

Abstract
Anidulafungin, which noncompetitively inhibits β-(1,3)-d-glucan synthase in fungal cell wall biosynthesis, is the newest antifungal drug to be developed. Echinocandin B deacylase from Actinoplanes utahensis NRRL 12052 catalyzes the cleavage of the linoleoyl group of echinocandin B, a key step in the process of manufacturing anidulafungin. Unfortunately, the natural yield of echinocandin B nucleus is low. In our study, the echinocandin B deacylase gene was systematically overexpressed by genetic engineering in its original producer, A. utahensis, and in the heterologous hosts Streptomyces lividans TK24 and Streptomyces albus. The introduction of additional copies of the gene, under the control of PermE* or its native promoter, into hosts showed significant increases in its transcription level and in the efficiency of the bioconversion of echinocandin B to its nucleus. The conditions for the cultivation and bioconversion of A. utahensis have been optimized further to improve production. As a result, while the wild-type strain initially produced 0.36 g/liter, a concentration of 4.21 g/liter was obtained after the generation of a strain with additional copies of the gene and further optimization of the reaction conditions. These results are useful for enhancing echinocandin B nucleus production in A. utahensis. Our study could enable the engineering of commercially useful echinocandin B nucleus-overproducing stains.
INTRODUCTION
Fungal infections are being seen in ever-increasing numbers, largely because of the increase in the size of the population at risk over the past 20 years. This population includes cancer patients, transplant recipients, and other individuals receiving immunosuppressive treatment. These people are at greater risk than others owing to their weakened immune systems and the chronic nature of diseases (1–3). The increased incidence of invasive fungal infections has created a major challenge for health care professionals. Since cell walls are present in fungal cells but absent in animal cells, the fungal cell wall perhaps represents the ideal target for the therapeutic treatment of fungal pathogens in humans (4, 5). The development of echinocandins, the first class of antifungals to target the fungal cell wall, was a milestone in antifungal chemotherapy (6, 7). Three semisynthetic echinocandin derivatives have been developed for clinical use: caspofungin, micafungin, and anidulafungin (8). Their strengths include low toxicity, rapid fungicidal activity against most isolates of Candida spp., and predictable, favorable kinetics allowing once-a-day dosing. Besides Candida spp., their inhibitory spectrum includes Aspergillus spp. and Pneumocystis jirovecii, but not Cryptococcus neoformans (9, 10).
Echinocandin B (ECB), obtained by the fermentation of Aspergillus nidulans and Aspergillus rugulosus, is known as one of the natural cyclic hexapeptides that have a linoleoyl side chain, which inhibits a crucial enzyme in fungal cell wall biosynthesis, β-(1,3)-d-glucan synthase (11). ECB can be modified by enzymatic deacylation to a cyclic hexapeptide without a linoleoyl side chain and by subsequent chemical reacylation to generate a few therapeutic antifungal agents for clinical practice, such as anidulafungin (12–15). A deacylase from Actinoplanes utahensis NRRL 12052 catalyzes the cleavage of the linoleoyl side chain from ECB (Fig. 1), an essential reaction for the three subsequent synthetic steps (16). The enzyme is a membrane-associated heterodimer composed of 63-kDa and 18- to-20-kDa subunits, and the expression of its activity is not affected by any cofactors, metal ion chelators, or reducing agents. In addition to that of ECB, this deacylase mediates the cleavage of aculeacin A, FR901379, various semisynthetic ECB derivatives, daptomycin and its three derivatives, teicoplanin, pseudomycin A, and capsaicins (17, 18). Thus, it may become increasingly significant as a pharmaceutical biocatalyst.
Fig 1.

ECB deacylase-catalyzed reaction.
However, enzymatic deacylation was rate-limiting when conducted with whole cells of A. utahensis. The low bioconversion yield was presumably related to inadequate production of the ECB deacylase. Because of the need for improved antifungal agents for the treatment of systemic fungal infections and the broad specificity of the enzyme, we were interested in constructing engineered strains with high production of the enzyme and in developing a better bioconversion method to further improve the efficiency of bioconversion of ECB. Here we describe the cloning of the gene encoding ECB deacylase, under the control of its native promoter or a PermE* promoter, into the original deacylase-producing strain and two Streptomyces strains by ΦC31-directed site-specific recombination in order to understand the effects of the promoters and gene dosage on the efficiency of the bioconversion of ECB to the ECB nucleus, particularly with regard to its potential biotechnological application.
MATERIALS AND METHODS
Bacterial strains, plasmids, and reagents.
The bacterial strains and plasmids used in this paper are listed in Table 1. Streptomyces lividans TK24, Streptomyces albus, and A. utahensis NRRL 12052 were obtained from our laboratory. Biochemicals, chemicals, media, restriction enzymes, and other molecular biological reagents were from standard commercial sources.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Host for general cloning | Invitrogen |
| ET12567(pUZ8002) | Donor strain for conjugation between E. coli and Actinomyces | 3 |
| A. utahensis | ECB deacylase-producing strain | NRRL 12052 |
| DYG2001 | A. utahensis derivative with two copies of the ECB deacylase gene (second copy under the control of PermE*) | This work |
| DYG2002 | A. utahensis derivative with two copies of the ECB deacylase gene under the control of its native promoter | This work |
| DYG2003 | A. utahensis derivative with three copies of the ECB deacylase gene (second and third copies under the control of PermE*) | This work |
| S. lividans TK24 | 19 | |
| DYG2004 | S. lividans derivative with a single copy of the ECB deacylase gene under the control of PermE* | This work |
| DYG2005 | S. lividans derivative with a single copy of the ECB deacylase gene under the control of its native promoter | This work |
| DYG2006 | S. lividans derivative with two copies of the ECB deacylase gene under the control of PermE* | This study |
| S. albus | 19 | |
| DYG2007 | S. albus derivative with a single copy of the ECB deacylase gene under the control of PermE* | This work |
| DYG2008 | S. albus derivative with a single copy of the ECB deacylase gene under the control of its native promoter | This work |
| DYG2009 | S. albus derivative with two copies of the ECB deacylase gene under the control of PermE* | This work |
| Plasmids | ||
| pSP72 | E. coli subcloning vector | Promega |
| pSET152 | E. coli replicon, Streptomyces ΦC31 attachment site; Aprr | 16 |
| pYG1003 | 2.0-kb fragment containing PermE*-controlled aveBIV in pSET152 | 20 |
| pYG2001 | 2.8-kb PCR fragment containing the ECB deacylase gene in pSP72 | This work |
| pYG2002 | 3.3-kb PCR fragment containing a PermE*-controlled ECB deacylase gene in pSP72 | This work |
| pYG2003 | 3.3-kb PCR fragment containing a PermE*-controlled ECB deacylase gene in pSET152 | This work |
| pYG2004 | 4.0-kb PCR fragment containing the ECB deacylase gene and its 5′ and 3′ regulatory sequences in pSP72 | This work |
| pYG2005 | 4.0-kb PCR fragment containing the ECB deacylase gene and its 5′ and 3′ regulatory sequences in pSET152 | This work |
| pYG2006 | 3.3-kb PCR fragment containing a PermE*-controlled ECB deacylase gene in pSP72 | This work |
| pYG2007 | Two copies of a 3.3-kb PCR fragment containing a PermE*-controlled ECB deacylase gene in pYG2003 | This work |
DNA isolation, manipulation, and sequencing.
DNA isolation and manipulation were performed by standard methods (21). PCR amplifications were conducted on an authorized Thermal Cycler (Eppendorf AG, Hamburg, Germany) using PrimerSTAR HS DNA polymerase (TaKaRa). Primer synthesis and DNA sequencing were carried out at Shanghai Invitrogen Biotechnology Co.
Plasmid construction.
To express the ECB deacylase gene under the control of a PermE* promoter, a 2.8-kb DNA fragment that contains only the gene encoding ECB deacylase was amplified by PCR from A. utahensis NRRL 12052 genomic DNA using primers 5′-AAAGAATTCGTGCGGGCCTGAAA-3′ and 5′-AAATCTAGAGACTGCGTGAGTTCTGC-3′ and was cloned into the pSP72 vector, yielding pYG2001. The identity of the PCR product with the gene encoding ECB deacylase (GenBank accession number BD226911) was confirmed by sequencing. A 0.5-kb fragment containing a PermE* promoter was PCR amplified from pYG1003 using primers 5′-AAAAGATCTTCTAGAAGCCCGACCCGAGCA-3′ and 5′-AAAGAATTCTCCGGAGGTCGCACC-3′ and was cloned into the BglII/EcoRI site of pYG2001, yielding pYG2002. (Underlined letters in primer sequences represent restriction sites.) The 3.3-kb XbaI/XbaI fragment containing the gene coding for ECB deacylase and a PermE* promoter from pYG2002 was inserted into the XbaI site of the pSET152 vector, yielding pYG2003 for the expression of the gene encoding ECB deacylase under the control of a PermE* promoter by a ΦC31-directed site-specific recombination event.
For the expression of the gene encoding ECB deacylase under the control of its native promoter, a 4.0-kb DNA fragment that contains the gene encoding ECB deacylase with approximately 1 kb of upstream sequence and 0.5 kb of downstream sequence was amplified from A. utahensis NRRL 12052 genomic DNA by PCR using primers 5′-ATAGAATTCCGTGCCCAGCTGTTC-3′ and 5′-AAATCTAGAGACTGCGTGAGTTCTGC-3′ and was cloned into the pSP72 vector, yielding pYG2004. The identity of the PCR product with the gene encoding ECB deacylase was also confirmed by sequencing. The 4.0-kb EcoRI/XbaI fragment from pYG2004 was inserted into the corresponding sites of pSET152, yielding pYG2005.
In order to express two copies of the gene encoding ECB deacylase under the control of a PermE* promoter, another 3.3-kb DNA fragment with terminal EcoRV/EcoRV sites containing the gene encoding ECB deacylase and a PermE* promoter was amplified by PCR from pYG2002 using primers 5′-AAAGATATCAGCCCGACCCGAGCA-3′ and 5′-AAAGATATCGACTGCGTGAGTTCTGC-3′ and was cloned into the pSP72 vector, yielding pYG2006. The identity of the PCR product with the gene encoding ECB deacylase was confirmed. The 3.3-kb EcoRV/EcoRV fragment containing the gene encoding ECB deacylase and a PermE* promoter from pYG2006 was inserted into the corresponding site of the recombinant vector pYG2003, yielding the last vector, pYG2007, containing two deacylase gene expression cassettes. The correct directions of the gene encoding ECB deacylase and the PermE* promoter in the pYG2003 or pYG2007 vector were verified by restriction enzyme digestion.
Conjugation and generation of recombinant strains.
For overexpression and heterologous expression of the ECB deacylase gene in A. utahensis NRRL 12052 and the two Streptomyces strains (S. lividans TK24 and the S. albus strain), expression vectors pYG2003, pYG2005, and pYG2007 were each introduced into hosts by intergeneric conjugation from Escherichia coli ET12567(pUZ8002) according to the standard procedure (19, 22). Transformants that were resistant to apramycin were identified as the recombinant strains, whose genomic DNAs were integrated with the deacylase gene and the apramycin resistance gene by ΦC31-directed site-specific recombination. The genotypes of the recombinant strains were further confirmed by PCR amplification with the vector-specific primer pair M13-47 and RV-M.
Culture growth and deacylation procedure.
Wild-type and recombinant strains were grown on agar plates with a medium consisting of 2% soluble starch, 0.05% NaCl, 0.05% K2HPO4·3H2O, 0.1% KNO3, 0.05% MgSO4·7H2O, 0.001% FeSO4·7H2O, and 2% agar powder (pH 7.4) at 28°C for sporulation. For the fermentation of A. utahensis NRRL 12052, an agar piece around 1 cm2 was inoculated into a 250-ml flask containing 50 ml of a seed medium consisting of 2.5% sucrose, 2.0% oatmeal, 0.25% yeast powder, 0.1% K2HPO4, 0.05% KCl, 0.05% MgSO4·7H2O, and 0.0002% FeSO4·7H2O and was incubated at 28°C and 220 rpm for 3 days. A 250-ml flask containing 50 ml of fresh fermentation medium, consisting of 2% sucrose, 1% peanut meal, 0.1% KH2PO4, and 0.025% MgSO4·7H2O, was then inoculated with 5 ml of the seed culture, and incubation was continued at 28°C and 220 rpm for 4 days. For the fermentation of S. lividans TK24 and S. albus, an agar piece around 1 cm2 was inoculated into a 250-ml flask containing 50 ml of a seed medium consisting of 1.0% glucose, 0.5% yeast powder, and 1% peptone and was incubated at 30°C and 220 rpm for 30 h. A 250-ml flask containing 50 ml of fresh fermentation medium consisting of 2.5% glucose, 1% bean flour, 0.3% NaCl, and 0.3% CaCO3 was then inoculated with 1 ml of the seed culture, and incubation was continued at 30°C and 220 rpm for 2 days.
The wet mycelia (15 g) were harvested by centrifugation, washed twice with double-distilled water, and then resuspended in a 50-ml biotransformation reaction mixture containing 0.1 M Na2HPO4/NaH2PO4 buffer (pH 6.8) in the presence of 2% dimethyl sulfoxide (DMSO). The enzymatic reaction was initiated by addition of the substrate ECB (1.5 g/liter) and was continued at 30°C and 220 rpm for 5 h before sample analysis.
Different experiments were subsequently conducted to optimize the cultivation medium of A. utahensis by changing the carbon and nitrogen sources and their proportions. Maltose, sucrose, glycerol, lactose, and soybean oil were used individually as the carbon source, at concentrations of 20 g/liter, instead of glucose. Cottonseed meal, soybean powder, and oatmeal were used individually as the nitrogen source, at concentrations of 10 g/liter, instead of peanut meal. The optimum concentration of sucrose or cottonseed meal was determined by dosing different amounts of sucrose (2 to 6%) or cottonseed meal (1 to 5%). The optimum pH for biotransformation was determined at 30°C. The optimum temperature for biotransformation was determined at pH 6.8. Ethanol was used as the cosolvent instead of DMSO. The optimum concentration of the ECB substrate was determined by dosing different amounts of ECB (1.5 to 12 g/liter) under the optimized conditions.
Analytical methods for the detection of ECB and the ECB nucleus.
The reaction was interrupted by the addition of methanol. After low-speed centrifugation to remove precipitated proteins and mycelia, the activity of the deacylase was determined by monitoring the formation of the cyclic hexapeptide (the ECB nucleus) by high-performance liquid chromatography (HPLC) with an Agilent C18 column (250 by 4.6 mm; Agilent, Palo Alto, CA). The column was equilibrated with 92% solvent A (H2O containing 2 mg/ml ammonium acetate) and 8% solvent B (60% CH3CN containing 2 mg/ml ammonium acetate), and the analytical method was developed with the following program: from 0 min to 15 min, a linear gradient from 92% A–8% B to 2% A–98% B; from 15 min to 25 min, a linear gradient from 2% A–98% B to 8% A–92% B; and from 25 min to 32 min, a constant proportion of 92% A–8% B. HPLC was carried out at a flow rate of 0.8 ml/min with UV detection at 222 nm using an Agilent 1120 HPLC system (Agilent Technologies, Palo Alto, CA). The identities of compounds were confirmed by liquid chromatography-mass spectrometry (LC-MS) analysis performed on an LCMS-2010A system (Shimadzu, Japan).
Assay of transcript levels by RT-PCR amplification.
Total RNAs of three wild-type strains and nine recombinant strains were isolated from mycelia in the fermentation cultures. An additional purification step was carried out by using TRIzol reagent (catalog no. 15596-026; Invitrogen). To obtain cDNAs, DNase treatment and reverse transcription were performed by using random primers (catalog no. C1181; Promega) and Moloney murine leukemia virus (M-MLV) reverse transcriptase (catalog no. M1701; Promega) according to the manufacturer's instructions. The transcript levels of the ECB deacylase gene were assayed on a Rotor-Gene RG-3000A instrument (Corbett Research Co., Australia), using real-time quantitative PCR and the 2−ΔΔCT method (23). Reverse transcription-PCR (RT-PCR) amplification was performed on each 25 μl of the mixture (consisting of 1 μg/ml of template cDNA, 2× SYBR Premix [Shanghai Xinghan Sci-Tech Co., Shanghai, China] and 0.4 μM forward and reverse primers) with the following program: 95°C for 3 min, followed by 30 cycles of 94°C for 20 s, 60°C for 20 s, and 72°C for 15 s. For assessment of the transcript level of the ECB deacylase gene, the 244-bp internal fragment was amplified from cDNA of the recombinant strains and wild-type A. utahensis NRRL 12052 using primers 5′-CGCATGTACGAGGACGTCAC-3′ and 5′-GCACAGCGGGGACGCTGA-3′. For assessment of the transcript level of the 16S rRNA gene (as a control), the 242-bp internal fragments were amplified from cDNA of S. lividans TK24 and its recombinant strains using primers 5′-GCAATCTGCCCTTCACTCTG-3′ and 5′-TATTCCCCACTGCTGCCTCC-3′; the 285-bp internal fragments were amplified from cDNA of S. albus and its recombinant strains using primers 5′-GCCGATACTGACGCTGAGGA-3′ and 5′-GGACCCTGTCTCCAGAGTTTTC-3′; and the 255-bp internal fragments were amplified from cDNA of A. utahensis NRRL 12052 and its recombinant strains using primers 5′-GGAGGCAGCAGTGGGGAAT-3′ and 5′-TGAGCCTCGGGATTTCACATT-3′.
RESULTS AND DISCUSSION
Qualitative and quantitative analysis of ECB and the ECB nucleus.
ECB and its nucleus were extracted from a bioconversion mixture, and their identities were determined according to the method described above. As shown in Fig. 2, analysis of the samples revealed that the product and the remaining substrate ECB had the same retention times as those of the standard ECB nucleus (6.75 min) and ECB (22.80 min), respectively. Their identities were further confirmed by electrospray ionization-quadrupole time of flight (ESI–Q-TOF)–MS analysis, showing (M + Na)+ ions at m/z 820.40, consistent with the molecular formula C34H51N7O15 for the ECB nucleus, and (M + Na)+ ions at m/z 1,082.61, consistent with the molecular formula C52H81N7O16 for ECB. The concentration of the ECB nucleus in the reaction mixture was calculated according to the standard curve of the reference ECB nucleus. The methods described here for qualitative and quantitative analysis of ECB and the ECB nucleus were applied to the biotransformation reaction mixture of A. utahensis, S. lividans, S. albus, and the recombinant strains obtained in this study.
Fig 2.

HPLC analysis of ECB nucleus production in a reaction system with whole cells of control strain A. utahensis NRRL 12052. Standard ECB (▽) and ECB nucleus (▼) were used as controls. (I) Standard ECB; (II) standard ECB nucleus; (III) A. utahensis NRRL 12052.
Overexpression of the ECB deacylase gene in A. utahensis NRRL 12052.
Enzymes can be overproduced by increasing the gene copy number. To investigate the effect of deacylase gene dosage, pYG2003 and pYG2007, which contain one and two deacylase gene expression cassettes, respectively, under the control of a PermE* promoter, were constructed in pSET152 and were introduced into A. utahensis NRRL 12052 by a ΦC31-directed site-specific recombination event, yielding recombinant strains DYG2001, containing two copies of the deacylase gene, and DYG2003, containing three copies of the deacylase gene. To express deacylase under the control of the native promoter, pTG2005 was constructed in pSET152 and was introduced into A. utahensis NRRL 12052, yielding recombinant strain DYG2002, which contained two copies of the deacylase gene (Fig. 3A). The 3.3-kb, 4.0-kb, and 6.6-kb fragments could be amplified separately from genomic DNAs of the recombinant strains mentioned above by PCR using primers M13-47 and RV-M. Sequencing showed that these recombinant strains had the designed genotypes (Fig. 4). Besides this, there were no apparent differences in growth characteristics or morphology.
Fig 3.

Genotypes and phenotypes of the recombinant strains. (A) Predicted fragment sizes for PCR analysis. Each wavy line indicates the DNA fragment of the vector integrated by ΦC31-directed site-specific recombination. (B) HPLC analysis of ECB nucleus production in a reaction system with ECB (▽) and the ECB nucleus (▼).
Fig 4.
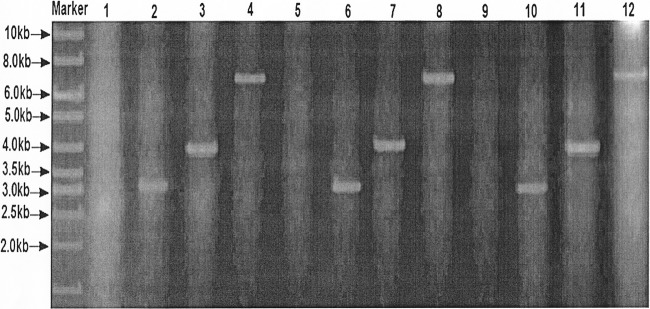
PCR analysis with genomic DNA from S. lividans TK24, S. albus, A. utahensis NRRL 12052, or mutants as the template. By use of primers M13-47 (5′-CGCCAGGGTTTTCCCAGTCACGAC-3′) and RV-M (5′-GAGCGGATAACAATTTCACACAGG-3′), the genotypes of the recombinant strains were confirmed by PCR to be well in agreement with their designed genetic patterns, demonstrating that the ΦC31-directed site-specific recombination event had taken place. Lane 1, no product for wild-type A. utahensis; lane 2, 3.3-kb product for DYG2001; lane 3, 4-kb product for DYG2002; lane 4, 6.6-kb product for DYG2003; lane 5, no product for wild-type S. lividans TK24; lane 6, 3.3-kb product for DYG2004; lane 7, 4-kb product for DYG2005; lane 8, 6.6-kb product for DYG2006; lane 9, no product for wild-type S. albus; lane 10, 3.3-kb product for DYG2007; lane 11, 4.0-kb product for DYG2008; lane 12, 6.6-kb product for DYG2003. All PCR products were sequenced to confirm each mutation.
Based on these results, we determined the bioconversion efficiency of each recombinant strain. As shown in Fig. 5, the efficiency of bioconversion of ECB was doubled in DYG2001 and DYG2002. These observations may mean that insufficient ECB deacylase activity was present in vivo. This idea is supported by other results. Even higher bioconversion efficiency was observed for DYG2003, containing three copies of the deacylase gene (Fig. 5). In our study, we found a decline in the level of the ECB substrate in the control system containing only the substrate and the bioconversion buffer, while the ECB nucleus product could be accumulated stably. In the A. utahensis NRRL 12052 bioconversion system, we found both the ECB nucleus product and the ECB substrate, while no visible ECB substrate was detected in the three recombinant strains upon HPLC analysis, as shown in Fig. 3B.
Fig 5.

Production of the ECB nucleus and ratio of the target gene transcript level to the transcript level of 16S rRNA, used as a control (relative mRNA), in reaction systems with whole cells of the different strains. Error bars represent standard deviations. For each strain, the experiment was carried out independently three times.
Another interesting question is the means of integrating the deacylase gene into A. utahensis and Streptomyces. As has been observed previously with 4′-epidaunorubicin production in Streptomyces coeruleorubidus (20), the ECB deacylase gene could be stably integrated into the genomes of host strains by a ΦC31-directed site-specific recombination event. Thus, it is not necessary to use a culture medium with apramycin resistance for the fermentation of the recombinant strains. Such a medium without apramycin helps cut the cost of ECB nucleus production on an industrial scale.
Heterologous expression of the ECB deacylase gene controlled by different promoters.
Streptomyces spp. are well-known producers of biologically active compounds and enzymes (24–26). Techniques for the cultivation of these organisms on an industrial scale are well established, so these species are especially attractive as hosts for heterologous products (27). Different species of Streptomyces were screened as heterologous expression hosts by using the same bioconversion method as A. utahensis. For the two species examined, S. lividans (strain TK24) and S. albus, no ECB nucleus or ECB from whole cells was detected in the reaction mixtures. The PermE* promoter has been used frequently as a strong constitutive promoter for native and heterologous genes in Streptomyces species and related bacteria (28). To compare the expression levels of the ECB deacylase gene under the control of its native promoter and under the control of a PermE* promoter, the genes were cloned into pSET152 and were then transformed into the two Streptomyces species by a ΦC31-directed site-specific recombination event to generate four new strains: DYG2004, DYG2005, DYG2007, and DYG2008. Another expression plasmid, pYG2007, with two ECB deacylase gene expression cassettes controlled by PermE*, was constructed and was introduced into S. lividans TK24 and S. albus, respectively, to yield DYG2006 and DYG2009, containing two copies of the deacylase gene (Fig. 3A). The genotypes of these recombinant strains were verified by PCR amplification, whereas no PCR products were isolated from the wild-type strains, as expected (Fig. 4). Subsequently, the bioconversion efficiencies of these recombinant strains were determined, and each reaction mixture was analyzed by HPLC. HPLC analysis revealed that the bioconversion efficiencies of these recombinant strains were enhanced over that of the original strain, A. utahensis NRRL 12052. More importantly, the bioconversion efficiencies of the recombinant strains DYG2004 and DYG2007, containing PermE*-controlled ECB deacylase, were higher than those of strains DYG2005 and DYG2008, in which the ECB deacylase gene was controlled by its native promoter. Strains DYG2006 and DYG2009, containing two copies of the deacylase gene, have higher bioconversion efficiencies than strains with one deacylase gene copy (Fig. 5). These findings demonstrated that PermE* was superior to the native promoter in the production of ECB deacylase for heterologous expression or overexpression and that there was potentially an enhancement of bioconversion efficiency and an improvement in the production of ECB deacylase due to the increase in gene copy number. As has been observed previously, the Streptomyces genome may contain multiple pseudo-attB sites. However, the frequency of integration of pSET152 into the ΔattB strain with additional pseudo-attB sites is reduced approximately 300-fold (29). In our study, there are strong positive correlations between the gene copy numbers of the recombinant strains and the transcript levels of the ECB deacylase gene. Therefore, there may be only one integrated gene. Of all the recombinant strains, DYG2003 from A. utahensis NRRL 12052 showed the highest bioconversion efficiency. A possible reason is that the ECB deacylase gene can be expressed better in A. utahensis NRRL 12052 and that DYG2003 contains more deacylase gene copies than NRRL 12052.
Optimal conditions for biotransformation of A. utahensis.
For the cultivation of A. utahensis and its recombinant strains, the new fermentation medium formulation based on 3.0% sucrose, 2.0% cottonseed meal, 0.12% K2HPO4·3H2O, and 0.05% KH2PO4·3H2O was a good alternative solution for the pigmentation of strain DYG2003 (unpublished data). As a result, the growth rate and mycelium volume of A. utahensis and its recombinant strains increased dramatically. Under the new culture conditions, the concentration of mycelium in the fermentation broth was 30% higher than that under the original conditions in the same fermentation time. For the biotransformation reaction, the optimal temperature was 25°C and the optimal pH was 4.5 (Fig. 6A and B). The optimum concentration of ECB in the reaction system was detected by additions of different ECB quantities under the optimized conditions (pH 4 and 25°C) for further evaluation of the bioconversion efficiency of the recombinant strain DYG2003 and the original strain A. utahensis NRRL 12052. With whole cells of NRRL 12052, the production of the ECB nucleus in reaction mixtures could rise as high as 4.21 g/liter when a suitable concentration of ECB (8 g/liter) was added to the reaction system (Fig. 6C). Additionally, ethanol is more economical and beneficial than DMSO as the cosolvent used in the reaction system to simplify the separation and purification of the product.
Fig 6.

Effects of temperature, pH, and the concentration of the ECB substrate on the biotransformation reaction. (A) Temperature course of the biotransformation reaction at pH 6.8. (B) pH course of the biotransformation reaction at 30°C. (C) Effects of different concentrations of the ECB substrate under optimized conditions (pH 4 and 25°C). Error bars represent standard deviations.
Assay of the transcript levels of the gene by RT-PCR amplification.
To assess the effects of different promoters and gene doses on the increase in the amount of enzyme produced, RT-PCR amplification for analysis of the transcription levels of the ECB deacylase gene was performed on the total RNAs extracted from cells of each strain investigated in this study. The transcript levels of the gene fragment that encodes 16S rRNA served as a control. As shown in Fig. 5, the introduction of one or two additional copies of the ECB deacylase gene caused significant increases in the transcript level of the gene. As expected, no ECB deacylase mRNA was detected for wild-type S. lividans and S. albus. The ratios of the transcript levels of the ECB deacylase gene under the control of PermE* in the S. lividans and S. albus heterologous expression hosts to the transcript level of the control 16S rRNA gene were as high as 0.66 and 0.87, respectively, while those of the gene controlled by its native promoter were as high as 0.46 and 0.84, respectively. Duplication and triplication of the gene resulted in increases as high as 2.00-fold and 6.94-fold in A. utahensis NRRL 12052. These results indicate that increasing the copy number of the gene encoding ECB deacylase leads to a significant increase in the transcript level of this gene, especially when it is controlled by the PermE* promoter. The transcript levels of the gene meet the expectation of increased efficiency of bioconversion of ECB to the ECB nucleus in recombinant strains (mentioned above).
The deacylation of ECB is the starting point for the semisynthesis of anidulafungin. The current annual production of anidulafungin is on the order of tons, and anidulafungin is widely used as an important anti-infective drug. Synthesis of the ECB nucleus by chemical deacylation is extremely difficult, and the biotransformation method described here avoids this problem. The findings of this study could enable the engineering of commercially useful ECB nucleus-overproducing strains.
ACKNOWLEDGMENTS
We thank Jia Zeng and S. Gabrielle Gladstone, Department of Biological Engineering, Utah State University, who helped with editing.
This work was supported in part by grants from the National Natural Science Foundation of China (grants 81172962, 30801449, and 81072557), the Science and Technology Commission of Shanghai Municipality (grant 11QB1406300), and the Ministry of Science and Technology of China (grant 2009ZX09301-007).
Footnotes
Published ahead of print 7 December 2012
REFERENCES
- 1. Clark TA, Hajjeh RA. 2002. Recent trends in the epidemiology of invasive mycoses. Curr. Opin. Infect. Dis. 15:569–574 [DOI] [PubMed] [Google Scholar]
- 2. Kriengkauykiat J, Ito JI, Dadwal SS. 2011. Epidemiology and treatment approaches in management of invasive fungal infections. Clin. Epidemiol. 3:175–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maertens J, Vrebos M, Boogaerts M. 2001. Assessing risk factors for systemic fungal infections. Eur. J. Cancer Care 10:56–62 [DOI] [PubMed] [Google Scholar]
- 4. Denning DW. 2003. Echinocandin antifungal drugs. Lancet 362:1142–1151 [DOI] [PubMed] [Google Scholar]
- 5. Georgopapadakou NH, Tkacz JS. 1995. The fungal cell wall as a drug target. Trends Microbiol. 3:98–104 [DOI] [PubMed] [Google Scholar]
- 6. Sucher AJ, Chahine EB, Balcer HE. 2009. Echinocandins: the newest class of antifungals. Ann. Pharmacother. 43:1647–1657 [DOI] [PubMed] [Google Scholar]
- 7. Wagner C, Graninger W, Presterl E, Joukhadar C. 2006. The echinocandins: comparison of their pharmacokinetics, pharmacodynamics and clinical applications. Pharmacology 78:161–177 [DOI] [PubMed] [Google Scholar]
- 8. Cleary JD. 2009. Echinocandins: pharmacokinetic and therapeutic issues. Curr. Med. Res. Opin. 25:1741–1750 [PubMed] [Google Scholar]
- 9. Denning DW. 2002. Echinocandins: a new class of antifungal. J. Antimicrob. Chemother. 49:889–891 [DOI] [PubMed] [Google Scholar]
- 10. Fera MT, La Camera E, De Sarro A. 2009. New triazoles and echinocandins: mode of action, in vitro activity and mechanisms of resistance. Expert Rev. Anti Infect. Ther. 7:981–998 [DOI] [PubMed] [Google Scholar]
- 11. Nyfeler R, Keller-Schierlein W. 1974. Metabolites of microorganisms. 143. Echinocandin B, a novel polypeptide-antibiotic from Aspergillus nidulans var. echinulatus: isolation and structural components. Helv. Chim. Acta 57:2459–2477 (In German.) [DOI] [PubMed] [Google Scholar]
- 12. Debono M, Abbott BJ, Turner JR, Howard LC, Gordee RS, Hunt AS, Barnhart M, Molloy RM, Willard KE, Fukuda D, Butler TF, Zeckner DJ. 1988. Synthesis and evaluation of LY121019, a member of a series of semisynthetic analogues of the antifungal lipopeptide echinocandin Ba. Ann. N. Y. Acad. Sci. 544:152–167 [DOI] [PubMed] [Google Scholar]
- 13. Onishi J, Meinz M, Thompson J, Curotto J, Dreikorn S, Rosenbach M, Douglas C, Abruzzo G, Flattery A, Kong L, Cabello A, Vicente F, Pelaez F, Diez MT, Martin I, Bills G, Giacobbe R, Dombrowski A, Schwartz R, Morris S, Harris G, Tsipouras A, Wilson K, Kurtz MB. 2000. Discovery of novel antifungal (1,3)-β-d-glucan synthase inhibitors. Antimicrob. Agents Chemother. 44:368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petraitiene R, Petraitis V, Groll AH, Candelario M, Sein T, Bell A, Lyman CA, McMillian CL, Bacher J, Walsh TJ. 1999. Antifungal activity of LY303366, a novel echinocandin B, in experimental disseminated candidiasis in rabbits. Antimicrob. Agents Chemother. 43:2148–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uzun O, Kocagöz S, Cetinkaya Y, Arikan S, Unal S. 1997. In vitro activity of a new echinocandin, LY303366, compared with those of amphotericin B and fluconazole against clinical yeast isolates. Antimicrob. Agents Chemother. 41:1156–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boeck LD, Fukuda DS, Abbott BJ, Debono M. 1989. Deacylation of echinocandin B by Actinoplanes utahensis. J. Antibiot. 42:382–388 [DOI] [PubMed] [Google Scholar]
- 17. Kreuzman AJ, Hodges RL, Swartling JR, Pohl TE, Ghag SK, Baker PJ, McGilvray D, Yeh WK. 2000. Membrane-associated echinocandin B deacylase of Actinoplanes utahensis: purification, characterization, heterologous cloning and enzymatic deacylation reaction. J. Ind. Microbiol. Biotechnol. 24:173–180 [Google Scholar]
- 18. Romano D, Gandolfi R, Guglielmetti S, Molinari F. 2011. Enzymatic hydrolysis of capsaicins for the production of vanillylamine using ECB deacylase from Actinoplanes utahensis. Food Chem. 124:1096–1098 [Google Scholar]
- 19. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 20. Shao L, Huang J, Jing L, Chen JY, Kan SD, Wang M, Li JA, Chen DJ. 2010. Overexpression of aveBIV leading to the improvement of 4′-epidaunorubicin production in Streptomyces coeruleorubidus strain SIPI-A0707. Appl. Microbiol. Biotechnol. 87:1057–1064 [DOI] [PubMed] [Google Scholar]
- 21. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 22. Luzhetskii AN, Ostash BE, Fedorenko VA. 2001. Intergeneric conjugation Escherichia coli–Streptomyces globisporus 1912 using integrative plasmid pSET152 and its derivatives. Russ. J. Genet. 37:1123–1129 [PubMed] [Google Scholar]
- 23. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding W, Lei C, He Q, Zhang Q, Bi Y, Liu W. 2010. Insights into bacterial 6-methylsalicylic acid synthase and its engineering to orsellinic acid synthase for spirotetronate generation. Chem. Biol. 17:495–503 [DOI] [PubMed] [Google Scholar]
- 25. Shao L, Qu XD, Jia XY, Zhao QF, Tian ZH, Wang M, Tang GL, Liu W. 2006. Cloning and characterization of a bacterial iterative type I polyketide synthase gene encoding the 6-methylsalicyclic acid synthase. Biochem. Biophys. Res. Commun. 345:133–139 [DOI] [PubMed] [Google Scholar]
- 26. Ueda S, Shibata T, Ito K, Oohata N, Yamashita M, Hino M, Yamada M, Isogai Y, Hashimoto S. 2011. Cloning and expression of the FR901379 acylase gene from Streptomyces sp. no. 6907. J. Antibiot. 64:169–175 [DOI] [PubMed] [Google Scholar]
- 27. Inokoshi J, Takeshima H, Ikeda H, Ōmura S. 1993. Efficient production of aculeacin A acylase in recombinant Streptomyces strains. Appl. Microbiol. Biotechnol. 39:532–536 [Google Scholar]
- 28. Pan HX, Li JA, He NJ, Chen JY, Zhou YM, Shao L, Chen DJ. 2011. Improvement of spinosad production by overexpression of gtt and gdh controlled by promoter PermE* in Saccharopolyspora spinosa SIPI-A2090. Biotechnol. Lett. 33:733–739 [DOI] [PubMed] [Google Scholar]
- 29. Combes P, Till R, Bee S, Smith MCM. 2002. The Streptomyces genome contains multiple pseudo-attB sites for the ΦC31-encoded site-specific recombination system. J. Bacteriol. 184:5746–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]