

Abstract
Human adenoviruses (HAdV) are important pathogens in both industrialized and developing nations. HAdV has been shown to be relatively resistant to monochromatic UVC light. Polychromatic UVC light, in contrast, is a more effective means of disinfection, presumably due to the involvement of viral proteins in the inactivation mechanism. Solar disinfection of HAdV, finally, is only poorly understood. In this paper, the kinetics and mechanism of HAdV inactivation by UVC light and direct and indirect solar disinfection are elucidated. PCR and mass spectrometry were employed to quantify the extent of genome and protein degradation and to localize the affected regions in the HAdV proteins. For this purpose, we used for the first time an approach involving stable isotope labeling by amino acids in cell culture (SILAC) of a human virus. Inactivation by UVC light and the full sunlight spectrum were found to efficiently inactivate HAdV, whereas UVA-visible light only caused inactivation in the presence of external sensitizers (indirect solar disinfection). Genome damage was significant for UVC but was less important for solar disinfection. In contrast, indirect solar disinfection exhibited extensive protein degradation. In particular, the fiber protein and the amino acids responsible for host binding within the fiber protein were shown to degrade. In addition, the central domain of the penton protein was damaged, which may inhibit interactions with the fiber protein and lead to a disruption of the initial stages of infection. Damage to the hexon protein, however, appeared to affect only regions not directly involved in the infectious cycle.
INTRODUCTION
As important human pathogens, human adenoviruses (HAdV) present a public health risk in both industrialized and developing countries, causing different types of illnesses. Besides severe respiratory diseases like pneumonia (HAdV serotypes B and C) and eye infections (serotypes B and D), some serotypes (F and G) cause viral gastroenteritis with the same symptoms as norovirus and rotavirus infections (1). Adenovirus gastroenteritis can eventually lead to death by dehydration in people who have limited access to safe drinking water, especially infants, young children, and disabled and elderly people in developing countries (2–4). Adenovirus is transmitted from person to person or through contaminated beverages, drinking water, food, and recreational water (2, 5, 6). As one of the few enteric viruses with double-stranded DNA, adenovirus is known for its stability and persistence in the aquatic environment compared to other enteric viruses (7–9).
Disinfection and control of enteric adenoviruses in water are therefore of vital importance. To date, research on the kinetics and mechanisms of virus inactivation has focused on drinking water treatment methods typically applied in industrialized countries. Adenoviruses generally show considerable resistance to disinfection with monochromatic UVC light and monochloramines (10–14) but are readily inactivated by free chlorine (12, 15).
Resistance to monochromatic UVC light is likely based on the fact that this treatment damages mainly DNA, while the proteins stay intact: adenoviruses are known to successfully infect and replicate inside a host even when their DNA is heavily damaged (16). The infectivity and replication capacity of adenoviruses therefore do not depend on an intact DNA. As long as the structural proteins associated with the infection apparatus (binding of the fiber head to the host receptor and downstream processes within the cell) are functional, adenovirus may survive extensive DNA damage, as the DNA damage can be repaired during host passage.
Only little information is available for disinfection of viruses during alternative, low-cost drinking water treatments, such as direct and indirect solar disinfection (17). Direct solar disinfection refers to inactivation due to the interaction of sunlight in the UVB range with the viral genome and proteins. Indirect solar disinfection relies on the production of oxidants upon irradiation of chromophores in solution (exogenous) or within the organism (endogenous) and subsequent energy or electron transfer to dissolved oxygen or other solution constituents. These oxidants, e.g., 1O2 (18), then react with the virus constituents and cause inactivation. An enhanced efficiency of solar disinfection compared to monochromatic UVC light may be found if, in addition to causing genome damage, solar disinfection damages vitally important proteins. A similar effect was previously observed for the inactivation of Escherichia coli in sunlight (19, 20) as well as for the inactivation of adenovirus by medium-pressure (polychromatic) UV light (21).
The goal of our work was to determine the efficiency and mechanisms of direct and indirect solar disinfection of human adenovirus type 2 in comparison with monochromatic UVC light. To obtain a complete picture of the molecular mechanism, we monitored genome damage and performed an in-depth analysis of protein damage. The novelty of our approach is that we not only quantified the extent of damage but also determined the location of the protein regions most affected by light treatment. To this end, we used a SILAC (stable isotope labeling by amino acids in cell culture) approach (22) to perform the quantification of our proteomic data. In the approach presented here, protein degradation of a “heavy” (i.e., isotopically labeled) virus upon treatment was monitored, whereas the “light” (i.e., native) virus served as an internal standard for quantification. Once combined, both heavy and light protein samples were enzymatically digested into small peptides and jointly analyzed by high-resolution, high-mass-accuracy mass spectrometry (MS). Pairs of chemically identical peptides of different stable isotope compositions can be distinguished by their accurate mass differences. Quantification of protein and peptide decay upon treatment was thus performed by comparing the peak intensity of every detected peptide of the treated, heavy labeled virus to that of the untreated, nonlabeled internal standard.
Infection by adenoviruses is mediated predominantly by the penton and the fiber capsid proteins (23). Thus, modifications to these structures are of particular interest to explain the mechanisms of adenovirus inactivation. Susceptible amino acids and peptides located in these structures may play an important role in adenovirus inactivation processes during irradiation with sunlight and UV light. Tyrosine, histidine, and lysine in the fiber head are crucial for host binding in adenovirus (24), and in particular, tyrosine is susceptible to degradation by light (25). Finally, the efficiency of adenovirus inactivation in a setting representative of solar disinfection of drinking water (SODIS), hence exposed to sunlight in a polyethylene terephthalate (PET) bottle, was investigated.
MATERIALS AND METHODS
Viruses, cells, and cultivation conditions.
Human adenovirus type 2 (HAdV-2) was cultivated and propagated on A549 human lung adenocarcinoma epithelial cells in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing penicillin (50 U/ml), streptomycin (50 μg/ml), and 10% fetal bovine serum (FBS) and incubated at 37°C in 5% CO2 and 95% humidity.
Propagation of HAdV-2.
A549 cells were cultivated to 95% confluence in a 10-ml cell culture flask. The medium was then discharged and replaced by medium containing 2% FBS. Viruses (108 to 109 PFU) were added to the culture. Cultures were incubated until cytopathic effects (CPE) indicated dying of cells and 80% of the cell layer was detached. After 3 to 5 days, cells were lysed by three freeze-thaw cycles, debris was centrifuged down at 13,000 × g, and the supernatant was membrane filtered (0.22 μm; Millipore). Viruses were washed three times with virus dilution buffer (5 mM NaHPO42−, 10 mM NaCl [pH 7.4]) and concentrated 10× with 15-ml centrifugal membrane filter units (100-kDa cutoff; Millipore). Virus stocks (1011 · PFU ml−1) were stored at 4°C.
MPNCU assay.
A 95% confluent A549 cell monolayer was prepared in 96-well plates with flat-bottom wells. HAdV-2 samples were diluted over a 10-fold dilution series in medium containing 2% FBS. Medium on cell monolayers was replaced with 150 μl of virus dilutions. After an incubation time of 7 to 10 days, cytopathic units (CU) were used to differentiate between positive and negative wells, and the corresponding HAdV-2 concentration was evaluated with a most probable number (MPN) table (MPNCU · ml−1). All samples were enumerated in triplicates.
Irradiation setting for sunlight experiments.
Two-milliliter samples containing 1 × 108 to 5 × 108 PFU · ml−1 in virus dilution buffer were exposed in 10-ml glass beakers to light emitted from a Sun 2000 solar simulator (Abet Technologies, Milford, CT) equipped with a 1-kW Xe lamp and an air mass (AM) 1.5 filter. Samples were constantly stirred during the experimental time, and at each time point, one beaker was withdrawn for analysis. A UVB cutoff filter was used for experiments considering sunlight in the UVA-visible range alone (here denoted UVA samples). The spectra of light produced by the solar simulator are displayed in Fig. 1. Rose Bengal (RB; Acros Organics) was added to some UVA samples as a 1O2 sensitizer, as described previously (26), to simulate indirect solar disinfection conditions (here denoted UVA+RB samples). Control experiments were conducted in the absence of RB or in the absence of light. For PET experiments, 200 ml of virus sample was exposed in a 500-ml transparent PET bottle. The incoming fluence was determined by a radiometer (model ILT-900-R; International Light) over a range of 280 to 800 nm. Samples aiming for a maximal reduction of 6 log10 units were exposed to doses of simulated sunlight of 66 kJ · m−2 for light in the UVA spectrum only (wavelengths greater 310 nm) and 17 kJ · m−2 for the full solar spectrum (including UVB).
Fig 1.
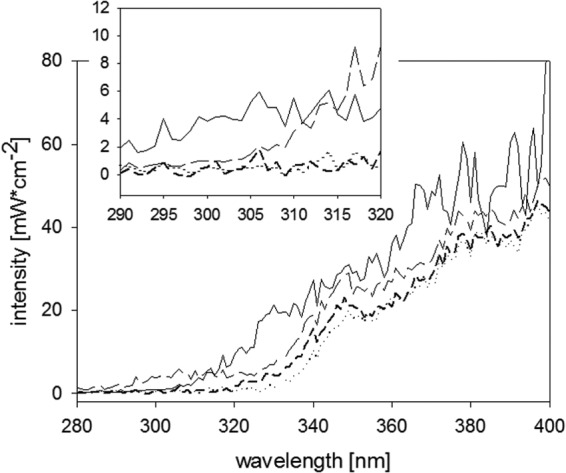
UVA/B portion of the solar spectrum in May in Lausanne, Switzerland (solid line), and solar simulator spectrum with an AM 1.5 filter (—— ——), with AM 1.5 and UVB-blocking filters (·····), and with an AM 1.5 filter and in a PET bottle (– – –). The inset shows an enhanced view of the UVB region. The natural atmosphere, especially ozone, cuts off UVB light at 310 nm, whereas PET has a cutoff of around 320 nm. Our solar simulator has a slightly higher UVB content than the natural spectrum.
Irradiation setting for UVC light.
Viruses were diluted to a concentration of 1 × 108 to 5 × 108 PFU · ml−1 in virus dilution buffer. Two-milliliter aliquots were distributed in black 10-ml glass beakers and placed into a custom-built device containing low-pressure 18-W UVC lamps (253.7 nm, model TUV T8; Philips) in a quasiparallel beam setup (27). Samples were constantly stirred during the experimental time, and at each time point, one beaker was withdrawn for analysis. The fluence was measured by actinometry (28) in parallel with inactivation experiments, and doses were corrected for light shielding by virus particles, as described previously (29). Samples aiming for a maximal reduction of 6 log10 units were exposed to a UVC dose of 3 kJ · m−2.
qPCR.
Viral DNA was extracted by using a PureLink viral RNA/DNA extraction kit (Invitrogen) according to the manufacturer's instructions. Primers were designed with the help of the Primer3 program (http://frodo.wi.mit.edu/primer3/). Two amplicons were chosen, which covered 317 to 330 bp close to the 3′ and 5′ ends of the adenoviral genome (forward primer 4719 [TGCCAGACTGCGGTATAATG] and reverse primer 5035 [CAGGGAAAACATGCAAGTCA], and forward primer 29948 [CATCGCCTTCATTCAGTTCA] and reverse primer 30277 [TATGGCTAGGGCAAAAATGG]). Quantitative PCR (qPCR) was performed on a RotorGene 3000 quantitative PCR platform (Corbett Life Science, Sydney, Australia). Each qPCR sample was run in a 15-μl total volume comprising 7.5 μl of 2× One Step SYBR RTPCR buffer III, 0.3 μl of TaKaRa ExTaqHS (5 U · μl−1), 0.3 μl of 10 μM forward and reverse primers, 3.6 μl of water, and 3 μl of sample (TaKaRa Bio, Shiga, Japan). The following thermocycling conditions were used: 20 s at 95°C and 45 cycles of 95°C for 15 s, 60°C for 20 s, and 72°C for 20 s, followed by a melting ramp from 72 to 95°C, holding for 45 s on the first step (72°C), followed by 5-s holds at all subsequent temperatures. The number of intact amplicons was determined relative to the untreated control. Specifically, the untreated samples were analyzed in undiluted and serially diluted (10- to 106-fold) forms, and a calibration curve was established. The qPCR signals obtained for light-treated samples were then quantified by means of this calibration curve. The extent of damage to each amplicon was determined by comparing the qPCR signal of the treated samples to that of the untreated control (qPCR/qPCR0). Two to three rounds of experiments were run for qPCR analysis of each treatment, with duplicate samples in each round. Different rounds exhibited very good reproducibility, and representative examples are shown here.
Cultivation of heavy adenovirus for quantitative SILAC analysis.
A549 cells were grown for six passages (10 days) using SILAC-DMEM containing 0.1 mg · ml−1 heavy [13C6]l-lysine–2HCl and 0.1 mg · ml−1 heavy [13C6]l-arginine–HCl or the light versions of these amino acids (Pierce, Thermo Fisher Scientific). Furthermore, the medium was supplemented with 10% dialyzed FBS. Viruses were propagated on these cells as mentioned above, yielding an incorporation of heavy amino acids of >99%. Heavy viruses were treated as described above, and the same amounts of light viruses were added as internal standard after the treatment. For each treatment, 5 replicate samples were prepared. Samples were stored at −20°C in SDS loading buffer before SDS-PAGE was performed.
SDS-PAGE and sample preparation for LC-MS/MS analysis.
A detailed protocol for SDS-PAGE and sample preparation for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was described previously (20). In short, samples from irradiation experiments were concentrated with 0.5-ml centrifugal membrane filter units (100-kDa cutoff; Millipore), and per lane, a volume corresponding to 1 × 109 to 5 × 109 PFU was mixed with SDS sample loading buffer, heated for 5 min to 95°C, and loaded onto small 12% SDS-PAGE gels. Instead of analyzing the whole molecular mass range, three regions were selected according to their molecular masses, with the aim to concentrate on the most abundant and interesting proteins: (i) 20 to 30 kDa, corresponding to minor capsid pVI, pVIII, and pVII; (ii) 60 to 75 kDa, corresponding to early E2A protein, fiber, penton, minor capsid pIIIa, and DNA terminal protein; and (iii) a single band at 109 kDa containing the hexon protein. The protein quantity in each band was determined by densitometry. Samples were reduced in 10 mM dithioerythritol (DTE; Fisher Scientific) and alkylated in 55 mM iodoacetamide (IAA; Sigma-Aldrich). Gel pieces were dried and then incubated overnight with trypsin (12.5 ng/μl; Promega, Madison, WI) at 37°C. Peptides were extracted from gel slices and dried in a Speed Vac.
Nano-LC-MS/MS and data analysis.
Tryptic peptides were resuspended and were analyzed by nano-LC-MS/MS using an LTQ-Orbitrap XL mass spectrometer (Thermo Fischer Scientific, San Jose, CA) equipped with an ultraperformance LC (UPLC) system (NanoAcquity; Waters, Hertfordshire, United Kingdom). Peptides were trapped on a homemade 3-μm 200-Å Magic C18 AQ 0.1- by 20-mm precolumn and separated on a homemade 5-μm 100-Å Magic C18 AQ 0.75- by 150-mm column. Mass spectra were acquired in the data-dependent mode with an automatic switch between a full scan and up to 10 data-dependent MS/MS scans. The survey scans were acquired at a resolution of 60,000 at m/z 400 in the Orbitrap analyzer, while up to 10 MS/MS spectra were acquired at low resolution in the linear ion trap. Data analysis was performed with MaxQuant (v1.2.2.5) (30). Proteins were identified by searching MS and MS/MS data of peptides against the Uniprot Human database, release 2011_06, containing human adenovirus 2 proteins. Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation and N-terminal acetylation were set as variable modifications. A maximum of 2 missed cleavages was allowed. Maximum false discovery rates (FDR) were set to 0.01 at both the peptide and the protein levels. The minimum required peptide length was 6 amino acids, and at least two (unique plus razor) peptides were used for protein identification.
Quantification of SILAC pairs was performed with MaxQuant using a minimum ratio count of 2, with the “requantify option” switched on. Contaminants and reverse hits were filtered as the proteins with a posterior error probability (PEP) of the identification of >0.1. Peptides with at least three quantifiable ratios per condition were kept for further statistical analyses with R (31). Normalized heavy/light peptide ratios before and after treatment were determined and compared by a pairwise t test (inequal variances; FDR of 0.05). Degradation of the whole protein was determined based on the decay of its individual peptides.
RESULTS
Inactivation kinetics of HAdV-2 during solar and UVC disinfection.
Inactivation was first order for all light sources and conditions (Fig. 2 and 3). The fastest inactivation was observed with UVC light, which caused a 99.99% (4-log10) reduction in MPNCU/ml of HAdV-2 after a dose of 2 kJ · m−2. Sunlight was slightly less efficient and reached this level of inactivation at a dose of 11 kJ · m−2. Samples irradiated only by light in the UVA range exhibited marginal inactivation, resulting in a 90% (1-log10) inactivation after a dose of 1,200 kJ · m−2. If the UVA samples were amended with RB, however, 90% and 99.99% inactivation were reached at light doses of 6 and 35 kJ · m−2, respectively (although this may depend not only on fluence but also on the concentration of RB). Indirect inactivation by singlet oxygen produced during the irradiation of RB thus increased the UVA treatment efficiency about 200-fold. HAdV-2 in local drinking water and stored in a PET bottle was inactivated by 90% after a 1-day exposure to sunlight, corresponding to a light dose of 1,200 kJ/m2 (Fig. 2 and 3).
Fig 2.
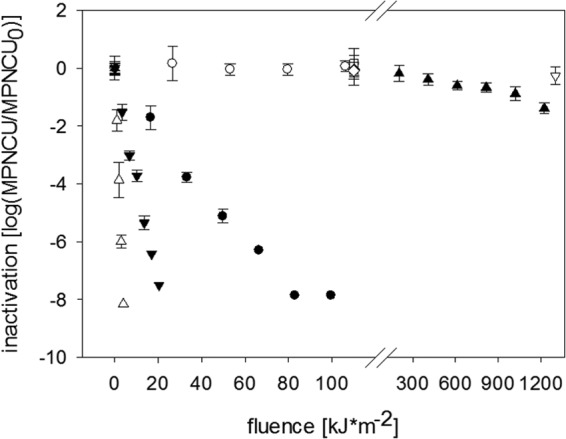
Inactivation kinetics of HAdV treated with UVA light (○), UVA+RB (●), full sunlight spectrum (▼), UVC light (△), and SODIS (full-spectrum illumination of sample in a PET bottle) (▲). Also shown are the corresponding dark controls (♢ and ▽) and RB-free controls (□). Error bars indicate standard deviations calculated from triplicate samples.
Fig 3.
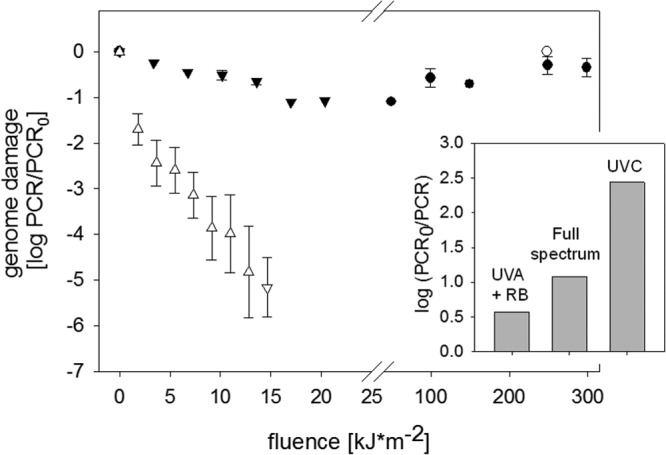
Decrease in PCR signal upon HAdV inactivation at different fluences. Symbols correspond to UVA light (○), UVA+RB (●), full sunlight spectrum (▽), and UVC light (△). The PCR signal corresponds to the average signal of the two amplicons considered. Error bars indicate the upper and lower measurements obtained by the two amplicons. The inset shows a comparison of the genome damage incurred upon a 7-log10 inactivation by indirect solar disinfection (UVA+RB), the full solar spectrum, and UVC light.
Quantification of genome damage during irradiation.
The degradation of the genome was determined by qPCR. Here it should be noted that the PCR assay covered only two small genome sections, which combined make up 2% of the entire viral genome. The measured genome damage is therefore not a direct reflection of the total genome damage incurred by the virus but rather of the damage incident to the small sections covered by the PCR assay.
If analyzed as a function of fluence, genome degradation was fastest for UVC light (Fig. 3). A difference of up to 2 log units was observed in the decay of the two amplicons. The faster decay of the amplicon closer to the 3′ end may result from its larger size as well its larger number of adjacent thymidines, which form thymidine dimers upon treatment by UVC light. A small yet discernible decay was also observed for inactivation by the full sunlight spectrum, whereas indirect solar disinfection led to only minor genome degradation. In the absence of RB, UVA light did not cause measurable genome degradation. This is in agreement with the inability of UVA light alone to cause inactivation (Fig. 2).
The fluences for these different treatments were calculated based on light of different wavelengths, which differ in their ability to induce genome damage (32). Fluence alone is therefore not an ideal measure to compare genome degradation among different light sources. For a more direct comparison, we additionally evaluated the extent of genome damage in each treatment for a given level of inactivation (ca. 7 log10 units). As can be seen in the inset of Fig. 3, a 7-log10 inactivation was accompanied by the most genome damage for the UVC treatment, followed by full-spectrum sunlight. For indirect solar disinfection, the same level of inactivation coincided with only slight genome damage.
Qualification and quantification of protein damage during irradiation.
On SDS-PAGE gels, bands containing the HAdV-2 proteins hexon, penton/fiber, and minor capsid were discernible. These proteins were affected to different degrees during inactivation by 6 log10 units, with UVA+RB having the most pronounced effects (Fig. 4 and Table 1). Compared to the nonirradiated control, bands containing hexon, penton/fiber, and minor capsid proteins were reduced to 73%, 82%, and 80%, respectively. Meanwhile, the effect of UVA light alone was comparable to that of sunlight and UVC light, for which more than 90% of proteins remained intact.
Fig 4.

Whole-protein damage (densitometry on SDS-PAGE gels) of HAdV inactivated by UVA light (A), UVA+RB (B), full sunlight spectrum (C), and UVC light (D). Boxes indicate the location of the hexon protein (solid line), penton and fiber proteins (dashed line), and minor capsid protein (dotted line). Outermost lanes are the protein standard, with the scale (kDa) indicated in panel A. Lanes a, c, h, and m are nonirradiated controls. The remaining lanes correspond to the following fluences (in kJ/m2): b, 100; c, 200; d, 25; e, 50; f, 100; g, 200; i, 10; j, 20; k, 30; l, 60; n, 0.9; o, 1.8; p, 3.6; q, 7.3.
Table 1.
Fraction of intact protein measured in gel after inactivation by approximately 6 log10 units and in the RB-free control (no inactivation)a
| Treatment | % intact protein |
||
|---|---|---|---|
| Hexon | Penton and fiber | Minor capsid | |
| UVA | 95.21 | 98.15 | 92.84 |
| UVA+RB | 72.94 | 81.64 | 80.01 |
| Full sunlight spectrum | 92.25 | 95.05 | 89.63 |
| UVC | 92.47 | 96.60 | 91.80 |
Values were measured in bands c, g, l, and q (Fig. 4). Note that the penton and fiber proteins could not be distinguished and therefore are presented jointly.
HAdV-2 irradiated to an inactivation of 6 log10 units was subjected to quantitative SILAC analysis in order to characterize the nature of the damage. Of the targeted proteins, only the three main structural proteins, hexon, fiber, and penton base, were detectable at a high-enough intensity and reproducibility to reliably quantify degradation by protein mass spectrometry. In total, 95 unique peptides were detected for these three proteins. Coverage (fraction of the total protein sequence detected by mass spectrometry) was at 74% for the hexon, 56% for the fiber, and 42% for the penton base protein sequences, with 55, 24, and 16 unique peptides detected, respectively. These proteins were subsequently subjected to in-depth analysis at the protein and peptide levels.
At the protein level, our analysis confirmed that full-spectrum sunlight, UVA light without sensitizers, and UVC light did not cause significant degradation (Fig. 5). Specifically, full-spectrum sunlight and UVC light degraded only 3 peptides and 1 peptide, respectively, corresponding to 3% and 1% of all unique peptides detected for hexon, penton base, and fiber protein. The boxes in Fig. 5 representing these treatments thus show an insignificant change in protein integrity compared to the untreated sample. In contrast, a total of 25 peptides were damaged when HAdV-2 was treated with UVA+RB, corresponding to 26% of all unique peptides. Under this condition, the ratios of degraded to conserved peptides were 17/24 (71%) in the fiber protein, 5/16 (31%) in the penton base protein, and 3/55 (5%) in the hexon protein. Correspondingly, the boxes in Fig. 5 representing the UVA+RB condition show a stark reduction in the intact fraction of the fiber and penton base proteins upon treatment. Individual box plots of the affected peptides during UVA+RB treatment are shown in Fig. 6, and the damaged regions are visualized graphically in Fig. 7.
Fig 5.
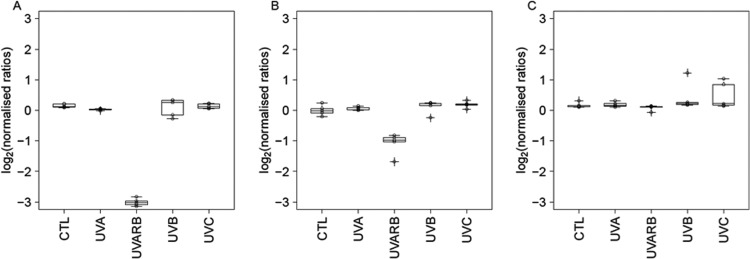
Integrity of the fiber (A), hexon (B), and penton (C) proteins in the absence of irradiation (control [CTL]) and upon treatment with UVA light, UVA+RB, full sunlight spectrum (denoted UVB), and UVC light. The graph shows the protein ratio between treated and untreated (inactivation of ca. 6 log10 units) samples on a log2 scale, measured using the SILAC method. The data are visualized as box plots based on at least four replicate samples, where the upper boundary represents the 75th percentile, the line represents the median, and the lower boundary represents the 25th percentile. A ratio of 0 indicates no change in protein integrity compared to the control sample; a ratio of <0 indicates protein degradation or modification.
Fig 6.

Summary of peptides susceptible to degradation by indirect solar disinfection (UVA+RB). The graph shows the ratios of the peptides from treated (UVA+RB) to untreated (CTL) samples, measured using the SILAC method. These peptides belong to three HAdV-2 proteins: fiber protein (A), hexon protein (B), and penton protein (C). Different data points indicate replicate samples. The data are visualized in box plots based on at least four replicate samples, where the upper boundary represents the 75th percentile, the line represents the median, and the lower boundary represents the 25th percentile.
Fig 7.
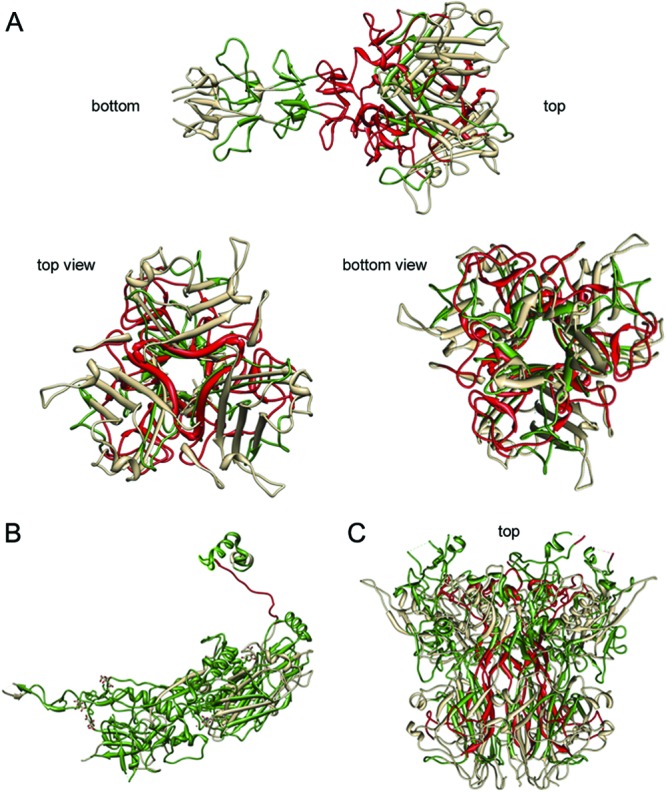
Visualizations of damaged peptides in protein structures of fiber (A), hexon (B), and penton (C) proteins after indirect sunlight disinfection (UVA+RB). Degraded peptides are shown in red, conserved peptides are shown in green, and peptides not covered by mass spectrometry are shown in gray. Images were created with Chimera (52).
DISCUSSION
Mechanisms contributing to solar disinfection of adenovirus.
HAdV-2 was readily inactivated by full-spectrum sunlight (Fig. 2), indicating immediate damage to virus components. Full-spectrum sunlight leads to direct inactivation without any need for endogenous or exogenous sensitizers. In contrast, UVA light alone showed no inactivation effect, indicating that HAdV-2 does not contain endogenous sensitizers that could contribute to endogenous indirect inactivation. With the addition of the external sensitizer Rose Bengal, HAdV were susceptible to UVA light, indicating that exogenous inactivation is taking place.
During SODIS, the water is exposed to UVA light alone, since the PET material of the bottles provides a neat UVB cutoff at 320 nm. SODIS with a 1-day exposure is thus inefficient for this virus. In comparison, E. coli is inactivated by 99.9999% (6 log10 units) with a comparable exposure (33). In some cases, natural waters might contain chromophores that act as external sensitizers, analogous to Rose Bengal, and thus facilitate a SODIS effect on HAdV-2. In addition, container materials other than PET, which are more transmissible for UVB light, may achieve more efficient virus inactivation.
Contribution of genome and protein damage to inactivation.
The different treatments exhibited different extents of genome damage when analyzed both as a function of fluence and as a function of the extent of inactivation (Fig. 3). The high damage induced by monochromatic UVC light compared to direct and indirect solar disinfection may result from two different causes. First, HAdV may be more capable of repairing UVC-induced DNA damage, such as pyrimidine dimers during host passage, than oxidative lesions induced by 1O2, such as 8-oxo-7,8-dihydroguanosine (34, 35). Hence, fewer oxidative lesions would be needed to cause inactivation than UVC-induced ones. Alternatively, the discrepancy in the extent of genome damage may point toward the contribution of protein damage to inactivation: as proposed previously by others, monochromatic UVC (254-nm, low-pressure) inactivation is caused almost exclusively by genome damage, whereas polychromatic UV (medium-pressure) inactivation additionally involved protein degradation and hence required a lesser extent of genome damage to achieve the same extent of inactivation (21, 36–41). In the treatment systems investigated here, this would thus indicate that the contribution of protein damage to inactivation was small for UVC light, greater for full-spectrum solar disinfection, and most prevalent for indirect solar disinfection. The characterization of the contribution of protein damage was thus crucial for a sound understanding of the molecular inactivation mechanism.
Our expectation regarding the involvement of proteins in the treatments investigated was corroborated by the protein loss observed on the SDS-PACE gel: only little protein degradation was found after a 6-log10 inactivation by the full sunlight spectrum or monochromatic UVC light. This finding differs from the results of others (21), who previously reported extensive protein damage in HAdV-2 upon irradiation by monochromatic UVC light at lower doses than the ones used here (ca. 7 kJ/m2). However, since the concurrent extent of inactivation was not reported, a direct comparison to our work is difficult. In contrast, protein damage was extensive for indirect solar disinfection (UVA+RB) (Fig. 4 and Table 1). However, as reported in a previous study (42), not all oxidative protein damage is of biological consequence, as viruses have been found to withstand a large extent of protein oxidation. It is thus not sufficient to quantify the extent of protein degradation to assess its impact on inactivation. Instead, it is important to relate the damaged protein regions to their location within the protein, which in turn may give an indication of the virus function affected by protein degradation.
The publicly available crystal structures of hexon, penton, and fiber proteins were retrieved from the Protein Data Bank (PDB) (accession numbers 1P2Z, 2C6S, and 1QIU) (43–45), and the functions of these proteins in virus assembly and infection were described in detail previously (46, 47). Penton and fiber capsid proteins are key players when HAdV infects host cells. They form the “vertex capsomer,” which mediates the initial stages of infection. The fiber contains the host binding sites and is responsible for binding to the host cell surface protein CAR (coxsackie and adenovirus receptor). The binding sites are most probably located on the lateral surfaces of the fiber knob (48, 49). Flexibility of the fiber shaft is crucial to facilitate orientation of the binding site toward the host cell surface. Therefore, the functionality of the fiber is central for infectivity of the virus. In the fiber, indirect solar disinfection damaged peptides containing amino acids D406, S408, P409, Y477, N482, S485, and P505 (Fig. 7). These amino acids are responsible for CAR host binding in the fiber knob of HAdV (50). In particular, tyrosine is susceptible to oxidation and degradation by light (25). Thus, indirect solar disinfection may hamper host binding and lower the infectivity of HAdV-2. The penton interacts with the integrins on the host cell surface (51) and is thus responsible for initialization of endocytosis, leading to viral uncoating, internalization, and infection of the host cell (47). Damage to the penton may prevent entry of HAdV into host cells. In our samples inactivated by indirect solar disinfection, the central domain of the penton was damaged (Fig. 7C). We therefore suggest that the interaction with the fiber protein was inhibited and that the vertex capsomer may have lost its function. Finally, the hexon proteins were damaged at a domain that likely contributes to hexon flexibility (47). These domains are important for hexon-hexon or hexon-minor coat protein IIIa and VIII interactions and therefore define the typical architecture of the virus capsid. As such, these domains are not involved in host infection mechanisms. We therefore assume that damage to these domains either has no effect on infectivity or contributes to a higher rigidity of the hexon, causing structural alterations in the capsid structure and thereby influencing virus shape and functionality in a more general way.
In summary, HAdV was susceptible to UVC light, full-spectrum sunlight, and exogenous indirect solar disinfection but not to light in the UVA-visible range only. Correspondingly, solar disinfection in PET bottles (SODIS), which are not transparent to light in the UVB range, was relatively inefficient when no external sensitizer was added. The different inactivating treatments acted by different mechanisms: while genome damage induced by UVC light caused efficient inactivation, the contribution of protein damage to the disinfection process was relevant for UVA+RB. Notably, protein damage occurred in the regions involved in host attachment and cell entry.
In this study, we describe for the first time the use of SILAC on a human virus. This method allowed us to qualitatively describe protein degradation damage and provide an unprecedented level of detail regarding the protein regions involved. In conjunction with the crystal structures and available information on protein function, we were able to identify biologically relevant effects of protein damage, leading to a sound understanding of the impact on inactivation. Overall, our results contribute to a more complete understanding of the HAdV-2 disinfection mechanism. Inclusion of other HAdV proteins not discussed in this study may augment the findings presented here.
ACKNOWLEDGMENTS
We thank the group of Rosina Girones (University of Barcelona) for providing HAdV-2 and its host cells. We acknowledge Aimee Gall and Michael Mattle for helpful advice with the experimental and culturing protocols. We acknowledge Marc Moniatte, manager of the PCF-PTP, for his support.
This work was supported by EPFL institutional funds. Extended bioinformatics work performed at the PCF-PTP was funded by institutional funding of the Life Science Faculty.
Footnotes
Published ahead of print 14 December 2012
REFERENCES
- 1. Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 2. Mena KD, Gerba CP. 2009. Waterborne adenovirus. Rev. Environ. Contam. Toxicol. 198:133–167 [DOI] [PubMed] [Google Scholar]
- 3. Zubarev R, Mann M. 2007. On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 6:377–381 [DOI] [PubMed] [Google Scholar]
- 4. Makarov A. 2006. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 78:2113–2120 [DOI] [PubMed] [Google Scholar]
- 5. Zubieta C, Schoehn G, Chroboczek J, Cusack S. 2005. The structure of the human adenovirus 2 penton. Mol. Cell 17:121–135 [DOI] [PubMed] [Google Scholar]
- 6. Wyn-Jones AP, Carducci A, Cook N, D'Agostino M, Divizia M, Fleischer J, Gantzer C, Gawler A, Girones R, Höller C, de Roda Husman AM, Kay D, Kozyra I, López-Pila J, Muscillo M, Nascimento MS, Papageorgiou G, Rutjes S, Sellwood J, Szewzyk R, Wyer M. 2011. Surveillance of adenoviruses and noroviruses in European recreational waters. Water Res. 45:1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thurston-Enriquez JA, Haas CN, Jacangelo J, Gerba CP. 2003. Chlorine inactivation of adenovirus type 40 and feline calicivirus. Appl. Environ. Microbiol. 69:3979–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bewley MC, Springer K, Zhang Y-B, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583 [DOI] [PubMed] [Google Scholar]
- 9. Ogorzaly L, Bertrand I, Paris M, Maul A, Gantzer C. 2010. Occurrence, survival, and persistence of human adenoviruses and F-specific RNA phages in raw groundwater. Appl. Environ. Microbiol. 76:8019–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sirikanchana K, Shisler JL, Marinas BJ. 2008. Effect of exposure to UV-C irradiation and monochloramine on adenovirus serotype 2 early protein expression and DNA replication. Appl. Environ. Microbiol. 74:3774–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yates MV, Malley J, Rochelle P, Hoffman R. 2006. Effect of adenovirus resistance on UV disinfection requirements: a report on the state of adenovirus science, vol 98 American Public Health Association, Washington, DC [Google Scholar]
- 12. Baxter CS, Hofmann R, Templeton MR, Brown M, Andrews RC. 2007. Inactivation of adenovirus types 2, 5, 41 in drinking water by UV light, free chlorine and monochloramine. J. Environ. Eng. 133:95–103 [Google Scholar]
- 13. Nwachuku N, Gerba CP, Oswald A, Mashadi FD. 2005. Comparative inactivation of adenovirus serotypes by UV light disinfection. Appl. Environ. Microbiol. 71:5633–5636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerba CP, Gramos DM, Nwachuku N. 2002. Comparative inactivation of enteroviruses and adenovirus 2 by UV light. Appl. Environ. Microbiol. 68:5167–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Page MA, Shisler JL, Mariñas BJ. 2009. Kinetics of adenovirus type 2 inactivation with free chlorine. Water Res. 43:2916–2926 [DOI] [PubMed] [Google Scholar]
- 16. Seth P. 1999. Adenoviruses: basic biology to gene therapy. RG Landes Company, Austin, TX [Google Scholar]
- 17. Love DC, Silverman A, Nelson KL. 2010. Human virus and bacteriophage inactivation in clear water by simulated sunlight compared to bacteriophage inactivation at a Southern California beach. Environ. Sci. Technol. 44:6965–6970 [DOI] [PubMed] [Google Scholar]
- 18. Kohn T, Grandbois M, McNeill K, Nelson KL. 2007. Association with natural organic matter enhances the sunlight-mediated inactivation of MS2 coliphage by singlet oxygen. Environ. Sci. Technol. 41:4626–4632 [DOI] [PubMed] [Google Scholar]
- 19. Bosshard F, Bucheli M, Meur Y, Egli T. 2010. The respiratory chain is the cell's Achilles' heel during UVA inactivation in Escherichia coli. Microbiology 156:2006–2015 [DOI] [PubMed] [Google Scholar]
- 20. Bosshard F, Riedel K, Schneider T, Geiser C, Bucheli M, Egli T. 2010. Protein oxidation and aggregation in UVA-irradiated Escherichia coli cells as signs of accelerated cellular senescence. Environ. Microbiol. 12:2931–2945 [DOI] [PubMed] [Google Scholar]
- 21. Eischeid AC, Linden KG. 2011. Molecular indications of protein damage in adenoviruses after UV disinfection. Appl. Environ. Microbiol. 77:1145–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1:376–378 [DOI] [PubMed] [Google Scholar]
- 23. Medina-Kauwe LK. 2003. Endocytosis of adenovirus and adenovirus capsid proteins. Adv. Drug Deliv. Rev. 55:1485–1496 [DOI] [PubMed] [Google Scholar]
- 24. Shannon MA, Bohn PW, Elimelech M, Georgiadis JG, Marinas BJ, Mayes AM. 2008. Science and technology for water purification in the coming decades. Nature 452:301–310 [DOI] [PubMed] [Google Scholar]
- 25. Wigginton KR, Menin L, Sigstam T, Gannon G, Cascella M, Hamidane HB, Tsybin YO, Waridel P, Kohn T. 2012. UV radiation induces genome-mediated, site-specific cleavage in viral proteins. Chembiochem 13:837–845 [DOI] [PubMed] [Google Scholar]
- 26. Wigginton K, Menin L, Montoya JP, Kohn T. 2010. Oxidation of virus proteins during UV254 and singlet oxygen mediated inactivation. Environ. Sci. Technol. 44:5437–5443 [DOI] [PubMed] [Google Scholar]
- 27. Roelvink PW, Lee GM, Einfeld DA, Kovesdi I, Wickham TJ. 1999. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing Adenoviridae. Science 286:1568–1571 [DOI] [PubMed] [Google Scholar]
- 28. Seiradake E, Henaff D, Wodrich H, Billet O, Perreau M, Hippert C, Mennechet F, Schoehn G, Lortat-Jacob H, Dreja H, Ibanes S, Kalatzis V, Wang JP, Finberg RW, Cusack S, Kremer EJ. 2009. The cell adhesion molecule “CAR” and sialic acid on human erythrocytes influence adenovirus in vivo biodistribution. PLoS Pathog. 5:e1000277 doi:10.1371/journal.ppat.1000277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kohn T, Nelson KL. 2007. Sunlight-mediated inactivation of MS2 coliphage via exogenous singlet oxygen produced by sensitizers in natural waters. Environ. Sci. Technol. 41:192–197 [DOI] [PubMed] [Google Scholar]
- 30. Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. 2009. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4:698–705 [DOI] [PubMed] [Google Scholar]
- 31. R Development Core Team 2008. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 32. Lytle CD, Sagripanti JL. 2005. Predicted inactivation of viruses of relevance to biodefense by solar radiation. J. Virol. 79:14244–14252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berney M, Weilenmann H-U, Egli T. 2006. Flow-cytometric study of vital cellular functions in Escherichia coli during solar disinfection (SODIS). Microbiology 152:1719–1729 [DOI] [PubMed] [Google Scholar]
- 34. Sinha RP, Hader D-P. 2002. UV-induced DNA damage and repair: a review. Photochem. Photobiol. Sci. 1:225–236 [DOI] [PubMed] [Google Scholar]
- 35. Ravanat J-L, Douki T, Cadet J. 2001. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 63:88–102 [DOI] [PubMed] [Google Scholar]
- 36. Eischeid AC, Meyer JN, Linden KG. 2009. UV disinfection of adenoviruses: molecular indications of DNA damage deficiency. Appl. Environ. Microbiol. 75:23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eischeid AC, Thurston JA, Linden KG. 2011. UV disinfection of adenovirus: present state of the research and future directions. Crit. Rev. Environ. Sci. Technol. 41:1375–1396 [Google Scholar]
- 38. Zimmer-Thomas JL, Slawson RM, Huck PM. 2007. A comparison of DNA repair and survival of Escherichia coli O157:H7 following exposure to both low- and medium-pressure UV irradiation. J. Water Health 5:407–415 [DOI] [PubMed] [Google Scholar]
- 39. Kalisvaart BF. 2001. Photobiological effects of polychromatic medium pressure UV lamps. Water Sci. Technol. 43(4):191–197 [PubMed] [Google Scholar]
- 40. Kalisvaart BF. 2004. Re-use of wastewater: preventing the recovery of pathogens by using medium-pressure UV lamp technology. Water Sci. Technol. 50(6):337–344 [PubMed] [Google Scholar]
- 41. Bosshard F, Berney M, Scheifele M, Weilenmann H-U, Egli T. 2009. Solar disinfection (SODIS) and subsequent dark storage of Salmonella typhimurium and Shigella flexneri monitored by flow cytometry. Microbiology 155:1310–1317 [DOI] [PubMed] [Google Scholar]
- 42. Wigginton K, Pecson BM, Sigstam T, Bosshard F, Kohn T. 2012. Virus inactivation mechanisms: impact of disinfectants on virus function and structural integrity. Environ. Sci. Technol. 46:12069–12078 [DOI] [PubMed] [Google Scholar]
- 43. Rux JJ, Kuser PR, Burnett RM. 2003. Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution X-ray crystallographic, molecular modeling, and sequence-based methods. J. Virol. 77:9553–9566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zubieta C, Blanchoin L, Cusack S. 2006. Structural and biochemical characterization of a human adenovirus 2/12 penton base chimera. FEBS J. 273:4336–4345 [DOI] [PubMed] [Google Scholar]
- 45. Van Raaij MJ, Mitraki A, Lavigne G, Cusack S. 1999. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401:935–938 [DOI] [PubMed] [Google Scholar]
- 46. Nemerow GR, Stewart PL, Reddy VS. 2012. Structure of human adenovirus. Curr. Opin. Virol. 2:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. San Martín C. 2012. Latest insights on adenovirus structure and assembly. Viruses 4:847–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nemerow GR, Pache L, Reddy V, Stewart PL. 2009. Insights into adenovirus host cell interactions from structural studies. Virology 384:380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith JG, Wiethoff CM, Stewart PL, Nemerow GR. 2010. Adenovirus. Curr. Top. Microbiol. Immunol. 343:195–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Law LK, Davidson BL. 2005. What does it take to bind CAR? Mol. Ther. 12:599–609 [DOI] [PubMed] [Google Scholar]
- 51. Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. 1993. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 73:309–319 [DOI] [PubMed] [Google Scholar]
- 52. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]