

Abstract
The amount of dietary sodium intake regulates the renin angiotensin system (RAS) and blood pressure, both of which play critical roles in atherosclerosis. However, there are conflicting findings regarding the effects of dietary sodium intake on atherosclerosis. This study applied a broad range of dietary sodium concentrations to determine the concomitant effects of dietary sodium intake on the RAS, blood pressure, and atherosclerosis in mice. Eight-week-old male low-density lipoprotein receptor −/− mice were fed a saturated fat-enriched diet containing selected sodium concentrations (Na 0.01%, 0.1%, or 2% w/w) for 12 weeks. Mice in these three groups were all hypercholesterolemic, although mice fed Na 0.01% and Na 0.1% had higher plasma cholesterol concentrations than mice fed Na 2%. Mice fed Na 0.01% had greater abundances of renal renin mRNA than those fed Na 0.1% and 2%. Plasma renin concentrations were higher in mice fed Na 0.01% (14.2±1.7 ng/ml/30 min) than those fed Na 0.1% or 2% (6.2±0.6 and 5.8±1.6 ng/ml per 30 min, respectively). However, systolic blood pressure at 12 weeks was higher in mice fed Na 2% (138±3 mm Hg) than those fed Na 0.01% and 0.1% (129±3 and 128±4 mmHg, respectively). In contrast, mice fed Na 0.01% (0.17±0.02 mm2) had larger atherosclerotic lesion areas in aortic roots than those fed Na 2% (0.09±0.01 mm2), whereas lesion areas in mice fed Na 0.1% (0.12±0.02 mm2) were intermediate between and not significantly different from those in Na 0.01% and Na 2% groups. In conclusion, while high dietary sodium intake led to higher systolic blood pressure, low dietary sodium intake augmented atherosclerosis in hypercholesterolemic mice.
Keywords: Dietary sodium, Atherosclerosis, Blood pressure, Renin, Angiotensin, Cholesterol
1. Introduction
Dietary salt (sodium chloride) is essential for the body to maintain homeostasis of water and sodium. It also plays an important role in the regulation of blood pressure. Although the mechanisms are poorly understood, epidemiological studies in humans and interventional experiments in animal models provide strong evidence that high sodium intake leads to increases of blood pressure [1–4]. High blood pressure is a well-recognized risk factor for atherosclerosis. However, manipulating dietary sodium has generated conflicting results in atherosclerosis studies using mouse models. While some studies have reported that high dietary sodium augments atherosclerosis [4,5], two studies have imparted that low dietary sodium increases atherosclerosis in hypercholesterolemic mice [6,7]. The discrepant findings of these studies infer a more complex role of dietary sodium intake in the development of atherosclerosis than a causal relationship mediated by blood pressure changes.
In addition to regulating blood pressure, high dietary sodium suppresses the renin angiotensin system (RAS), while restricted sodium intake activates the RAS, as well-documented in both human and animal studies [8–10]. Activation of the RAS increases blood pressure and contributes to the development of atherosclerosis. Direct evidence shows that angiotensin II (AngII) infusion causes a marked increase of systolic blood pressure and accelerates atherosclerotic formation in hypercholesterolemic mice [11,12]. Conversely, pharmacological inhibitions and genetic disruptions of the RAS decrease blood pressure and attenuate atherosclerosis in these animal models [13–19]. Although high blood pressure is a critical risk factor for atherosclerosis, there is accumulating evidence that activation of the RAS contributes to atherosclerosis through mechanisms not involving blood pressure changes [11,13,14,16,17,20–23].
Still unknown is whether the inconsistent results from previous studies regarding dietary sodium intake on atherosclerosis reflect differential effects of dietary sodium intake on blood pressure and the RAS. To address this question, we randomized hypercholesterolemic mice to one of three dietary sodium concentrations. Our hypothesis was that low dietary sodium intake would contribute to the development of atherosclerosis via activation of the RAS. The dietary requirements for “normal” sodium concentrations in mice are unknown. A saturated fat-enriched diet used by many laboratories[14,21,24,25] to stimulate hypercholesterolemia and induces atherosclerosis in mice contains 0.1% weight/weight (w/w) of sodium (Na 0.1%). Adult mice (8 weeks old) fed this diet steadily gain body weight at a rate of 2–3 g per week. Therefore, we used Na 0.1% as an approximation to the “normal” dietary sodium concentration for this study. We then selected Na 2% and 0.01% as the high and low dietary sodium concentrations, respectively. To determine the effects of dietary sodium intake on the development of atherosclerosis, we examined atherosclerotic lesions throughout the aortic root after 12 weeks of feeding the selected diets to low-density lipoprotein (LDL) receptor −/− male mice.
2. Methods and materials
2.1. Mice and diet
Male LDL receptor −/− (B6.129S7-Ldlrtm1Her; Stock# 002207) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and randomized to three groups. All mice were maintained in a barrier animal facility at the University of Kentucky and fed a normal mouse laboratory diet containing Na 0.2% (Diet number 2918; Harlan Teklad, Madison, WI, USA). To induce hypercholesterolemia, the diet was changed to one supplemented with same amount of saturated fat (milk fat 21% w/w) and cholesterol (0.2% w/w) but containing various concentrations of sodium (Na 0.01%, 0.1%, or 2% w/w; Harlan Teklad). The duration of the selected diet feeding was 12 weeks beginning when the mice were 8 weeks old. The study was performed with the approval of the University of Kentucky Institutional Animal Care and Use Committee.
2.2. Blood pressure measurements
Systolic blood pressure was measured using a non-invasive tail cuff system (Coda 6; Kent Scientific Corp., Torrington, CT, USA). Measurements were performed for 5 consecutive days prior to the selected diet feeding to generate a baseline blood pressure (Week 0), and at Weeks 4, 8, and 12, respectively, during fed the selected diets [26].
2.3. Measurement of urine sodium concentrations
Twenty four-hour urine was collected after 10 weeks of the selected diet feeding using a metabolic cage system (Model MMC100; Hatteras Instruments, Cary, NC, USA) and stored at −80°C. Urine samples were shipped on dry ice to the University of Missouri for measurements of urine sodium concentrations. Urine sodium excretion rates were calculated using urine sodium concentrations and the 24-h urine volume.
2.4. Measurements of plasma cholesterol concentrations and lipoprotein distributions
Plasma cholesterol concentrations were measured using a commercial kit (Catalog number 439-17501; Wako Chemicals USA, Richmond, VA, USA). Plasma lipoprotein distributions (five to six plasma samples from each group) were determined by size exclusion method using a fast performance liquid chromatographic machine. Thirty-two fractions from each plasma sample were collected and subsequently measured using the enzyme-based assay kit for plasma cholesterol concentrations. Plasma cholesterol concentrations of very low-density lipoprotein (VLDL-C), intermediate/low density lipoprotein (I/LDL-C), and high density lipoprotein (HDL-C) in the three groups were analyzed using PeakFit software (SeaSolve Software, San Jose, CA, USA). This software allowed us to analyze the chromatographic data using an automated nonlinear peak separation function as described previously [27].
2.5. Measurements of plasma renin and aldosterone concentrations
Plasma renin concentrations were measured by radioimmunoassay (RIA). Briefly, plasma samples (8 μl) were incubated with an excess of rat angiotensinogen in the presence of EDTA (0.02 M) for 30 min at 37°C. AngI generated in the samples was quantified by RIA using a commercial kit (Catalog number 1553; DiaSorin, Stillwater, MN, USA). Plasma aldosterone concentrations were determined with a commercial kit (Catalog number DSL-8600; Diagnostic Systems Laboratories, Webster, TX, USA) as described previously [14].
2.6. Quantification of atherosclerotic lesions
Atherosclerotic lesions were quantified throughout the aortic root as described previously [28,29].
2.7. Real-time polymerase chain reaction (PCR) of renin in kidneys
Total RNA was harvested from mouse kidneys using the SV Total RNA Isolation System (Catalog number Z3100; Promega, Madison, WI, USA). Real-time PCR was performed as described previously [30]. Abundance of renin mRNA in kidneys was calculated with normalization to 18S rRNA using ΔΔ Ct method.
2.8. Immunostaining of macrophages
Immunostaining was performed on sections of aortic roots as described previously [31]. Macrophages in atherosclerotic lesions were detected using rat anti-mouse CD68 antibody (Clone FA-11, Catalog number MCA1957, AbDSerotec, Raleigh, NC, USA) with appropriate negative controls [31].
2.9. Statistical analyses
SAS version 9.2 (SAS Institute, Cary, NC, USA) and SigmaPlot version 11 (SYSTAT Software, San Jose, CA, USA) were used for statistical analyses. To compare the three groups on a continuous response variable, we used one-way analysis of variance (ANOVA) or an appropriate methodological alternative as described previously [14]. Systolic blood pressure data were analyzed using one-way repeated measures ANOVA. The relationship between mean atherosclerotic lesion areas and systolic blood pressure at week 12 was analyzed using Pearson correlations and linear regression models. Data were presented as mean±S.E.M. P<.05 was considered statistically significant.
3. Results
3.1. Characterization of study mice
As expected, urine sodium excretion rates in the study mice were positively related to dietary sodium concentrations (Table 1). Water consumption and urine volume at a 24-h period were greater in mice fed Na 2% than in those fed Na 0.01% or Na 0.1% (Table 1). Although food consumption was similar, mice fed Na 2% gained less body weight (Table 1) compared to the other two groups. In contrast to urine sodium excretion rates, plasma aldosterone concentrations were negatively related to dietary sodium concentrations (Table 1).
Table 1.
Characterization of male LDL receptor −/− mice fed a saturated fat-enriched diet containing selected sodium concentrations
| Sodium concentrations in the diet (w/w) |
0.01% | 0.1% | 2% |
|---|---|---|---|
| Number per group | 18 | 16 | 18 |
| Body weight (baseline; g) | 21.4±0.7 | 21.5±0.6 | 21.5±0.7 |
| Body weight (termination; g) | 33.8±0.6 * | 34.8±1.0 * | 28.5±0.8 |
| Plasma cholesterol concentrations (mg/dl) |
1625±92 * | 1612±78 * | 1264±74 |
| Plasma aldosterone concentrations (ng/ml) |
253±24 * | 150±36 *,# | 53±6 |
| Water consumption (ml/24 h) | 1.9±0.1 * | 2.5±0.3 * | 5.1±0.2 |
| Urine volume (ml/24 h) | 0.70±0.04 * | 0.87±0.11 * | 2.13±0.15 |
| Urine Na excretion rate (μmol/24 h) |
8.8±0.7 * | 29.7±3.1 *,# | 770.2±93.3 |
Values are represented as mean±S.E.M.
P<.01 for comparisons to the mice fed Na 2%, and
P<.01 for comparisons to the mice fed Na 0.01%.
Our previous study showed that plasma cholesterol concentrations were less than 100 mg/dl in LDL receptor +/+ (C57BL/6 strain) mice fed a normal laboratory rodent diet containing Na 0.2% (Diet number 2918; Harlan Teklad) [32]. In the present study, the saturated fat-enriched diet feeding increased plasma cholesterol concentrations to more than 1,000 mg/dl in the LDL receptor −/− mice from all three study groups (Table 1). Compared to mice fed Na 2%, those fed Na 0.01% or Na 0.1% had moderately higher plasma cholesterol concentrations. Resolution of lipoproteins through size exclusion chromatography (Fig. 1A) followed by non linear curve fitting analysis demonstrated that plasma VLDL and I/LDL cholesterol concentrations were higher in mice fed Na 0.01% or Na 0.1% than in those fed Na 2% (Fig. 1B). HDL cholesterol concentrations were not significantly different among the three study groups.
Fig. 1.

Low dietary sodium led to higher plasma VLDL and IDL/LDL cholesterol concentrations in hypercholesterolemic mice. (A) Lipoproteins in plasma were resolved by size exclusion chromatography and measured using an enzymatic method. Triangles represent the mean values from 5-6 individual mice and bars represent S.E.M. (B) Plasma cholesterol concentrations of lipoprotein fractions were calculated using a non linear curve fitting method. Histobars represent means and bars represent S.E.M. Statistical analysis was performed using one way ANOVA. *P<.05 for comparisons with mice fed Na 2% (n=16–18/group).
3.2. High dietary sodium eventually led to increased systolic blood pressure
Systolic blood pressure in all study groups prior to the selected dietary sodium feeding was similar (Fig. 2). Among the three groups, only mice fed Na 2% had significantly increased systolic blood pressure during the 12-week selected diet feeding.
Fig. 2.
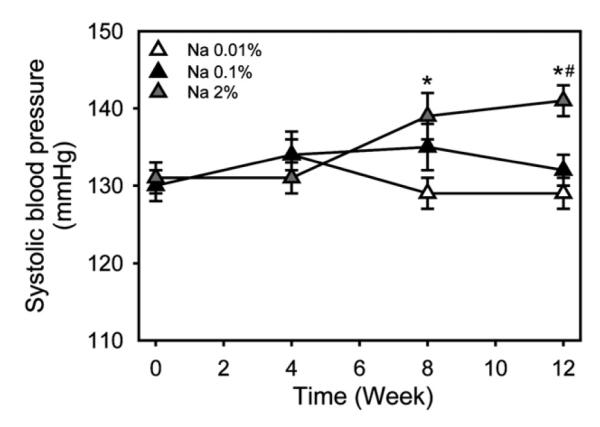
High dietary sodium increased systolic blood pressure in hypercholesterolemic mice. Systolic blood pressure was measured using a tail-cuff system at Week 0, 4, 8, and 12 during the selected dietary sodium feeding. Triangles represent means of weekly observations and bars represent S.E.M. As analyzed with one way repeated measures ANOVA, mice fed Na 2%, but not mice fed either Na 0.01% or Na 0.1%, had increased systolic blood pressure during the 12-week selected diet feeding. * and #P<.05 for comparisons with mice fed Na 0.01% and Na 0.1%, respectively (n=16-18/group).
The saturated fat-enriched diet feeding containing selected sodium concentrations did not produce an overall statistically significant difference on systolic blood pressure during the first 4 weeks among the three study groups. In addition, no significant difference was detected between mice fed Na 0.01% and Na 0.1% during the 12 weeks.
At week 8, mice fed Na 2% had significantly higher (mean of 9 mmHg) systolic blood pressure compared to mice fed Na 0.01%, but were not significantly different compared to mice fed Na 0.1% (Fig. 2). Systolic blood pressures at week 12 (from low to high dietary sodium) were 129±3, 128±4, and 138±3 mmHg, respectively. The mean difference between mice fed Na 2% and Na 0.1% at week 12 was 10 mmHg, and between mice fed Na 2% and Na 0.01% was 11 mmHg (Fig. 2).
3.3. Low dietary sodium resulted in increased abundance of renal renin mRNA and plasma renin concentrations
Renin is a sensitive parameter in determining circulating hormonal changes of the RAS. Renal renin mRNA was more abundant in mice fed Na 0.01% than in those fed Na 2% or Na 0.1% (Fig. 3A). Consistent with the mRNA change, mice fed Na 0.01% showed higher concentrations of plasma renin than those fed Na 2% or Na 0.1% (Fig. 3B).
Fig. 3.

Low dietary sodium resulted in higher renal renin mRNA abundances and plasma renin concentrations. (A) mRNA abundance of renal renin was quantified with real-time PCR using ΔΔCt method (n=5/group). (B) Plasma renin concentrations were measured using radioimmunoassay in 9-10 mice/group. Data are mean± S.E.M. Statistical analysis was performed using one way ANOVA. * P<.01 for comparisons with mice fed Na 0.01%.
3.4. Low dietary sodium concentrations augmented atherosclerotic lesions
Atherosclerotic lesion areas were measured in serial sections of aortic roots. Consistent with our previous studies [14,21], the fat-enriched diet feeding for 12 weeks led to development of profound atherosclerotic lesions in LDL receptor −/− mice. Mean lesion areas of aortic roots in mice fed Na 0.01% were approximately 90% greater than in mice fed Na 2%. Despite a sodium concentration-dependent relationship with mean lesion areas of atherosclerosis, it did not reach a significant difference when mice fed Na 0.1% were compared with those fed either Na 0.01% or Na 2% (Fig. 4). In addition, irrespective of the difference of the atherosclerotic lesion areas in aortic roots between mice fed the low and the high dietary sodium concentrations, lipid-laden macrophages were the predominant cell type in all lesions as determined by immunostaining of CD68 (data not shown).
Fig. 4.
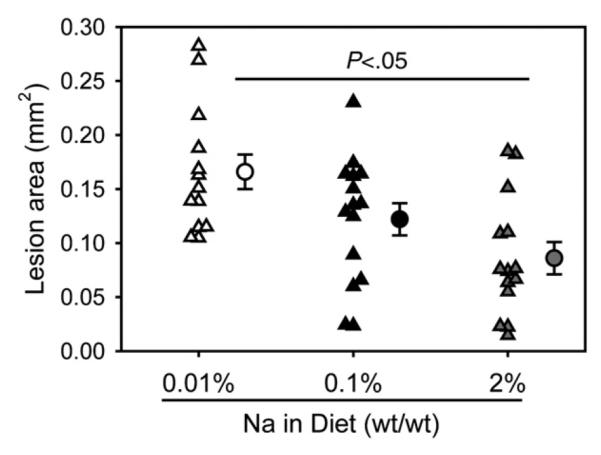
Low dietary sodium led to greater atherosclerotic lesion areas in aortic roots. Atherosclerotic lesion areas were measured in aortic roots. Triangles represent average lesion areas throughout the aortic root (eight serial sections/aortic root; n=13–15/group) of each mouse. Circles represent the means, and bars are S.E.M. Statistical analysis was performed using one way ANOVA. There were no significant differences of mean lesion areas between mice fed Na 0.1% and mice fed either Na 0.01% or Na 2%.
We also analyzed the association between systolic blood pressure and atherosclerotic lesion size using Pearson correlations and linear regression models. No significant correlation between systolic blood pressure and lesion size was observed.
4. Discussion
In this study, selected dietary sodium intakes displayed differential effects on systolic blood pressure and atherosclerotic lesions. While high dietary sodium increased systolic blood pressure, it decreased both plasma renin concentrations and atherosclerotic lesion area in aortic roots of male LDL receptor −/− mice fed a saturated fat-enriched diet for 12 weeks.
Consistent with previous observations in humans and animal models including both normal C57BL/6 mice and hypercholesterolemic mice [1–4], male LDL receptor −/− mice fed high dietary sodium (Na 2%) in this study had higher systolic blood pressure than those fed either a normal (Na 0.1%) or low (Na 0.01%) dietary sodium. However, systolic blood pressure was similar between mice fed Na 0.1% and Na 0.01%, although dietary sodium concentrations were 10-fold different. Notably, the changes of systolic blood pressure did not parallel the changes of atherosclerotic lesion size in aortic roots. Despite a significant difference of systolic blood pressure, mice fed high dietary sodium (Na 2%) had smaller atherosclerotic lesions in aortic roots, compared to those fed low dietary sodium (Na 0.01%). In contrast, mice fed normal dietary sodium (Na 0.1%) had lower systolic blood pressure but similar lesion size, compared to those fed high dietary sodium (Na 2%). In addition to our findings, a previous study reported that low dietary sodium (Na 0.01%) in both male and female apolipoprotein (apo)E −/− mice led to more augmented atherosclerotic lesions in thoracic aorta in the absence of changes of mean arterial pressure, compared to mice fed Na 0.25% [7]. These results implicate that dietary sodium-induced changes of blood pressure in hypercholesterolemic mice do not directly contribute to the development of atherosclerotic lesions. Indeed, there is a debate on the correlations of dietary sodium intake with cardiovascular diseases in humans. A recent prospective population study has reported that high sodium excretion modestly increases systolic blood pressure, whereas low sodium excretion is associated with a higher mortality due to cardiovascular events [33].
In contrast to the lower systolic blood pressures, plasma renin concentrations were higher in mice fed low dietary sodium than in those fed normal or high dietary sodium (Na 0.1% and Na 2%). Although the mechanisms are not yet established, these results agree with findings reported in both humans and animal models[6,7,9,10,34]. In addition to this study, there is other evidence of RAS activation in relation to dietary sodium intake. More specifically, apoE −/− mice fed a normal laboratory rodent diet containing Na 0.01% had higher plasma AngII concentrations compared to those fed Na 0.25% [7]. Our results, combined with the evidence from the literature, imply that low dietary sodium activates the RAS in hypercholesterolemic mice, which may contribute to the development of atherosclerosis.
The present study used a combination of selected dietary sodium concentrations and a saturated fat-enriched diet that mimics the human “western diet” in the industrialized countries. Male LDL receptor −/− mice fed this saturated fat-enriched diet have a remarkable increase of plasma cholesterol concentrations that contributes to atherosclerosis [14,21]. Consistent with the previous reports, all mice in this study were hypercholesterolemic. However, mice fed the diet containing Na 2% had lower plasma cholesterol concentrations than those fed the same fat-enriched diet containing either Na 0.01% or Na 0.1%. Similar findings have also been reported in both human and animal studies [6,7,34–36]. The adverse lipid effects of low dietary sodium include elevations in plasma total cholesterol, VLDL, and I/LDL cholesterol concentrations, as also observed in this study. The inverse correlation between dietary sodium intake and plasma cholesterol concentrations indicates a potentially important role of dietary sodium in the regulation of plasma lipid metabolism. However, the mechanism by which low dietary sodium increases plasma cholesterol concentrations has not been defined. Hypercholesterolemia and activation of the RAS are two critical components in the development of atherosclerosis. There is compelling evidence that hypercholesterolemia and AngII synergistically promote the progression of atherosclerosis [11,12,37]. We have also found that hypercholesterolemia activates the RAS, leading to augmentation of atherosclerotic lesions in mice fed a saturated fat-enriched diet [21], although the causal relationship between the RAS and plasma cholesterol concentrations in mice fed selected dietary sodium has not been determined.
This study also found that mice fed the high dietary sodium had less body weight gain compared to those fed either Na 0.01% or Na 0.1%. While this has not been reported elsewhere, a recent study found a divergent effect of dietary sodium intake on body weight change [38]. ApoE −/− female mice fed either Na 0.03% or Na 3.2% gained more weight compared to those fed Na 0.28%, but the mechanism was not investigated. In our study, we did not see any difference on food consumption, although water consumption and urine volume were increased in mice fed Na 2%. It has been reported that reduction of the RAS leads to less body weight gain in mice [39]. While this topic is beyond the scope of the purpose of the present study, it will be interesting to explore the mechanism by which the amount of dietary sodium intake regulates body weight in a future study.
In conclusion, selected dietary sodium manipulation has differential effects on systolic blood pressure regulation and the development of atherosclerosis in hypercholesterolemic mice.
Acknowledgments
This study was supported by the National Institutes of Health (HL062846). We acknowledge the skilled technical assistance of Jessica Moorleghen, Debra L. Rateri, and Victoria English.
Footnotes
Funding Sources: the National Institutes of Health (HL062846).
References
- [1].Intersalt cooperative Research Group Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ. 1988;297:319–28. doi: 10.1136/bmj.297.6644.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- [3].Horie R, Yamori Y, Nara Y, Sawamura M, Mizushima S, Mano M. Blood pressure levels in the elderly with or without nutritional intervention. J Cardiovasc Pharmacol. 1990;16(Suppl 8):S57–8. [PubMed] [Google Scholar]
- [4].Ketonen J, Merasto S, Paakkari I, Mervaala EM. High sodium intake increases vascular superoxide formation and promotes atherosclerosis in apolipoprotein E-deficient mice. Blood Press. 2005;14:373–82. doi: 10.1080/08037050500383687. [DOI] [PubMed] [Google Scholar]
- [5].Ketonen J, Mervaala E. Effects of dietary sodium on reactive oxygen species formation and endothelial dysfunction in low-density lipoprotein receptor-deficient mice on high-fat diet. Heart Vessels. 2008;23:420–9. doi: 10.1007/s00380-008-1066-5. [DOI] [PubMed] [Google Scholar]
- [6].Catanozi S, Rocha JC, Passarelli M, Guzzo ML, Alves C, Furukawa LN, et al. Dietary sodium chloride restriction enhances aortic wall lipid storage and raises plasma lipid concentration in LDL receptor knockout mice. J Lipid Res. 2003;44:727–32. doi: 10.1194/jlr.M200330-JLR200. [DOI] [PubMed] [Google Scholar]
- [7].Ivanovski O, Szumilak D, Nguyen-Khoa T, Dechaux M, Massy ZA, Phan O, et al. Dietary salt restriction accelerates atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 2005;180:271–6. doi: 10.1016/j.atherosclerosis.2004.12.020. [DOI] [PubMed] [Google Scholar]
- [8].Ingert C, Grima M, Coquard C, Barthelmebs M, Imbs JL. Effects of dietary salt changes on renal renin-angiotensin system in rats. Am J Physiol Renal Physiol. 2002;283:F995–1002. doi: 10.1152/ajprenal.00321.2001. [DOI] [PubMed] [Google Scholar]
- [9].Carillo BA, Beutel A, Mirandola DA, Vidonho AF, Jr, Furukawa LN, Casarini D, et al. Differential sympathetic and angiotensinergic responses in rats submitted to low- or high-salt diet. RegulPept. 2007;140:5–11. doi: 10.1016/j.regpep.2006.11.007. [DOI] [PubMed] [Google Scholar]
- [10].Abiko H, Konta T, Hao Z, Takasaki S, Suzuki K, Ichikawa K, et al. Factors correlated with plasma renin activity in general Japanese population. Clin Exp Nephrol. 2009;13:130–7. doi: 10.1007/s10157-008-0114-x. [DOI] [PubMed] [Google Scholar]
- [11].Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103:448–54. doi: 10.1161/01.cir.103.3.448. [DOI] [PubMed] [Google Scholar]
- [12].Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–12. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wassmann S, Czech T, Van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/Angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110:3062–7. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- [14].Lu H, Rateri DL, Feldman DL, CharnigoJr RJ, Fukamizu A, Ishida J, et al. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest. 2008;118:984–93. doi: 10.1172/JCI32970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Candido R, Jandeleit-Dahm KA, Cao ZM, Nesteroff SP, Burns WC, Twigg SM, et al. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation. 2002;106:246–53. doi: 10.1161/01.cir.0000021122.63813.32. [DOI] [PubMed] [Google Scholar]
- [16].Candido R, Allen TJ, Lassila M, Cao Z, Thallas V, Cooper ME, et al. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation. 2004;109:1536–42. doi: 10.1161/01.CIR.0000124061.78478.94. [DOI] [PubMed] [Google Scholar]
- [17].da Cunha V, Tham DM, Martin-McNulty B, Deng G, Ho JJ, Wilson DW, et al. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005;178:9–17. doi: 10.1016/j.atherosclerosis.2004.08.023. [DOI] [PubMed] [Google Scholar]
- [18].Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Takarada S, Kitabata H, et al. Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension. 2008;52:563–72. doi: 10.1161/HYPERTENSIONAHA.108.111120. [DOI] [PubMed] [Google Scholar]
- [19].Weiss D, Taylor WR. Deoxycorticosterone acetate salt hypertension in apolipoprotein E−/− mice results in accelerated atherosclerosis: the role of angiotensin II. Hypertension. 2008;51:218–24. doi: 10.1161/HYPERTENSIONAHA.107.095885. [DOI] [PubMed] [Google Scholar]
- [20].Lu H, Cassis LA, Daugherty A. Atherosclerosis and arterial blood pressure in mice. Curr Drug Targets. 2007;8:1181–9. doi: 10.2174/138945007782403829. [DOI] [PubMed] [Google Scholar]
- [21].Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation. 2004;110:3849–57. doi: 10.1161/01.CIR.0000150540.54220.C4. [DOI] [PubMed] [Google Scholar]
- [22].Takaya T, Kawashima S, Shinohara M, Yamashita T, Toh R, Sasaki N, et al. Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis. 2006;186:402–10. doi: 10.1016/j.atherosclerosis.2005.08.009. [DOI] [PubMed] [Google Scholar]
- [23].Doran DE, Weiss D, Zhang Y, Griendling KK, Taylor WR. Differential effects of AT(1) receptor and Ca(2+) channel blockade on atherosclerosis, inflammatory gene expression, and production of reactive oxygen species. Atherosclerosis. 2007;195:39–47. doi: 10.1016/j.atherosclerosis.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].VanderLaan PA, Reardon CA, Thisted RA, Getz GS. VLDL best predicts aortic root atherosclerosis in LDL receptor deficient mice. J Lipid Res. 2009;50:376–85. doi: 10.1194/jlr.M800284-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Babaev VR, Ishiguro H, Ding L, Yancey PG, Dove DE, Kovacs WJ, et al. Macrophage expression of peroxisome proliferator activated receptor-{alpha} reduces atherosclerosis in low-density lipoprotein receptor deficient mice. Circulation. 2007;116:1404–12. doi: 10.1161/CIRCULATIONAHA.106.684704. [DOI] [PubMed] [Google Scholar]
- [26].Daugherty A, Rateri D, Lu H, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. J Vis Exp. 2009:1291. doi: 10.3791/1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang S, Subramanian V, Lu H, Howatt DA, Moorleghen JJ, Charnigo R, et al. Deficiency of receptor-associated protein attenuates angiotensin II-induced atherosclerosis in hypercholesterolemic mice without influencing abdominal aortic aneurysms. Atherosclerosis. 2012;220:375–80. doi: 10.1016/j.atherosclerosis.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. doi: 10.1385/1-59259-340-2:293. [DOI] [PubMed] [Google Scholar]
- [29].Daugherty A, Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods. 2005;36:129–38. doi: 10.1016/j.ymeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- [30].Lu H, Boustany-Kari CM, Daugherty A, Cassis LA. Angiotensin II. increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab. 2007;292:E1280–7. doi: 10.1152/ajpendo.00277.2006. [DOI] [PubMed] [Google Scholar]
- [31].Lu H, Rateri DL, Daugherty A. Immunostaining of mouse atherosclerosis lesions. Methods Mol Med. 2007;139:77–94. doi: 10.1007/978-1-59745-571-8_4. [DOI] [PubMed] [Google Scholar]
- [32].Uchida HA, Poduri A, Subramanian V, Cassis LA, Daugherty A. Urokinase-type plasminogen activator deficiency in bone marrow-derived cells augments rupture of angiotensin II-induced abdominal aortic aneurysms. Arterio Thromb Vasc Biol. 2011;31:2845–52. doi: 10.1161/ATVBAHA.111.234997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stolarz-Skrzypek K, Kuznetsova T, Thijs L, Tikhonoff V, Seidlerova J, Richart T, et al. Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. JAMA. 2011;305:1777–85. doi: 10.1001/jama.2011.574. [DOI] [PubMed] [Google Scholar]
- [34].Nakandakare ER, Charf AM, Santos FC, Nunes VS, Ortega K, Lottenberg AM, et al. Dietary salt restriction increases plasma lipoprotein and inflammatory marker concentrations in hypertensive patients. Atherosclerosis. 2008;200:410–6. doi: 10.1016/j.atherosclerosis.2007.12.034. [DOI] [PubMed] [Google Scholar]
- [35].Alderman MH, Cohen H, Madhavan S. Dietary sodium intake and mortality: the National Health and Nutrition Examination Survey (NHANES I) Lancet. 1998;351:781–5. doi: 10.1016/S0140-6736(97)09092-2. [DOI] [PubMed] [Google Scholar]
- [36].Catanozi S, Rocha JC, Nakandakare ER, Passarelli M, Mesquita CH, Silva AA, et al. The rise of the plasma lipid concentration elicited by dietary sodium chloride restriction in Wistar rats is due to an impairment of the plasma triacylglycerol removal rate. Atherosclerosis. 2001;158:81–6. doi: 10.1016/s0021-9150(01)00415-4. [DOI] [PubMed] [Google Scholar]
- [37].Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Ann NY Acad Sci. 1999;892:108–18. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- [38].Johansson ME, Bernberg E, Andersson IJ, Bie P, Skott O, Gan LM, et al. High-salt diet combined with elevated angiotensin II accelerates atherosclerosis in apolipoprotein E-deficient mice. J Hypertens. 2009;27:41–7. doi: 10.1097/hjh.0b013e328318697b. [DOI] [PubMed] [Google Scholar]
- [39].Kouyama R, Suganami T, Nishida J, Tanaka M, Toyoda T, Kiso M, et al. Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a receptor. Endocrinology. 2005;146:3481–9. doi: 10.1210/en.2005-0003. [DOI] [PubMed] [Google Scholar]