 2010. Mixed genotype transmission bodies and virions contribute to the maintenance of diversity in an insect virus. Proc. R. Soc. Lond. B 277:943–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simón O, Williams T, López-Ferber M, Caballero P. 2005. Functional importance of deletion mutant genotypes in an insect nucleopolyhedrovirus population. Appl. Environ. Microbiol. 71:4254–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. LeOra Software 1987. POLO-PC. A user's guide to Probit or Logit analysis. LeOra Software Inc., Berkeley, California [Google Scholar]
- 30. Crawley MJ. 1993. GLIM for ecologists. Blackwell Scientific Publications, Oxford, UK [Google Scholar]
- 31. Kikhno I, Gutiérrez S, Croizier L, Croizier G, Ferber ML. 2002. Characterization of pif, a gene required for the per os infectivity of Spodoptera littoralis nucleopolyhedrovirus. J. Gen. Virol. 83:3013–3022 [DOI] [PubMed] [Google Scholar]
- 32. Pijlman GP, Pruijssers AJ, Vlak JM. 2003. Identification of pif-2, a third conserved baculovirus gene required for per os infection of insects. J. Gen. Virol. 84:2041–2049 [DOI] [PubMed] [Google Scholar]
- 33. Lu A, Miller LK. 1995. The roles of eighteen baculovirus late expression factor genes in transcription and DNA-replication. J. Virol. 69:975–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simón O, Williams T, López-Ferber M, Caballero P. 2004. Virus entry or the primary infection cycle are not the principal determinants of host specificity of Spodoptera spp. nucleopolyhedroviruses. J. Gen. Virol. 85:845–2855 [DOI] [PubMed] [Google Scholar]
- 35. O'Reilly DR. 1997. Auxiliary genes of baculoviruses. In Miller LK. (ed), The baculoviruses. Plenum Press, New York, NY [Google Scholar]
- 36. Turner PE, Chao L. 1999. Prisoner's dilemma in an RNA virus. Nature 398:441–443 [DOI] [PubMed] [Google Scholar]
- 37. Bull JC, Godfray HC, O'Reilly DR. 2001. Persistence of an occlusion-negative recombinant nucleopolyhedrovirus in Trichoplusia ni indicates high multiplicity of cellular infection. Appl. Environ. Microbiol. 67:5204–5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simón O, Williams T, Caballero P, López-Ferber M. 2006. Dynamics of deletion genotypes in an experimental insect virus population. Proc. Biol. Sci.. 273:783–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. López-Ferber M, Simón O, Williams T, Caballero P. 2003. Defective or effective? Mutualistic interactions between virus genotypes. Proc. Biol. Sci. 270:2249–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clavijo G, Williams T, Simón O, Muñoz D, Cerutti M, López-Ferber M, Caballero P. 2009. Mixtures of complete and pif1- and pif2- deficient genotypes are required for increased potency of an insect nucleopolyhedrovirus. J. Virol. 83:5127–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muñoz D, Caballero P. 2000. Persistence and effects of parasitic genotypes in a mixed population of the Spodoptera exigua nucleopolyhedrovirus. Biol. Control. 19:259–264 [Google Scholar]
- 42. van der Werf W, Hemerik L, Vlak JM, Zwart MP. 2011. Heterogeneous host susceptibility enhances prevalence of mixed-genotype micro-parasite infections. PLoS Comput. Biol. 7:e1002097 doi:10.1371/journal.pcbi.1002097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hitchman RB, Hodgson DJ, King LA, Hails RS, Cory JS, Possee RD. 2007. Host mediated selection of pathogen genotypes as a mechanism for the maintenance of baculovirus diversity in the field. J. Invertebr. Pathol. 94:153–162 [DOI] [PubMed] [Google Scholar]
- 44. Hodgson DJ, Hitchman RB, Vanbergen AJ, Hails RS, Possee RD, Cory JS. 2004. Host ecology determines the relative fitness of virus genotypes in mixed-genotype nucleopolyhedrovirus infections. J. Evol. Biol. 17:1018–1025 [DOI] [PubMed] [Google Scholar]
- 45. Muñoz D, Ruiz de Escudero I, Caballero P. 2000. Phenotypic characteristics and relative proportions of three genotypic variants isolated from a nucleopolyhedrovirus of Spodoptera exigua. Entomol. Exp. Appl. 97:75–282 [Google Scholar]