

Abstract
Methods for genetic manipulation of Clostridium ljungdahlii are of interest because of the potential for production of fuels and other biocommodities from carbon dioxide via microbial electrosynthesis or more traditional modes of autotrophy with hydrogen or carbon monoxide as the electron donor. Furthermore, acetogenesis plays an important role in the global carbon cycle. Gene deletion strategies required for physiological studies of C. ljungdahlii have not previously been demonstrated. An electroporation procedure for introducing plasmids was optimized, and four different replicative origins for plasmid propagation in C. ljungdahlii were identified. Chromosomal gene deletion via double-crossover homologous recombination with a suicide vector was demonstrated initially with deletion of the gene for FliA, a putative sigma factor involved in flagellar biogenesis and motility in C. ljungdahlii. Deletion of fliA yielded a strain that lacked flagella and was not motile. To evaluate the potential utility of gene deletions for functional genomic studies and to redirect carbon and electron flow, the genes for the putative bifunctional aldehyde/alcohol dehydrogenases, adhE1 and adhE2, were deleted individually or together. Deletion of adhE1, but not adhE2, diminished ethanol production with a corresponding carbon recovery in acetate. The double deletion mutant had a phenotype similar to that of the adhE1-deficient strain. Expression of adhE1 in trans partially restored the capacity for ethanol production. These results demonstrate the feasibility of genetic investigations of acetogen physiology and the potential for genetic manipulation of C. ljungdahlii to optimize autotrophic biocommodity production.
INTRODUCTION
Homoacetogenic microorganisms have unique metabolic pathways and energy conservation mechanisms that could substantially enhance microbial strain design options for the production of fuels and other biocommodities. Furthermore, homoacetogens play an important role in the carbon cycle of a diversity of anaerobic environments (1, 2). However, the understanding of homoacetogen physiology and the development of a homoacetogenic microorganism as a chassis for the production of biocommodities have been limited by a lack of methods for genetic manipulation.
The homoacetogen Clostridium ljungdahlii has been proposed as a potential chassis for biocommodity production (3). Like other acetogens, C. ljungdahlii metabolizes sugars through the Embden-Meyerhof-Parnas pathway, and CO2 released during glycolysis is fixed via the Wood-Ljungdahl pathway, which makes it possible to recover completely the carbon from sugars in organic end products (4, 5), i.e., C6H12O6 + 2H2O → 2CH3COOH + 2CO2 + 8H+ + 8e− and 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O. Depending on the growth conditions, other organic products such as ethanol and 2,3-butanediol are also generated (3, 6).
C. ljungdahlii can grow autotrophically using H2 and/or CO as the electron donor, reducing CO2 via the Wood-Ljungdahl pathway to produce organic products (3, 5, 6). The ability of C. ljungdahlii to use CO as an electron donor is significant because CO is a waste product of steel manufacturing and a major component of the syngas produced from the gasification of municipal waste and other organic feedstocks (7, 8). Biocommodity production using syngas as an intermediate is an attractive strategy because organic feedstocks, such as lignocellulosic biomass, municipal waste, and plastics, are difficult for microorganisms to degrade directly (9–11).
Furthermore, C. ljungdahlii was able to grow with electrons derived directly from an electrode as the electron donor coupled to reduction of CO2 to produce acetate (12). Electrode-driven reduction of carbon dioxide via acetogenic microorganisms, known as microbial electrosynthesis (13, 14), is a strategy for conversion of CO2 to organic commodities without a biomass intermediate. Biofilms of acetogenic microorganisms colonize surfaces of cathodes and directly convert CO2 to organic products that are excreted from the cells. When microbial electrosynthesis is powered with electricity derived from solar technology, it is an artificial form of photosynthesis that converts CO2 to desired products much more efficiently and in a more environmentally sustainable manner than biomass-based approaches (13, 15).
The development of C. ljungdahlii as a chassis for production of biocommodities will require strategies for genetic manipulation. Although heterologous gene expression by introduction of a plasmid in C. ljungdahlii was reported (3), the efficiency of plasmid transformation by the reported method was low (M. Köpke, personal communication). In general, genetic manipulation of clostridia has been difficult (8, 16–18). Limiting factors have been a strong restriction-modification system, high nuclease activity that can degrade foreign DNA, and the thick outer layers of these Gram-positive organisms. Even when the restriction-modification system barrier has been overcome by protecting DNA with in vivo or in vitro methylation (19, 20), homologous recombination frequencies have been low, with single-crossover recombination as the predominant event (21–23). Consequently, not many Clostridium mutants have been produced in the last 20 years (21, 22, 24–26). However, with renewed interest in biotechnological applications of Clostridium species there have been renewed efforts to develop strategies for genetic modification, leading to such recent developments as the following: replicative plasmids for gene deletion (27); counterselection methods to improve the efficiency of gene deletion and to select for double-crossover events (28–31); the use of a promoterless antibiotic resistance cassette in conjunction with a constitutively expressed promoter to select for double-crossover events (32); the use of the Bacillus subtilis recU gene, which codes for resolvase, to increase homologous recombination frequencies (33–36); the use of the bacterial mobile group II intron as an alternative to homologous recombination to disrupt a gene (16, 37–40); and the use of antisense RNA to downregulate a target gene product (41, 42). Here we report on a more efficient electroporation protocol for C. ljungdahlii and demonstrate that chromosomal gene deletion is feasible for C. ljungdahlii. These results open windows for biotechnological applications of C. ljungdahlii and for investigation of basic acetogen physiology.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli strain NEB 10-beta [araD139 Δ(ara-leu)7697 fhuA lacX74 galK (ϕ80 Δ(lacZ)M15) mcrA galU recA1 endA1 nupG rpsL (Strr) Δ(mrr-hsdRMS-mcrBC)] (New England BioLabs) was used for general plasmid propagation and cloning. Plasmid DNA to be electroporated into C. ljungdahlii was isolated from E. coli strain NEB Express [fhuA2 [lon] ompT gal sulA11 R(mcr-73::miniTn10-Tets)2 [dcm] R(zgb-210::Tn10-Tets) endA Δ(mcrC-mrr)114::IS10] (New England BioLabs). E. coli strain ER2275 (recA1 hsdR mcrBC) was the host strain for in vivo methylation of plasmid DNA prior to transformation of C. ljungdahlii when the effect of in vivo methylation on transformation efficiency was evaluated, as previously described (3, 20). E. coli cells were cultivated in LB medium supplemented with the appropriate antibiotic, if necessary, at 37°C with shaking.
C. ljungdahlii DSM 13528 (ATCC 55383) was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ). For growth studies and phenotype characterization, C. ljungdahlii cells were grown anaerobically at 37°C in DSMZ 879 medium supplemented with 0.04% l-cysteine, 0.04% sodium sulfide, 0.1% sodium bicarbonate, and 5 g liter−1 fructose. For general propagation, cells were grown anaerobically at 37°C in PETC 1754 medium (American Type Culture Collection [ATCC]) supplemented with 1 mM l-cysteine (pH 7) and 5 g liter−1 fructose. The original protocol includes both cysteine and sodium sulfide as reductants, but we found no significant difference in growth if the sulfide was omitted.
Cells were grown on agar plates (1.5% Difco Noble agar) at 35°C in a heated anaerobic glove bag containing an N2-CO2-H2 (83:10:7) atmosphere. The medium was either a modified reinforced clostridial medium (RCM) (43) supplemented with 5 mM l-cysteine (pH 7) or YTF medium (10 g liter−1 yeast extract, 16 g liter−1 Bacto tryptone, 4 g liter−1 sodium chloride, 5 g liter−1 fructose, pH 6) supplemented with 2 mM l-cysteine (pH 7).
For motility assays, C. ljungdahlii wild-type and mutant cells were spotted on YTF soft agar plates with 0.3% Difco Noble agar and incubated at 35°C in an anaerobic chamber.
C. ljungdahlii strains were maintained by freezing mid-log-phase cultures at −80°C with 10% dimethyl sulfoxide (DMSO) for long-term storage.
Thiamphenicol was dissolved in dimethylformamide (DMF). Clarithromycin was first dissolved in acidic MilliQ water (pH 2) and then adjusted to pH 6.5 with sodium hydroxide. Both antibiotic solutions were made anoxic by sparging and overlaying with an N2 atmosphere.
DNA manipulation and construction of E. coli-C. ljungdahlii shuttle vectors.
C. ljungdahlii genomic DNA was prepared with the Epicenter MasterPure DNA purification kit. All enzymes for DNA manipulation were purchased from New England BioLabs, unless stated otherwise. Qiagen Taq polymerase was used for all DNA amplifications, unless stated otherwise. Plasmids, PCR products, and DNA fragments from agarose gels were purified with Qiagen mini/midiprep, PCR purification, and gel extraction kits, respectively.
Plasmid pCL1 was constructed by replacing the kanamycin resistance cassette (EcoRI-PstI) of pIKM1 (44) with an EcoRI-SmaI-BamHI-SalI-PstI polylinker (see Table S1 in the supplemental material). Plasmids pQexp and pQint were generous gifts from G. M. Church (40). Plasmid pJIR750ai was purchased from Sigma. Plasmid pCL2 was constructed by digesting pJIR750ai with PvuI and religating the vector portion with T4 DNA ligase.
In vivo methylation of plasmid DNA was carried out when necessary as previously described (20). E. coli strain ER2275 harboring plasmid pAN1 for in vivo methylation was provided by E. T. Papoutsakis.
Preparation of electrocompetent C. ljungdahlii cells.
The procedure for making C. ljungdahlii electrocompetent cells was modified from a protocol reported previously (3). All manipulations except centrifugation were carried out on ice in an anaerobic chamber. All buffers and centrifuge tubes were made ice cold and anoxic before use. All plasticware was placed in the anaerobic chamber at least 24 h before use in order to eliminate any residual oxygen. Competent cells were maintained in SMP buffer (270 mM sucrose, 1 mM MgCl2, 7 mM sodium phosphate, pH 6) with 10% DMSO at −80°C until use.
Electrocompetent C. ljungdahlii cells were prepared from cultures that were freshly inoculated from frozen stocks and then transferred twice in PETC liquid medium. PETC medium can be replaced with YTF medium without significant loss of transformation efficiencies. About 15 to 16 h before preparation of competent cells, a mid- to late-log-phase culture was transferred into two serum bottles containing 100 ml fresh PETC medium supplemented with 40 mM dl-threonine (final optical density at 600 nm [OD600], ∼0.004). After overnight growth at 37°C, early-log-phase cells (OD600, ∼0.2 to 0.3; 200 ml, 3 × 107 to 3.5 × 107 cells/ml) were harvested by centrifugation at 10,000 rpm for 10 min at 4°C. The cells were washed twice with 200 ml of SMP wash buffer and resuspended in the same buffer at a final concentration of ∼1010 to 1011 cells/ml. Antifreezing buffer (60% DMSO–40% SMP, pH 6) was added to the competent cells at one-fifth of the final resuspension volume to achieve a final concentration of 10% DMSO. The resulting competent cells (25 μl/tube) were stored at −80°C for future use. The competence of these frozen competent cells remained stable for about 1 month.
Electrotransformation procedures for C. ljungdahlii.
All procedures were carried out in an anaerobic chamber. Electrocompetent C. ljungdahlii cells (25 μl) were quickly transferred on ice from a −80°C freezer to an anaerobic chamber. After thawing on ice (about 1 min), the cells were mixed with 1 to 5 μg DNA and transferred to a prechilled, 0.1-cm-gap Gene Pulser cuvette (Bio-Rad). Cells were pulsed at 0.625 kV with resistance at 600 Ω and a capacitance of 25 μF by using a Gene Pulser Xcell microbial electroporation system (Bio-Rad). Immediately after the pulse, cells were recovered with 0.5 ml of fresh PETC medium, transferred to a pressure tube containing 10 ml of PETC medium supplemented with 5 mM l-cysteine (pH 7), and incubated at 37°C. The electroporated cells were allowed to recover at 37°C until their cell densities were higher than immediately after the electroporation (about 9 to 12 h). Five-milliliter volumes of the outgrowth cultures or appropriately diluted cultures were mixed with 20 ml of RCM molten agar (1.5%) containing an appropriate antibiotic and poured into a petri dish. RCM medium can be replaced with YTF medium without significant loss of transformation efficiencies. After the agar mixtures were solidified, plates were incubated upside down in a secondary container with a petri dish containing palladium pellets in order to eliminate any residual oxygen.
Verification of the presence of plasmids in C. ljungdahlii transformants was carried out by either of two methods: (i) restriction analysis of plasmid DNA isolated from an E. coli strain transformed with plasmid DNA isolated from C. ljungdahlii transformants or (ii) colony PCR analysis of C. ljungdahlii transformants to detect the catP gene (primer sequences are listed in Table S1 in the supplemental material).
Construction of a mutant allele to disrupt the fliA gene.
All primers are listed in Table S1 in the supplemental material. The gene fliA (CLJU_c10410) was replaced with the gene ermC, such that the coding region from 83 Lys to 220 Val was deleted. DNA fragments containing the upstream (883 bp) and downstream (882 bp) regions of fliA were amplified by PCR with chromosomal DNA as a template. The ermC cassette was amplified by PCR with pCL1 as a template. The PCR products were cloned separately in a plasmid, and their sequences were confirmed. The upstream region of the fliA gene, the downstream region of the fliA gene, and the ermC gene were prepared by digesting the plasmids with XbaI and EcoRI, HindIII and XhoI, and EcoRI and HindIII, respectively, and were cloned in the XbaI and XhoI sites of pBluescript II KS(−) (Stratagene). The plasmid (pBuTU-ΔfliA-ermC) thus constructed was electroporated into the wild-type C. ljungdahlii strain as described above. Transformants resistant to clarithromycin were selected as described above. The genotypes were examined by PCR amplification with primers fliA-1 and fliA-4.
Construction of mutant alleles to disrupt adhE1, adhE2, or both.
The coding region from 327 Glu to 685 Pro of the gene adhE1 (CLJU_c16510) was replaced with the gene ermC. Upstream (977 bp) and downstream (1,024 bp) regions of adhE1 were amplified by PCR with wild-type genomic DNA as a template. The coding region of the ermC gene was amplified by PCR with pCL1 as a template. The SacI site within the multiple-cloning sites in pBluescript II KS(−) was deleted by digestion of the vector with SacI, filling in with Klenow fragment, and vector ligation. The resulting vector, designated pBuKsII-SacI, was sequenced to confirm that the SacI site was absent. The upstream and downstream regions and the ermC coding region were digested with XbaI and SacI, HindIII and XhoI, and SacI and HindIII, respectively, and were cloned into the XbaI and XhoI sites of pBuKsII-SacI.
The coding region from 34 Val to 775 Arg of adhE2 (CLJU_c16520) was replaced with the gene ermC. For construction of a mutant allele to disrupt adhE2, the upstream (977 bp) and downstream (997 bp) regions of adhE2 were amplified by PCR. The ermC cassette was amplified with the same primers as for the fliA mutant allele. The three pieces were digested with the respective restriction enzymes and cloned into pBluescript II KS(−) as described above.
For disruption of both adhE1 and adhE2, the coding region from 34 Val of adhE1 to 775 Arg of adhE2 was replaced with the gene ermC. The 904-bp upstream region of adhE1 and the 997-bp downstream region of adhE2 were amplified by PCR and the mutant allele was constructed as described above.
Expression of adhE1 in trans.
The complete coding sequence of adhE1 was amplified with Phusion DNA polymerase (New England BioLabs). The adhE1 coding sequence was digested with NdeI and BamHI and inserted into the NdeI and BamHI sites of the expression vector pMTL83152 (45) to generate plasmid pMTL83152-adhE1. The adhE1 gene was then sequenced to confirm that no mutations were introduced during amplification. The pMTL83152-adhE1 plasmid was then electroporated into either the adhE1 or the adhE1 adhE2 deletion mutants. The presence of the plasmid was confirmed by PCR and plasmid preparation.
Analytical techniques.
Acetate was measured via high-performance liquid chromatography (HPLC) as previously described (12). Ethanol was measured with a gas chromatograph as previously described (46). Cell growth was monitored using a Genesys 2 spectrophotometer (Spectronic Instruments, Rochester, NY) at 600 nm. Cell numbers were determined by epifluorescence microscopy using acridine orange staining (47).
Transmission electron microscopy.
Both wild-type and fliA deletion mutant cells grown in PETC medium were placed on 400-mesh carbon-coated copper grids, incubated for 5 min, and then stained with 2% uranyl acetate. Cell appendages were observed using a Tecnai 12 transmission electron microscope at an accelerating voltage of 100 kV. Images were taken digitally with the Teitz TCL camera system.
RESULTS AND DISCUSSION
Growth on solid medium.
Conditions for growth of C. ljungdahlii on solidified media were evaluated in order to obtain single colonies of C. ljungdahlii with a high plating efficiency for isolation of mutant strains. The PETC medium (ATCC 1754) that was used routinely for maintenance of C. ljungdahlii cultures was unsuitable for sustaining growth on solidified medium. On average, fewer than 5 colonies were obtained when ∼100 cells were plated on the PETC agar medium. The poor plating efficiencies might be due to a shift in pH caused by differences in gas compositions between the culture tube containing the liquid medium (100% CO2) and the anaerobic chamber (10% CO2, 7% H2, and 83% N2), where the plates were incubated. In contrast, the plating efficiency with RCM medium was ∼30%. Furthermore, if cells were added to molten agar (∼45°C), plating efficiencies were 72% ± 10% (mean ± standard deviation; n = 3). Single colonies were visible within 3 days.
The MICs of thiamphenicol and clarithromycin were determined with the molten agar plating method. The growth of ca. 108 cells on solid medium was inhibited by thiamphenicol (5 μg/ml) or clarithromycin (4 μg/ml).
Development of an electroporation protocol and identification of E. coli-C. ljungdahlii shuttle vectors.
A transformation protocol for C. ljungdahlii was recently reported (3, 48). However, the reported protocol did not consistently yield transformants with the E. coli-Clostridium shuttle vector pIMP1 (M. Köpke, personal communication). After the evaluation of the protocol, several changes were made, including changes in (i) the pH of the wash buffer, (ii) the density of competent cells, and (iii) the electroporation procedures (Table 1). With this revised protocol, transformants were consistently obtained with plasmid pCL1, a derivative of pIMP1 (0.2 transformants/μg DNA) (Table 2). However, the transformation efficiency was still poor, possibly due to the plasmid not being stably maintained.
Table 1.
Changes in the optimized transformation protocol for C. ljungdahlii
| Step or parameter | Original procedure (3, 48) | Optimized procedure |
|---|---|---|
| Preparation of competent cells | ||
| Growth phase for harvesting (OD600) | 0.3–0.7 | 0.2–0.3 |
| pH of wash buffer | 7.4 | 6 |
| Resuspension buffer | SMPa (pH 7.4) | SMPa (pH 6) with 10% DMSOb |
| Cell density of competent cells | ∼80× concentrated from the original cultures | 1010–1011 cells/ml (∼1,000× concentrated) |
| Freeze-thaw | No | Yes |
| Thawing of cells | No | 1 min on ice |
| Preparation of plasmids | ||
| In vivo methylation | Yes (20) | No |
| E. coli strain for plasmid preparation | Kc strain (ER2275) | Bc strain (NEB Expressd) |
| Electroporation procedures | ||
| Cell vol (μl) | 600 | 25 |
| Preincubation with plasmid | 5 min on ice | No |
| Amount of plasmid DNA (μg) | 0.1–1.5 | 1–5 |
| Electroporation cuvette gap (cm) | 0.4 | 0.1 |
| Electric pulse | 2.5 kV, 600 Ω, 25 μF | 0.625 kV, 600 Ω, 25 μF |
| Recovery | 5 ml PETCe; 37°C, until clear growth occurs | 10 ml PETC; 37°C, 9–12 h |
| Plating | Liquid cultures on a solid agar plate | Liquid cultures mixed with molten agar |
| Antibiotic concn (μg/ml) | Clarithromycin (5), thiamphenicol (20) | Clarithromycin (4), thiamphenicol (5) |
SMP, 270 mM sucrose, 1 mM MgCl2, and 7 mM phosphate buffer.
DMSO, dimethyl sulfoxide.
K strain, Dcm+ Dam−; B strain, Dcm− Dam+.
New England Biolabs, Ipswich, MA.
PETC, American Type Culture Collection (ATCC) PETC 1754 medium.
Table 2.
Transformation of C. ljungdahlii with different plasmids
| Plasmid | Origins of replication (E. coli, Clostridium)a | Antibiotic resistance gene(s)b | Transformation efficiency (transformants/μg DNA), mean ± SD (n) |
|
|---|---|---|---|---|
| First optimizationc | Final protocol | |||
| pCL1 | pMB1, pIM13 | bla, ermC | 0.2 ± 0 (5) | 1.1 ± 0.1 (3) |
| pQexp | pMB1, pAMβ1 | ermB | 0.7 ± 0.5 (3) | 14.9 ± 4.9 (6) |
| pJIR750ai | pMB1, pIP404 | catP | 7 (1) | NDd |
| pCL2 | pMB1, pIP404 | catP | ND | (1.7 ± 0.6) × 104 (5) |
| pMTL82151e | ColE1, pBP1 | catP | ND | (3.8 ± 0.2) × 103 (3) |
| pMTL83151e | ColE1, pCB102 | catP | ND | (3.1 ± 1.8) × 103 (3) |
All plasmids are E. coli-Clostridium shuttle vectors. E. coli origins of replication, pMB1 and ColE1; Clostridium origins of replication, pIM13, pAMβ1, pIP404, pBP1, and pCB102.
bla, ampicillin resistance; ermC or ermB, clarithromycin/erythromycin resistance; catP, thiamphenicol/chloramphenicol resistance.
The first optimization included the changes of wash buffer pH, cell density of the competent cells, and electroporation procedures (details are listed in Table 1).
ND, not determined.
The plasmid became available after the optimized protocol was developed.
In order to determine whether plasmids known to be stably maintained in other Clostridium species (49) might be more effectively propagated in C. ljungdahlii, plasmids pQexp (40) and pJIR750ai (38), with origins of replication from pAMβ1 and pIP404, respectively, were tested. Both plasmids were more efficiently transformed than pIMP1 or pCL1 (Table 2). The transformation efficiencies for pQexp and pJIR750ai were 0.7 and 7 transformants/μg plasmid DNA, respectively (Table 2). The presence of these plasmids in C. ljungdahlii was confirmed with restriction analysis and/or colony PCR (Fig. 1). These results indicate that replication origins from pQexp and pJIR750ai are functional in C. ljungdahlii.
Fig 1.
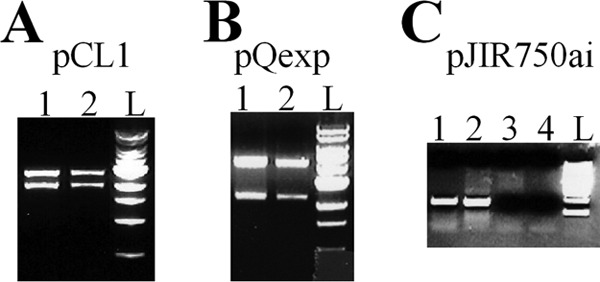
Restriction and PCR analyses of various plasmid DNAs from C. ljungdahlii. (A) Plasmid pCL1. Plasmid DNA was digested with HindIII and analyzed by agarose gel electrophoresis. Lane 1, plasmid preparation from E. coli which was transformed with a plasmid preparation from a C. ljungdahlii transformant; lane 2, plasmid used to transform C. ljungdahlii; lane L, 1-kb DNA ladders (New England BioLabs). (B) HindIII-digested pQexp. Lane 1, plasmid preparation from E. coli which was transformed with a plasmid preparation from a C. ljungdahlii transformant; lane 2, plasmid used to transform C. ljungdahlii; lane L, 1-kb DNA ladders. (C) Colony PCR amplification of the catP gene from pJIR750ai. Lane 1, a C. ljungdahlii transformant; lane 2, plasmid preparation from E. coli as the positive control; lane 3, C. ljungdahlii genomic DNA as a negative control; lane 4, no DNA as a negative control for the PCR amplification.
The transformation protocol was further optimized with pCL2, which was constructed from pJIR750ai by deleting the region for the group II intron insertion that is not required for its propagation in C. ljungdahlii. One modification in the optimized protocol (Table 1) was to change the growth phase at which cells were harvested to prepare competent cells, because it is known that growth phase can affect competence and the optimal growth phase must be decided empirically (50). Furthermore, the cells were frozen prior to electroporation, which may have weakened the cell wall structure, and the 10% DMSO in the storage buffer may facilitate DNA-cell membrane contact (50). The methods for isolating plasmid DNA for electroporation were also modified (Table 1). With the optimized conditions, the transformation efficiency of pCL2 was 1.73 × 104 ± 0.57 × 104/μg DNA (n = 5) (Table 2). The transformation efficiency of pCL1 using the final optimized conditions was 1.1 ± 0.1/μg DNA (n = 3) (Table 2). Similar transformation efficiency was also obtained for a plasmid (pCL1-catP) that has the same origin of replication as pCL1 but carries the same antibiotic resistance gene as pCL2 (data not shown), suggesting that the poor transformation efficiency of pCL1 was not due to selections with different antibiotics. The competence of these frozen competent cells remains consistent for about 1 month (data not shown).
After the optimization of the transformation protocol with pCL2, plasmids from the pMTL80000 modular system (45) with origins of replication other than pAMβ1 and pIP404 were made available to us. The pBP1 and pCB102 origins of replication were functional in C. ljungdahlii, whereas the pCD6 origin of replication was not. The transformation efficiency for plasmids with either a pBP1 or pCB102 origin of replication was somewhat lower than that of pCL2 (Table 2).
In vivo or in vitro methylation of plasmid DNA to be transformed stimulates the transformation efficiency in some Clostridium species (19, 20), and this procedure was employed in C. ljungdahlii in the previously reported protocol (3). However, in vivo methylation had a negative impact on the transformation efficiency in C. ljungdahlii with our protocols (data not shown). For C. thermocellum, transformation efficiency was increased when plasmid DNA was isolated from an E. coli Dcm− Dam+ strain (e.g., the B strain) instead of a Dcm+ Dam− (K) strain (51, 52). This was also the case for C. ljungdahlii (data not shown). The B and K strains differ in their endogenous methylation sites. The endogenous adenine methylation at GATC sequences is abolished in the K strain, whereas the cytosine methylation at CCWGG sequences is abolished in the B strain. Therefore, methylation plays a role in transformation of foreign DNA in C. ljungdahlii. Analysis of the C. ljungdahlii genome sequence identified homologs of a type I restriction-modification system (CLJU_c03310-03320-03330). However, the specificity of the C. ljungdahlii restriction-modification system has yet to be characterized, and elucidation of the restriction-modification system may further improve the transformation efficiency.
Gene replacement via homologous recombination.
In order to expand the available genetic tools for C. ljungdahlii, genetic modification of the C. ljungdahlii chromosome was examined with the improved transformation protocol described above. Homologous recombination was evaluated with the fliA gene (CLJU_c10410), which encodes a putative sigma factor of RNA polymerase known to control the expression of genes involved in flagellar biosynthesis and motility in other bacteria (53, 54). Deletion of fliA was expected to result in the loss of flagella and motility in C. ljungdahlii.
Initial attempts to obtain a fliA deletion mutant with introduction of a linear DNA fragment prepared by digesting the plasmid pBuTU-ΔfliA-ermC with XhoI were not successful. No transformants were obtained. Transformants were obtained only with the introduction of the intact plasmid pBuTU-ΔfliA-ermC into C. ljungdahlii competent cells. PCR analysis demonstrated both single- and double-crossover homologous recombination (Fig. 2B). The frequencies of the double-crossover events were ca. 30%. This result demonstrated that a strategy for mutagenesis via homologous recombination with a suicide vector was possible.
Fig 2.
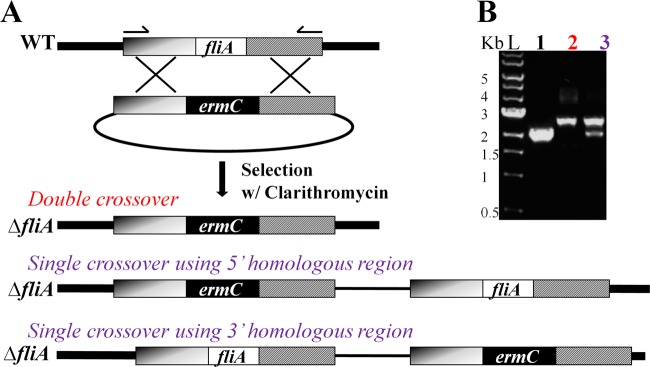
Construction of a fliA deletion mutant. (A) Schematic diagram showing the mutagenesis strategy employed to construct the fliA deletion mutant of C. ljungdahlii. Homologous recombination via a suicide vector, pBuTU-ΔfliA-ermC, resulted in three possible genotypes: double crossover, single crossover at the 5′ homologous region, and single crossover at the 3′ homologous region. (B) PCR analysis of representative fliA deletion mutants. Genotypes were verified by PCR amplification with distal primers indicated by arrows in panel A. The PCR product from the wild-type locus (lane 1) is smaller than that from the mutant locus (lanes 2 and 3) because the replaced region in the fliA gene is smaller than the ermC gene. Lane L, 1-kb DNA ladders (New England BioLabs); lane 1, wild type; lane 2, double-crossover fliA mutant; lane 3, single-crossover fliA mutant.
Transmission electron microscopy revealed that wild-type cells produced multiple flagella, whereas the mutant did not (Fig. 3A). Deletion of the fliA gene did not affect cell growth in the PETC liquid medium with fructose. However, when the wild-type and the mutant cells were spotted onto YTF soft agar plates, the mutant cells were nonmotile, whereas the wild-type cells were motile and formed a larger growth zone than the mutant cells (Fig. 3B). These results demonstrated that it was possible to create a deletion mutant of C. ljungdahlii by double-crossover homologous recombination.
Fig 3.

Characterization of the fliA deletion mutant. (A) Production of flagella. Transmission electron micrographs of negatively stained C. ljungdahlii wild-type cells (left panel) and the fliA deletion mutant (right panel) showed that the mutant did not produce any flagella, whereas the wild-type cells produced flagella as indicated by arrows. Scale bars in both panels represent 2,000 nm. (B) Motility assay. The fliA deletion mutant was not motile when spotted on a YTF soft agar plate on which the wild-type cells were motile and thus exhibited a larger growth zone. The scale bar represents 3 cm.
Identification of a bifunctional aldehyde/alcohol dehydrogenase that contributes to ethanol production in C. ljungdahlii.
In order to determine whether carbon and electron flow could be altered by gene deletion, the possibility of diminishing ethanol production was evaluated for proof of concept. CLJU_c16510 and CLJU_c16520, designated adhE1 and adhE2, respectively, have been annotated to code for bifunctional aldehyde/alcohol dehydrogenases (3) and thus have the potential to promote ethanol production. These genes are located next to each other and possibly are transcribed from the same promoter. Therefore, care was taken when designing the mutagenesis cassette to avoid polar effects. The mutagenesis cassette for deletion of adhE1 was designed to introduce two stop codons (TAA and TAA) after the sequence for amino acid 326 (Leu), followed by a promoterless ermC and then the downstream homologous region. This ensured that adhE2 was transcribed from the original promoter sequence. The mutagenesis cassettes for deletion of either adhE2 or both adhE1 and adhE2 were designed as described above for deletion of fliA because polar effects were not a concern. All three mutants were isolated from agar plates supplemented with fructose and clarithromycin. Double-crossover events for all three mutants were confirmed by PCR analysis (data not shown).
The adhE2 deletion mutant grew on fructose as well as the wild type and produced similar amounts of ethanol and acetate (Fig. 4; Table 3). Deleting adhE1, either alone or in combination with adhE2, slightly diminished the growth yield, increased the doubling time, and significantly inhibited ethanol production while increasing acetate production (Fig. 4; Table 3). The amounts of ethanol produced by the adhE1 deletion strain and the adhE1 adhE2 double deletion strain were similar. In both strains, ethanol production was decreased to ca. 15% of that in the wild type (Table 3). Within the error of the measurements, the decreased carbon recovery in ethanol in the adhE1 deletion or the adhE1 adhE2 double deletion strain could be accounted for by an increase in carbon recovery in acetate over that produced by the wild type (Table 3). The slightly decreased cell yields and increased doubling times in the absence of AdhE1 or both AdhE1 and AdhE2 may suggest that these mutants were not able to dispose of reducing equivalents as efficiently as the wild type.
Fig 4.
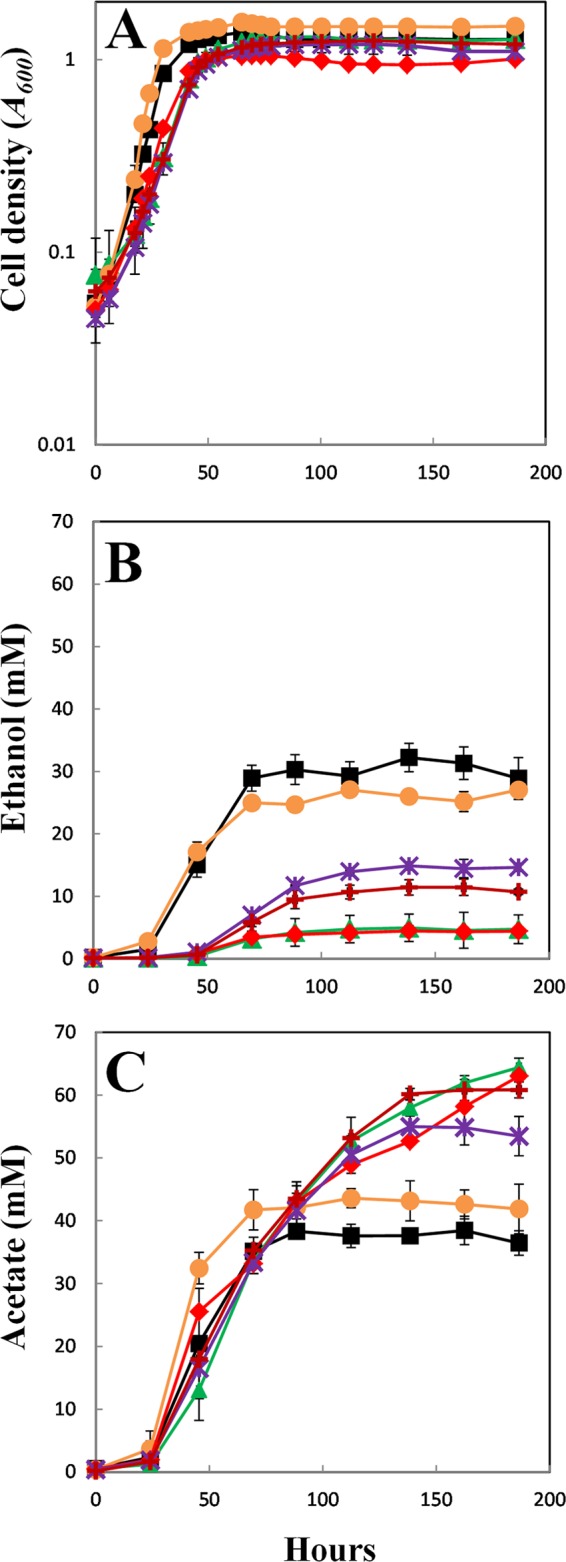
Cell growth and production of ethanol and acetate by the C. ljungdahlii wild-type strain, bifunctional aldehyde/alcohol dehydrogenase deletion mutants (ΔadhE1, ΔadhE2, and ΔadhE1 ΔadhE2), and the complemented strains (ΔadhE1/adhE1 and ΔadhE1 ΔadhE2/adhE1). (A) Cell growth (A600). (B) Ethanol production (mM). (C) Acetate production (mM). Symbols: filled squares, wild type; green triangles, ΔadhE1; orange circles, ΔadhE2; red diamonds, ΔadhE1 ΔadhE2; purple crosses, ΔadhE1/adhE1; and maroon crosses, ΔadhE1 ΔadhE2/adhE1. Data are the means ± standard deviations of quadruplicates.
Table 3.
Production of acetate, ethanol, and biomass by the wild type, bifunctional aldehyde/alcohol dehydrogenase gene deletion mutants, and complemented strains
| Parameter | Value for straina: |
|||||
|---|---|---|---|---|---|---|
| WT | ΔadhE1 | ΔadhE2 | ΔadhE1 ΔadhE2 | ΔadhE1/adhE1 | ΔadhE1 ΔadhE2/adhE1 | |
| Ethanol production (mM) | 28.9 ± 3.4 | 4.7 ± 2.3 | 27.1 ± 0.5 | 4.4 ± 0.6 | 14.7 ± 0.4 | 11.5 ± 1.3 |
| Acetate production (mM) | 38.5 ± 2.3 | 64.4 ± 1.5 | 41.9 ± 3.9 | 63.0 ± 1.8 | 53.4 ± 3.1 | 60.8 ± 1.7 |
| Cell yield (A600) | 1.4 ± 0.1 | 1.3 ± 0.2 | 1.5 ± 0.1 | 1.1 ± 0.0 | 1.2 ± 0.1 | 1.2 ± 0.1 |
| Doubling time (h) | 5.8 ± 0.2 | 8.4 ± 0.4 | 5.7 ± 0.1 | 7.3 ± 0.2 | 8.7 ± 0.5 | 9.5 ± 1.0 |
Data are the means ± standard deviations of quadruplicates. WT, wild-type C. ljungdahlii cells; ΔadhE1, adhE1 deletion mutant; ΔadhE2, adhE2 deletion mutant; ΔadhE1 ΔadhE2, adhE1 and adhE2 double deletion mutant; ΔadhE1/adhE1, adhE1 deletion mutant with the adhE1 gene expressed in trans; ΔadhE1 ΔadhE2/adhE1, adhE1 and adhE2 double deletion mutant with the adhE1 gene expressed in trans.
When adhE1 was expressed in trans under the control of a C. acetobutylicum thiolase gene promoter on a plasmid, ethanol production in the adhE1-deficient strain was tripled, with a corresponding decrease in acetate production, maintaining the carbon balance in ethanol and acetate comparable to that for the wild type and the uncomplemented adhE1-deficient strain (Table 3; Fig. 4). In a similar manner, expressing adhE1 in trans in the strain deficient in both adhE1 and adhE2 increased ethanol production and decreased acetate production, with an overall carbon recovery in acetate and ethanol comparable to that for the wild type (Fig. 4; Table 3). The longer doubling time of the complemented strain may be due to the addition of thiamphenicol to the medium, which was required in order to maintain the plasmid carrying adhE1.
These results suggest that AdhE1 is an important bifunctional aldehyde/alcohol dehydrogenase in C. ljungdahlii. This finding demonstrates that it is now possible to elucidate gene function in C. ljungdahlii via genetic manipulations. The results suggest that AdhE2 does not play a role in ethanol production, at least under the conditions evaluated, even though AdhE2 shares 88% identity with AdhE1. This finding emphasizes the importance of functional genomic studies in order to understand cellular metabolism. The results also demonstrate that it is possible to redirect carbon and electron flow in C. ljungdahlii with a gene deletion without significantly impairing overall growth. This indicates that the metabolic engineering of C. ljungdahlii is now feasible.
Conclusions.
The ability to effectively express foreign genes in C. ljungdahlii and to delete genes via homologous recombination is expected to substantially promote the development of this organism as a chassis for the production of biocommodities. For example, the wild-type C. ljungdahlii can produce multiple organic products from acetyl coenzyme A (acetyl-CoA), the central intermediate in the Wood-Ljungdhal pathway. Efficient production of desired commodities from acetyl-CoA will require disabling these competing pathways. As demonstrated in this study, deletion of the ethanol production pathway led to increased production of acetate in C. ljungdahlii. The homologous recombination method described here should also be applicable for introducing desired metabolic genes or reporters into the chromosome.
Furthermore, the ability to genetically manipulate C. ljungdahlii should aid in basic studies of homoacetogen physiology. Although important aspects of the physiology of acetogens have been elucidated with elegant biochemical investigations, much is still unknown about mechanisms for energy conservation and regulation of gene expression in these organisms (1, 55, 56). Tools for facile genetic manipulation of C. ljungdahlii should enhance further investigation of homoacetogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Advanced Research Projects Agency-Energy (ARPA-E), U.S. Department of Energy, under award no. DE-AR0000087, and the Office of Science (BER), U.S. Department of Energy, cooperative agreement no. DE-FC02-02ER63446.
We thank Peter Dürre and Michael Köpke for discussions on C. ljungdahlii genetics. We thank Eleftherios T. Papoutsakis and his lab members for valuable discussions and providing expertise on clostridia and the plasmid pAN1 as well as E. coli strain ER2275. Plasmid pQexp and pMTL80000 modular systems were generously provided by George M. Church and Nigel P. Minton, respectively. We thank Muktak Aklujkar for proofreading the manuscript. We are grateful to Joy Ward, Manju L. Sharma, Cory Gillis, Trevor Woodard, Jaclyn Izbicki, and Courtney Thompson for technical support.
Footnotes
Published ahead of print 30 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02891-12.
REFERENCES
- 1. Drake HL, Gößner AS, Daniel SL. 2008. Old acetogens, new light. Ann. N. Y. Acad. Sci. 1125:100–128 [DOI] [PubMed] [Google Scholar]
- 2. Ragsdale SW, Pierce E. 2008. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784:1873–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kopke M, Held C, Hujer S, Liesegang H, Wiezer A, Wollherr A, Ehrenreich A, Liebl W, Gottschalk G, Durre P. 2010. Clostridium ljungdahlii represents a microbial production platform based on syngas. Proc. Natl. Acad. Sci. U. S. A. 107:13087–13092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ljungdahl LG. 1986. The autotrophic pathway of acetate synthesis in acetogenic bacteria. Annu. Rev. Microbiol. 40:415–450 [DOI] [PubMed] [Google Scholar]
- 5. Tanner RS, Miller LM, Yang D. 1993. Clostridium ljungdahlii sp. nov., an acetogenic species in clostridial rRNA homology group I. Int. J. Syst. Bacteriol. 43:232–236 [DOI] [PubMed] [Google Scholar]
- 6. Kopke M, Mihalcea C, Liew F, Tizard JH, Ali MS, Conolly JJ, Al-Sinawi B, Simpson SD. 2011. 2,3-Butanediol production by acetogenic bacteria, an alternative route to chemical synthesis, using industrial waste gas. Appl. Environ. Microbiol. 77:5467–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tirado-Acevedo O, Chinn MS, Grunden AM. 2010. Production of biofuels from synthesis gas using microbial catalysts. Adv. Appl. Microbiol. 70:57–92 [DOI] [PubMed] [Google Scholar]
- 8. Tracy BP, Jones SW, Fast AG, Indurthi DC, Papoutsakis ET. 2012. Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr. Opin. Biotechnol. 23:364–381 [DOI] [PubMed] [Google Scholar]
- 9. Henstra AM, Sipma J, Rinzema A, Stams AJ. 2007. Microbiology of synthesis gas fermentation for biofuel production. Curr. Opin. Biotechnol. 18:200–206 [DOI] [PubMed] [Google Scholar]
- 10. Mohammadi M, Najafpour GD, Younesi F, Lahijani P, Uzir MH, Mohamed AR. 2011. Bioconversion of synthesis gas to second generation biofuels: a review. Renew. Sust. Energ. Rev. 15:4255–4273 [Google Scholar]
- 11. Munasinghe PC, Khanal SK. 2010. Biomass-derived syngas fermentation into biofuels: opportunities and challenges. Bioresour. Technol. 101:5013–5022 [DOI] [PubMed] [Google Scholar]
- 12. Nevin KP, Hensley SA, Franks AE, Summers ZM, Ou J, Woodard TL, Snoeyenbos-West OL, Lovley DR. 2011. Electrosynthesis of organic compounds from carbon dioxide is catalyzed by a diversity of acetogenic microorganisms. Appl. Environ. Microbiol. 77:2882–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lovley DR, Nevin KP. 2011. A shift in the current: new applications and concepts for microbe-electrode electron exchange. Curr. Opin. Biotechnol. 22:441–448 [DOI] [PubMed] [Google Scholar]
- 14. Garner LE, Park J, Dyar SM, Chworos A, Sumner JJ, Bazan GC. 2010. Modification of the optoelectronic properties of membranes via insertion of amphiphilic phenylenevinylene oligoelectrolytes. J. Am. Chem. Soc. 132:10042–10052 [DOI] [PubMed] [Google Scholar]
- 15. Lovley DR. 2012. Electromicrobiology. Annu. Rev. Microbiol. 66:391–409 [DOI] [PubMed] [Google Scholar]
- 16. Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 70:452–464 [DOI] [PubMed] [Google Scholar]
- 17. Papoutsakis ET. 2008. Engineering solventogenic clostridia. Curr. Opin. Biotechnol. 19:420–429 [DOI] [PubMed] [Google Scholar]
- 18. Shao L, Hu S, Yang Y, Gu Y, Chen J, Jiang W, Yang S. 2007. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 17:963–965 [DOI] [PubMed] [Google Scholar]
- 19. Jennert KC, Tardif C, Young DI, Young M. 2000. Gene transfer to Clostridium cellulolyticum ATCC 35319. Microbiology 146:3071–3080 [DOI] [PubMed] [Google Scholar]
- 20. Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 59:1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Green EM, Bennett GN. 1996. Inactivation of an aldehyde/alcohol dehydrogenase gene from Clostridium acetobutylicum ATCC 824. Appl. Biochem. Biotechnol. 57–58:213–221 [DOI] [PubMed] [Google Scholar]
- 22. Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN. 1996. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology 142:2079–2086 [DOI] [PubMed] [Google Scholar]
- 23. Nair RV, Green EM, Watson DE, Bennett GN, Papoutsakis ET. 1999. Regulation of the sol locus genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 by a putative transcriptional repressor. J. Bacteriol. 181:319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liyanage H, Kashket S, Young M, Kashket ER. 2001. Clostridium beijerinckii and Clostridium difficile detoxify methylglyoxal by a novel mechanism involving glycerol dehydrogenase. Appl. Environ. Microbiol. 67:2004–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, Cheung JK, Rood JI. 2006. Construction and analysis of chromosomal Clostridium difficile mutants. Mol. Microbiol. 61:1335–1351 [DOI] [PubMed] [Google Scholar]
- 26. Wilkinson SR, Young M. 1994. Targeted integration of genes into the Clostridium acetobutylicum chromosome. Microbiology 140:89–95 [Google Scholar]
- 27. Harris LM, Welker NE, Papoutsakis ET. 2002. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J. Bacteriol. 184:3586–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl. Environ. Microbiol. 78:8112–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl. Environ. Microbiol. 78:4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nariya H, Miyata S, Suzuki M, Tamai E, Okabe A. 2011. Development and application of a method for counterselectable in-frame deletion in Clostridium perfringens. Appl. Environ. Microbiol. 77:1375–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, McCool JD, Warner AK, Rajgarhia VB, Lynd LR, Hogsett DA, Caiazza NC. 2010. Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl. Environ. Microbiol. 76:6591–6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP. 2012. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res. 40:e59 doi:10.1093/nar/gkr1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bi C, Jones SW, Hess DR, Tracy BP, Papoutsakis ET. 2011. SpoIIE is necessary for asymmetric division, sporulation, and expression of σF, σE, and σG but does not control solvent production in Clostridium acetobutylicum ATCC 824. J. Bacteriol. 193:5130–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones SW, Tracy BP, Gaida SM, Papoutsakis ET. 2011. Inactivation of sigmaF in Clostridium acetobutylicum ATCC 824 blocks sporulation prior to asymmetric division and abolishes σE and σG protein expression but does not block solvent formation. J. Bacteriol. 193:2429–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tracy BP, Jones SW, Papoutsakis ET. 2011. Inactivation of σE and σG in Clostridium acetobutylicum illuminates their roles in clostridial-cell-form biogenesis, granulose synthesis, solventogenesis, and spore morphogenesis. J. Bacteriol. 193:1414–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tracy BP, Papoutsakis ET. March 2010. Methods and compositions for genetically engineering clostridia species. US patent 2010/0075424 A1
- 37. Cai GQ, Jin B, Saint C, Monis P. 2011. Genetic manipulation of butyrate formation pathways in Clostridium butyricum. J. Biotechnol. 155:269–274 [DOI] [PubMed] [Google Scholar]
- 38. Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl. Environ. Microbiol. 71:7542–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li YC, Tschaplinski TJ, Engle NL, Hamilton CY, Rodriguez M, Liao JC, Schadt CW, Guss AM, Yang YF, Graham DE. 2012. Combined inactivation of the Clostridium cellulolyticum lactate and malate dehydrogenase genes substantially increases ethanol yield from cellulose and switchgrass fermentations. Biotechnol. Biofuels 5:2 doi:10.1186/1754-6834-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tolonen AC, Chilaka AC, Church GM. 2009. Targeted gene inactivation in Clostridium phytofermentans shows that cellulose degradation requires the family 9 hydrolase Cphy3367. Mol. Microbiol. 74:1300–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Desai RP, Papoutsakis ET. 1999. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl. Environ. Microbiol. 65:936–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tummala SB, Welker NE, Papoutsakis ET. 2003. Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J. Bacteriol. 185:1923–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cotter JL, Chinn MS, Grunden AM. 2009. Ethanol and acetate production by Clostridium ljungdahlii and Clostridium autoethanogenum using resting cells. Bioprocess Biosyst. Eng. 32:369–380 [DOI] [PubMed] [Google Scholar]
- 44. Mai V, Lorenz WW, Wiegel J. 1997. Transformation of Thermoanaerobacterium sp. strain JW/SL-YS485 with plasmid pIKM1 conferring kanamycin resistance. FEMS Microbiol. Lett. 148:163–167 [Google Scholar]
- 45. Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 78:79–85 [DOI] [PubMed] [Google Scholar]
- 46. Morita M, Malvankar NS, Franks AE, Summers ZM, Giloteaux L, Rotaru AE, Rotaru C, Lovley DR. 2011. Potential for direct interspecies electron transfer in methanogenic wastewater digester aggregates. mBio 2(4):e00159–11 doi:10.1128/mBio.00159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lovley DR, Phillips EJP. 1988. Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl. Environ. Microbiol. 54:1472–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kopke M. 2009. Genetische Veränderung von Clostridium ljungdahlii zur Produktion von 1-Butanol aus Synthesegas. Ph.D. thesis University of Ulm, Ulm, Germany [Google Scholar]
- 49. Davis IJ, Carter G, Young M, Minton NP. 2005. Gene clonging in clostridia, p 37–52 In Dürre P. (ed), Handbook on clostridia. CRC Press, Boca Raton, FL [Google Scholar]
- 50. Aune TE, Aachmann FL. 2010. Methodologies to increase the transformation efficiencies and the range of bacteria that can be transformed. Appl. Microbiol. Biotechnol. 85:1301–1313 [DOI] [PubMed] [Google Scholar]
- 51. Guss AM, Olson DG, Caiazza NC, Lynd LR. 2010. Dcm methylation is detrimental to plasmid replication in Clostridium thermocellum, abstr P01, p 17 Clostridium 11, San Diego, CA: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tyurin MV, Desai SG, Lynd LR. 2004. Electrotransformation of Clostridium thermocellum. Appl. Environ. Microbiol. 70:883–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCarter LL. 2006. Regulation of flagella. Curr. Opin. Microbiol. 9:180–186 [DOI] [PubMed] [Google Scholar]
- 54. Smith TG, Hoover TR. 2009. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv. Appl. Microbiol. 67:257–295 [DOI] [PubMed] [Google Scholar]
- 55. Ragsdale SW. 2008. Enzymology of the Wood-Ljungdahl pathway of acetogenesis. Ann. N. Y. Acad. Sci. 1125:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schiel-Bengelsdorf B, Dürre P. 2012. Pathway engineering and synthetic biology using acetogens. FEBS Lett. 586:2191–2198 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.