

Abstract
Cerebrospinal fluid (CSF) is a low protein content biological fluid with dynamic range spanning at least nine orders of magnitude in protein content and is in direct contact with the brain. A modified IgY-14 immunodepletion treatment was performed to enhance analysis of the low volumes of CSF that are obtainable from mice. As a model system in which to test this approach, we utilized transgenic mice that over-express the intermediate filament glial fibrillary acidic protein (GFAP). These mice are models for Alexander disease (AxD), a severe leukodystrophy in humans. From the CSF of control and transgenic mice we report the identification of 289 proteins, with relative quantification of 103 proteins. Biological and technical triplicates were performed to address animal variability as well as reproducibility in mass spectrometric analysis. Relative quantitation was performed using distributive normalized spectral abundance factor (dNSAF) spectral counting analysis. A panel of biomarker proteins with significant changes in the CSF of GFAP transgenic mice has been identified with validation from ELISA and microarray data, demonstrating the utility of our methodology and providing interesting targets for future investigations on the molecular and pathological aspects of AxD.
Keywords: Cerebrospinal fluid, proteomics, spectral counting, Alexander disease, mass spectrometry, immunodepletion, transgenic, GFAP overexpressor
INTRODUCTION
Alexander disease (AxD) is a fatal neurogenetic disorder caused by heterozygous point mutations in the coding region of GFAP (encoding glial fibrillary acidic protein).1 The hallmark diagnostic feature of this disease is the accumulation of astrocytic cytoplasmic inclusions known as Rosenthal fibers, containing GFAP, Hsp27, αB-crystallin, and other components.2–5 Although several potential treatment strategies6–8 are under investigation, clinical trial design is hampered by the absence of a standardized clinical scoring system, or means to quantify lesions in MRI, that could serve to monitor severity and progression of disease. One solution to this problem would be the identification of biomarkers in readily sampled body fluids as indirect indicators of disease.
Cerebrospinal fluid (CSF) has a long history as a surrogate biopsy site for brain or spinal cord in evaluating diseases of the central nervous system. The protein composition of CSF is well defined, at least for the most abundant species of proteins, and numerous studies exist that characterize individual biomarkers or complex patterns of biomarkers in various diseases.9, 10 GFAP itself is present in CSF (albeit at much lower levels than in brain parenchyma), and in one study of three Alexander disease patients its levels were markedly increased11. Whether an increase in CSF GFAP will be a consistent finding in Alexander disease, or whether other useful biomarkers for this disease could be identified through an unbiased analysis of the CSF proteome, is not yet known.
The rarity of Alexander disease makes analysis of human samples difficult. However, mouse models exist that replicate key features of the disease such as formation of Rosenthal fibers. Unfortunately, mouse CSF poses particular problems for proteomic studies, and there is an urgent need for technical improvements for dealing with this fluid. For instance, collection from an adult mouse typically yields ~10 μL of CSF, often with some contamination by blood.12 To further complicate analysis, CSF has an exceedingly low protein content (~0.4 μg/μL), with over 60% of the total protein content consisting of a single protein, albumin.13, 14 A number of techniques have been developed to remove albumin from biological samples, including Cibacron Blue,15 IgG immunodepletion,16 and IgY immunodepletion.17–19 IgY, which is avian in origin, offers reduced non-specific binding and increased avidity when compared to IgG antibodies from rabbits, goats, and mice.20–23 One widely used IgY cocktail is IgY-14, which contains fourteen specific antibodies specific for albumin, IgG, transferrin, fibrinogen, α1-antitrypsin, IgA, IgM, α2-macroglobulin, haptoglobin, apolipoproteins A-I, A-II, and B, complement C3, and α1-acid glycoprotein. Since existing protocols for IgY-14 depletion are optimized for use with large volumes of serum, new protocols must be developed to permit its use with the low volumes of a low protein fluid represented by mouse CSF.
Various improvements have also taken place in the field of proteomic analysis that could facilitate analysis of mouse CSF. Data dependent tandem mass spectrometry followed by quantification of proteins is used in standard shotgun proteomics.24–29 Several methods now exist for introducing quantitation into mass spectrometry, including stable isotope labeling,30–32 isobaric tandem mass tags,33, 34 and spectral counting.35, 36 Spectral counting, which is a frequency measurement that uses MS/MS counts of identified peptides as the metric to enable protein quantitation, is attractive because it is label-free and requires no additional sample preparation. Finally, recent advances in spectral counting has produced a data refinement strategy termed normalized spectral abundance factor (NSAF)37, 38 and further developed into distributive NSAF (dNSAF) to address issues with peptides shared by multiple proteins.39
To identify potential biomarkers in AxD, we report a novel scaled-down version of IgY antibody depletion strategy to reduce the complexity and remove high abundance proteins in mouse CSF samples. The generated spectral counts data were then subjected to dNSAF, natural log data transformation, and t-test analysis to determine which proteins differ in abundance when comparing GFAP transgenics and controls with multiple biological and technical replicates.
EXPERIMENTAL DETAILS
Chemicals
Acetonitrile (HPLC grade), urea (>99%), sodium chloride (99.7%), and ammonium bicarbonate (certified) were purchased from Fisher Scientific (Fair Lawn, NJ). Deionized water (18.2 MΩ·cm) was prepared with a Milli-Q Millipore system (Billerica, MA). Optima LC/MS grade acetonitrile and water were purchased from Fisher Scientific (Fair Lawn, NJ). DL-Dithiothreitol (DTT) and sequencing grade modified trypsin were purchased from Promega (Madison, WI). Formic acid (≥98%) was obtained from Fluka (Buchs, Switzerland). Iodoacetamide (IAA), tris(hydroxymethyl)aminomethane (Tris, ≥99.9% ), ammonium formate (≥99.995%), glycine (≥98.5%), and IgY-14 antibodies were purchased from Sigma-Aldrich (Saint Louis, MO).
Mice
Transgenic mice over-expressing the human GFAP gene (Tg73.7 line) were maintained as hemizygotes in the FVB/N background and genotyped via PCR on DNA prepared from tail samples as described previously.40 The mice were housed on a 14–10 light-dark cycle with ad libitum access to food and water. All procedures were conducted using protocols approved by the UW-Madison IACUC.
CSF collection
CSF was collected from mice as described previously.12 Briefly, mice were anesthetized with avertin (400–600 mg/kg i.p.). A midline sagittal incision was made over the dorsal aspect of the hindbrain and three muscle layers carefully peeled back to expose the cisterna magna. The membrane covering the cisterna magna was pierced with a 30 gauge needle, and CSF was collected immediately using a flexible plastic pipette. Approximately ten microliters of CSF was collected per animal. All samples used for MS analysis showed no visible contamination of blood.
Enzyme-linked immunosorbent assay (ELISA)
A sandwich ELISA was used to quantify GFAP.8 Briefly, a microtiter plate was coated with a cocktail of monoclonal antibodies (Covance SMI-26R). Plates were blocked with 5% milk before addition of sample or standards diluted in PBS with Triton-X and BSA. A rabbit polyclonal antibody (DAKO Z334) was used to detect the GFAP followed by a peroxidase conjugated anti-rabbit IgG antibody (Sigma A6154) secondary antibody. The peroxidase activity was detected with SuperSignal ELISA Pico Chemiluminescent Substrate (PIERCE) and quantified with a GloRunner Microplate Luminometer. Values below the biological limit of detection (16ng/L) were given the value 16ng/L before multiplying by the dilution factor.
Immunodepletion of abundant proteins
Currently, there are no commercial immunodepletion products available for use with CSF and to address the low protein content of this fluid (~0.4 μg/μL). Therefore, 100 μL of purchased IgY-14 resin was coupled with a Pierce Spin Cup using a paper filter from Thermo Scientific (Waltham, MA). CSF samples from either transgenics or controls were first pooled to 100 μL (CSF from ten mice was pooled for each control sample whereas two sets of twelve GFAP overexpressor transgenic mice and one set of eleven mice overexpressing GFAP were pooled and collected respectively to achieve three 100 μL of CSF samples respectively). The pooled CSF sample had100 μL of 20 mM Tris-HCl, 300 mM, pH 7.4 (2x dilution buffer) and allowed to mix with the custom IgY-14 depletion spin column on an end-over-end rotor for 30 minutes at 4°C. The sample was then centrifuged at 0.4 rcf for 45 seconds with an Eppendorf Centrifuge 5415D (Hamburg, Germany). The antibodies were then washed with 50 μL 1x dilution buffer, vortexed for 5 minutes, centrifuged at 0.4 rcf for 45 seconds and the flow through was collected for tryptic digestion. The antibodies were then stripped of the bound proteins with four 0.25 mL washes of 0.1 M glycine, pH 2.5, neutralized with two 0.25 mL washes of 0.1 M Tris-HCl, pH 8.0, and washed once with 0.25mL of 1x dilution buffer. The immunodepletion protocol was repeated for each transgenic or control pooled 100 μL sample (N=3).
Preparation of tryptic digests
The immunodepleted pooled mouse CSF samples (200 μL total volume) were concentrated to 10 μL using a Thermo Scientific Savant SpeedVac (SVC100, Waltham, MA). To each sample, 8 μL of 13.3 M urea and 1 μL of 0.50 M DTT were added and allowed to incubate at 37°C for one hour. After incubation, 2.7 μL of 0.55 M IAA was added for carboxymethylation, and the sample was allowed to incubate for 15 minutes in the dark. To quench the IAA, 1 μL of 0.50 M DTT was added and allowed to react for 10 minutes. To perform trypsin digestion, 70 μL of 50 mM NH4HCO3 was then added along with 0.25 μg trypsin which had previously been dissolved in 50 mM acetic acid at a concentration of 0.5 μg/μL. Digestion was performed overnight at 37°C and quenched by addition of 2.5 μL of 10% formic acid. The tryptic peptides were then subjected to solid phase extraction using a Varian Omix Tip, C18, 100 μL (Palo Alto, CA). Peptides were eluted with 50% ACN in 0.1% formic acid, concentrated, and reconstituted in 30 μL H2O in 0.1% formic acid.
RP nanoLC separation
The setup for nanoLC consisted of an Eksigent nanoLC Ultra (Dublin, CA) with a 10 μL injection loop. Mobile phase A consisted of H2O in 0.1% formic acid and mobile phase B consisted of ACN. Injections consisted of 5 μL of prepared CSF tryptic peptides onto an Agilent Technologies Zorbax 300 SB-C18 5 μm, 5×0.3 mm trap cartridge (Santa Clara, CA) at a flow rate of 5 μL/min for 5 minutes at 95% A 5% B. Separation was performed on a Waters 3 μm Atlantis dC18 75 μm × 150 mm (Milford, MA) using a gradient from 5 to 45% mobile phase B at 250 nL/min, over 90 minutes at room temperature. Emitter tips were pulled from 75 μm inner diameter 360 μm outer diameter capillary tubing (Polymicro Technologies, Phoenix, AZ) using an in-house model P-2000 laser puller (Sutter Instrument Co., Novato, CA).
Mass spectrometry data acquisitions
An ion trap mass spectrometer (amaZon ETD, Bruker Daltonics, Bremen, Germany) equipped with an on-line nanospray source was used for mass spectrometry data acquisition. Hystar (Version 3.2, Bruker Daltonics, Bremen, Germany) was used to couple and control Eksigent nanoLC software (Dublin, CA; Version 3.0 Beta, Build 080715) to MS acquisition for all experiments. Smart parameter setting (SPS) was set to 700 m/z, compound stability and trap drive level was set at 100%. Optimization of the nanospray source resulted in dry gas temperature of 125°C, dry gas flow of 4.0 L/min, capillary voltage of −1300 V, end plate offset of −500 V, MS/MS fragmentation amplitude of 1.0V, and SmartFragmentation set at 30–300%.
Data were generated in data dependent mode with strict active exclusion set after two spectra and released after one minute. MS/MS spectra were obtained via collision induced dissociation (CID) fragmentation for the six most abundant MS ions with a preference for doubly charged ions. For MS generation the ion charge control (ICC) target was set to 200,000, maximum accumulation time, 50.00 ms, one spectral average, rolling average, 2, acquisition range of 300–1500 m/z, and scan speed (enhanced resolution) of 8,100 m/z s−1. For MS/MS generation the ICC target was set to 300,000, maximum accumulation time, 50.00 ms, two spectral averages, acquisition range of 100–2000 m/z, and scan speed (Ultrascan) of 32,000 m/z.
Data analysis
MS data were processed with DataAnalysis (Version 4.0, Bruker Daltonics Bremen, Germany). Deviations in parameters from the default Protein Analysis in DataAnalysis were as follows: intensity threshold 1000, maximum number of compounds 1E9, and retention time window 0.001 minutes. These parameter changes were required for spectral counting to prevent loss of spectra. Identification of peptides were performed using Mascot41 (Version 2.3, Matrix Science, London, U.K.). Database searching was performed against SwissProt mus musculus (house mouse) database (version 57.5). False positive analyses42 were calculated using an automatic decoy option of Mascot. Results from the Mascot results were reported using Proteinscape 2.1 and technical replicates were combined and reported as a protein compilation using ProteinExtractor (Bruker Daltonics, Bremen, Germany).
Mascot search parameters were as follows: Allowed missed cleavages, 2; enzyme, trypsin; variable modifications, carboxyamidomethylation (C) and oxidation (M); peptide tolerance, ±1.2 Da; maximum number of 13C, 1; MS/MS tolerance, ±0.5 Da; instrument type, ESI-Trap. Reported proteins required a minimum of one peptide with a Mascot score ≥30.0 with bold red characterization. Spectral counts were determined from the number of MS/MS spectra identified from accepted proteins. A bold red peptide combines a bold peptide, which represents the first query result from a submitted MS/MS spectrum with the red peptide, which indicates the top peptide for the identified protein. Requiring one bold red peptide assists in removal of homologous redundant proteins and further improves protein results. In addition, requiring one peptide to be identified by a score >30.0 removes the ability for proteins to be identified by multiple low Mascot scoring peptides.
Each immunodepleted biological replicate had technical triplicates performed and the technical triplicates were summed together by ProteinExtractor. Peptide spectral counts were then summed for each protein and subjected to dNSAF analysis. Details for this method can be found elsewhere,37, 39 but briefly peptide spectral counts are summed per protein (SpC) based on unique peptides and a weighted distribution of any shared peptides with homologous proteins. To address non-unique homologous peptides between proteins, distributive spectral counting was performed, as described elsewhere.39 The dSpC is divided by the protein’s length (L), and then divided by the summation of the dSpC/L from all proteins in the experiment (N), to produce each protein’s specific dNSAF value.
The resulting data were then transformed by taking the natural log of the dNSAF value. The means for each protein were calculated using the ln(dNSAF) from N=3 biological replicates and the whole data set was subjected to the Shapiro-Wilk test for normality/Gaussian distribution performed on the software PAST (Version 1.98, University of Oslo, Norway, Osla). The Shapiro-Wilk test was repeated multiple times to determine a non-zero value for zero spectral counts. A non-zero value is required to alleviate the errors of dividing by zero which was experimentally determined to be 0.43. The Gaussian data were then subjected to the t-test to identify statistically significant changes in protein expression.
RESULTS AND DISCUSSION
General workflow
Individual CSF samples were manually inspected and samples were only selected that showed no visual blood contamination. Preliminary experiments showed that the maximum degree of blood contamination, estimated from counts of red blood cells in the CSF, that was not visible to the eye, was less than 0.05% by total mass. Approximately 10 individual mouse CSF samples were pooled to achieve the desired 100 μL volume for a single biological replicate. The CSF samples were IgY immunodepleted, followed by digestion with trypsin. The resulting digested samples were then de-salted, concentrated, reconstituted in 30 μL of 0.1% formic acid and 5 μL was loaded onto a reversed phase nanoflow HPLC column, subjected to a 90 minute gradient, and infused into the amaZon ETD ion trap mass spectrometer. The workflow for mouse CSF analysis is shown in Figure 1. The remainder of the desalted sample was saved for technical replicates.
Figure 1.
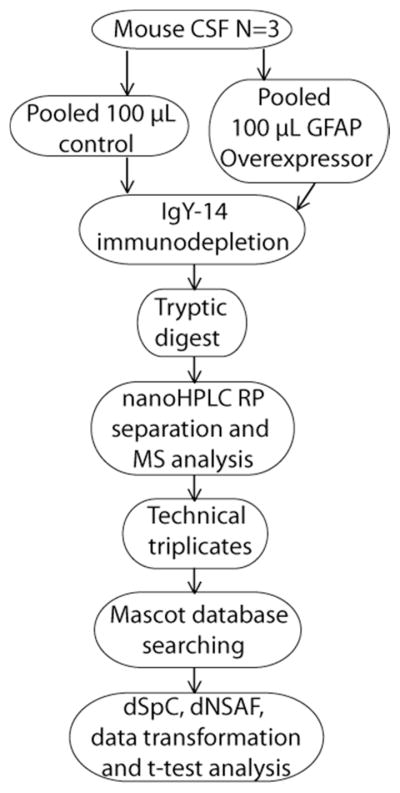
The general workflow indicating the major steps involved in sample collection, sample processing, mass spectrometric data acquisition and analysis of mouse CSF samples.
Immunodepletion for CSF
Currently there are no immunodepletion techniques specifically designed for CSF. Nonetheless, the protein profiles between CSF and serum are similar enough to use currently available immunodepletion techniques designed for serum as a starting point. The smallest commercially available IgY-14 immunodepletion kit is for 15 μL of serum which is equivalent in protein content to ~1.5 mL of CSF. Therefore, for 100 μL CSF one fifteenth of the total IgY-14 beads would be used for the equivalent immunodepletion which is 66 μL of proprietary bead slurry. The potential for irreversible binding of abundant proteins to their respective IgY antibody, even after an extra stripping wash, and low amounts of total beads made using 66 μL of IgY-14 bead slurry unrealistic. In practice, almost doubling the amount of IgY-14 beads (100 μL) used was not sufficient, and resulted in albumin and transferrin peptides to be observed in high abundance (data not shown). The most important protein to immunodeplete is albumin and it has been reported to be a greater percentage of total CSF protein content (~60%) than serum (~49%) in humans.14 The difference in albumin percentage supports the results that proprietary blends of immunodepletion beads for high abundance proteins, such as albumin, cannot be scaled down on a strict protein scale and further modifications to the serum immunodepletion protocol need to be made.
Since IgY-14 beads were developed for use with serum, all of its protocols need to be taken into account to modify the protocol for CSF. Serum samples should be diluted fifty times before mixing with the IgY-14 beads, but CSF protein concentration is at least one hundred times lower than serum. Therefore, CSF is below half the recommended diluted protein concentration for IgY immunodepletion. Consequently, multiple steps have been devised to address this limitation. First, the binding time between the proteins targeted for removal from the CSF and IgY-14 beads was doubled during the end-over-end rotor depletion step from the recommended 15 minutes to 30 minutes. Second, to prevent non-specific protein binding to the antibodies, the CSF samples were diluted with an equal amount (100 μL) of 2X dilution buffer. The dilution buffer of NaCl and Tris-HCl was needed to assist in reducing non-specific binding of proteins to the 14 antibodies, and ensuring the sample is held at physiological pH. In addition to these modifications, a doubling of the starting IgY-14 bead slurry to 200 μL (equal to 100 μL IgY-14 resin) provided the desired results. Overall, this modified protocol results in effective depletion of CSF abundant proteins using only one-fifth of the antibodies provided by the smallest commercially available platform.
Data Analysis
Spectral counting technique for relative quantitation provides numerous benefits for the study of mouse CSF due to its label-free advantage. In contrast, isotopic labeling method often involves additional sample processing that could cause sample loss, which is highly undesirable for low protein content and low volume samples. Labeling methods also require a mixing of two sets of isotopically labeled samples, which would effectively increase the sample complexity and reduce the amount of sample that can be loaded onto the nanoLC column by half. In addition, more than two sets of samples can be compared by label-free methods. The use of label-free spectral counting method does not lead to an increase in sample complexity or interference in quantitation from peptides in the m/z window selected for tandem MS. Using spectral counting for relative quantitation however, is dependent on reproducible HPLC separation and careful mass spectrometry operation to minimize technical variability during the study. To address concerns of analytical reliability and run to run deviations, base peak chromatograms from two transgenic IgY-14 immunodepleted biological replicates, including two technical replicates of each, were shown to be highly reproducible (Figure 2). In addition, the coefficients of variance were calculated using the summation of all the ln(dNSAF) values for each run from Supplemental Table 3 for which produced a run-to-run variability of 32.7% for all the control samples and 18.3% for all the transgenic runs. As a whole, the coefficient of variance was 28.6% for all the runs including the transgenic and control CSF samples.
Figure 2.

Assessment of run to run variability of the base peak chromatograms within and between two biological and technical replicates. The peak profile and intensity scale is consistent between the four chromatograms. The four panels show two biological replicates (Tg 4 and Tg5) with two technical replicates for each biological sample.
Control and transgenic samples were both analyzed in triplicate using the same protocols on the amaZon ETD. From the three technical replicates for each biological replicate the spectral counts of the peptides for the proteins identified were summed. The results from these mouse CSF biological triplicates are shown in Figure 3A for GFAP overexpressor and Figure 3B for control. The summation of spectral counts for each biological replicate was performed to remove the inherent bias related to data dependent analysis for protein identification. One concern in grouping technical replicates is a potential loss of information regarding analytical variability. Figure 4 provides a graphical representation of variability of technical replicates illustrating the standard deviation of technical replicates with error bars. Figure 4 shows that a statistically changed protein, creatine kinase M (A), and an unchanged protein, kininogen-1 (B), have similar variability within (technical replicates) and between samples (biological replicates) for each protein. In addition, Figure 4B illustrates that even with the variability of kininogen-1, the resulting mean, shown by the dashed line, of control and transgenic samples were almost equal, whereas Figure 4A shows significantly different expression level of creatine kinase M. Performing replicate analysis of each biological sample (n=3) helps correct for systematic errors deriving from sample handling, and pooling of samples helps reduce random error during the CSF sample collection process.
Figure 3.

A: Venn-diagram of the biological triplicates (#1, #2, and #3) of transgenic mouse CSF showing 78 proteins identified in all three experiments. B: Venn-diagram of the biological triplicates (#1, #2, and #3) of control mouse CSF having 61 proteins identified by all three replicates. C: The overlap between control and transgenic CSF proteomic analysis showing 139 proteins identified by both groups, and 73 and 54 uniquely identified by respective groups.
Figure 4.

Assessment of technical replicate variability between biological replicates. The error bars in both A and B are the standard deviation derived from the technical triplicates for each biological replicate. Panel A shows creatine kinase M having more or equal variability in the biological triplicates than each technical triplicate. The means of the biological triplicates are illustrated by the dashed line. Panel B represents kininogen-1 which is unchanged between control (Ctl) and transgenic (Tg) samples. The biological variability coupled with the technical replicates provides a barely noticeable difference in the pooled mean between control and transgenic spectral counts. The difference in means is contrasted with the three fold change observed from creatine kinase M (A).
Protein Identification and Spectral Counting Analysis
The data for dNSAF analysis, like any mass spectrometry proteomics experiment, requires multiple layers of verification to ensure reliable data. Our initial protein identifications were subjected to a database search using a decoy database from Mascot, which resulted in an average false positive rate below 1% for all the proteins identified. Representative MS/MS spectra between control and GFAP transgenic samples are illustrated in Figure 5. Overall, 289 proteins were identified in a combination of control and transgenic samples (Supplemental Table 1). The next level of quality control was to only include ln(dNSAF) values from proteins identified by combining 2 or more unique peptides (Mascot score of ≥30.0) from all three analytical runs and observed at least in two out of three biological replicates in either control or transgenic samples. These selection parameters resulted in 103 proteins remaining after dNSAF analysis (Supplemental Table 3). The resulting spectral counts were then subjected to dSpC in order to account and correct for the systematic error of peptides shared by multiple proteins (Supplemental Table 3).
Figure 5.
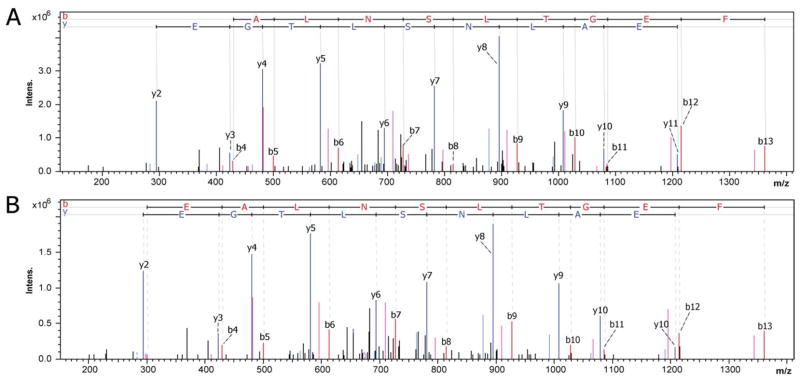
MS/MS profile of the tryptic peptide LSVEALNSLTGEFK from creatine kinase M. (A) The top MS/MS is from a control sample with an elution at 46.3 minutes. (B) The bottom MS/MS is from the GFAP transgenic sample with an elution at 46 minutes. These tandem MS spectra show instrument reliability and consistent fragmentation patterns which are necessary for spectral counting analysis.
It is inevitable in large scale and complex proteomics experiments that some proteins will be seen in some samples and not others. In addition, when controls were compared to transgenic samples, as shown in Figure 3C, 73 and 54 proteins were unique to control or GFAP transgenic samples respectively, whereas 139 proteins were observed in both samples. From dNSAF equation, if the spectral count is zero the numerator is zero and the value will not be normalized between runs. In order to circumvent the zero spectral counts in dNSAF analysis, zero spectral counts must be replaced by an experimentally determined non-zero value, determined to be 0.43. The 0.43 spectral counts for zero spectral counts was calculated by serial Shapiro-Wilk tests to determine the lowest value (0.43) for zero spectral counts and maintain a Gaussian distribution for all data sets. The 0.43 value for zero spectral counts in the current study was higher than the 0.16 reported value for zero spectral counts in the original NSAF spectral counting study.37 Our study may have a higher zero spectral count value than the previous study because the spectral counting data were an addition of three technical replicates and three times 0.16 is close to 0.43. The normalized Gaussian data were then subjected to t-test analysis and P-values below 0.05 are considered as statistically significant and are presented in Table 1. The proteins with significant up or down regulation from Table 1 can be further evaluated as how close significant proteins were to a p-value of 0.05, such as ganglioside GM2 activator, serine protease inhibitor A3N, and collagen alpha-2(I) chain having t-test scores of 0.045, 0.047, and 0.043 respectively. Proteins exhibiting a P-value close to 0.05 were more likely to be highly variable proteins or have smaller fold changes between control and transgenic samples, and thus provide less biological relevancy to future studies. The modest change in antithrombin-III, which is reduced by 1.4-fold in transgenic, is included due a low pooled standard deviation in spectral counts.
Table 1.
Statistically changed proteins between transgenic and control mouse CSF using dNSAF analysis
| Accession | Protein | Pa | SC%b | Fold Changec | Control dSpCd | Transgenic dSpCd |
|---|---|---|---|---|---|---|
| KCRM_MOUSE | Creatine kinase M-type | 0.034 | 42.5 | 3.0 | 18.2 | 54.1 |
| HEXB_MOUSE | eta-hexosaminidase subunit β | 0.012 | 18.3 | ↑ | 0 | 5.9 |
| CMGA_MOUSE | Chromogranin-A | 0.00078 | 15.3 | ↓ | 4.4 | 0 |
| ANT3_MOUSE | Antithrombin-III | 0.017 | 17.6 | −1.4 | 6.7 | 4.7 |
| SAP3_MOUSE | Ganglioside GM2 activator | 0.045 | 41.5 | ↓ | 2.8 | 0 |
| SPA3N_MOUSE | Serine protease inhibitor A3N | 0.047 | 17.0 | 4.2 | 1.0 | 4.2 |
| CO1A2_MOUSE | Collagen alpha-2(I) chain | 0.043 | 5.6 | ↓ | 1.9 | 0 |
| BLVRB_MOUSE | Flavin reductase | 0.0054 | 20.4 | ↑ | 0 | 1.2 |
| CATS_MOUSE | Cathepsin S | 0.0032 | 23.2 | ↑ | 0 | 7.3 |
| GFAP_MOUSE | Glial fibrillary acidic protein | 0.021 | 10.7 | ↑ | 0 | 2.1 |
| RET4_MOUSE | Retinol-binding protein 4 | 0.040 | 18.9 | ↓ | 1.3 | 0 |
| CBPE_MOUSE | Carboxypeptidase E | 0.043 | 13.9 | ↓ | 1.1 | 0 |
| CATL1_MOUSE | Cathepsin L1 | 0.015 | 8.7 | 9.4 | 0.2 | 1.9 |
The statistics are performed using the t-test from the ln(dNSAF) Gaussian data.
P: p-value of the t-test, where the null hypothesis states that there was no change in expression between control and transgenic GFAP overexpressor. P-value was calculated using the control means of ln(dNSAF) and overexpressor ln(dNSAF) using a pooled standard deviation from each individual protein.
SC%: percentage of sequence coverage of the listed protein from sequenced tryptic peptides.
Fold Change: positive values are indicative of a fold change for transgenic CSF, negative values are fold changes for control CSF (i.e., down-regulated in transgenic CSF), ↑ indicates the protein was only detected in transgenic CSF, and ↓ indicates the protein was only detected in control CSF.
dSpC: distributive spectral counts which represent the average spectral counts observed per run analysis on the mass spectrometer and corrected using distributive analysis for peptides shared by more than one protein.
Spectral counting has been analyzed with fold changes derived directly from the average spectral counts from the technical replicates and then the average of the three biological replicates. We decided to perform additional analysis using fold changes to dig deeper into proteins that statistically support the null hypothesis from the dNSAF analysis. To only pull out highly confident protein identifications we used the same strict cut-off of two unique peptides identified per protein as in dNSAF analysis. We only accepted proteins with greater than threefold change in total spectral counts listed in Table 2. Two proteins, contactin-1 (cntn1) and cathepsin B (CB), illustrate the potential biomarkers missed by dNSAF analysis. Cntn1 had zero spectral count in the transgenic sample, and had an average spectral count of 4.1 in control samples. The lack of any spectral counts in one biological control for cntn1 resulted in a large standard deviation in the ln(dNSAF) means for the control which resulted in the t-test supporting the null hypothesis. Another example is CB, which was detected by numerous spectral counts in every GFAP transgenic biological replicate with an average spectral count of 9.7 (Table 2). The presence of CB in one biological control sample (2.3 average spectral counts) resulted in a high standard deviation in the mean of the control samples. These examples exhibit a limitation of dNSAF analysis which could cause a loss of potentially useful information.
Table 2.
Proteins showing greater than three-fold changes with at least two unique peptides identified for each protein.
| Accession | Protein | SC (%)a | Fold Change b | Control dSpC c | Transgenic dSpC c |
|---|---|---|---|---|---|
| MUG1_MOUSE | Murinoglobulin-1 precursor | 14.7 | 4.2 | 15.5 | 3.7 |
| CO4B_MOUSE | Complement C4-B | 11.3 | 5.4 | 2.2 | 11.8 |
| PRDX6_MOUSE | Peroxiredoxin-6 | 57.6 | 4.6 | 1.4 | 6.4 |
| CNTN1_MOUSE | Contactin-1 | 6.5 | ↓ | 4.1 | 0 |
| CATB_MOUSE | Cathepsin B | 26.3 | 4.2 | 2.3 | 9.7 |
| CAH3_MOUSE | Carbonic anhydrase 3 | 39.6 | 7.6 | 1.1 | 8.4 |
| APOH_MOUSE | Beta-2-glycoprotein 1 | 12.8 | 4.4 | 4.4 | 1 |
| CSF1R_MOUSE | Macrophage colony-stimulating factor 1 receptor | 8.0 | 4.4 | 1.4 | 6.2 |
| PGK1_MOUSE | Phosphoglycerate kinase 1 | 35.5 | 3.6 | 1.7 | 6.1 |
| NDKB_MOUSE | Nucleoside diphosphate kinase B | 40.8 | 3.4 | 1.3 | 4.4 |
| FHL1_MOUSE | Four and a half LIM domains protein 1 | 24.3 | 3.9 | 1.3 | 5.1 |
| NELL2_MOUSE | Protein kinase C-binding protein NELL2 | 4.5 | −4.3 | 1.3 | 0.3 |
| MDHM_MOUSE | Malate dehydrogenase, mitochondrial | 38.5 | 4.1 | 1.2 | 4.9 |
| CSF1R_MOUSE | Macrophage colony-stimulating factor 1 receptor | 8.0 | 4.4 | 1.4 | 6.2 |
SC%: percentage of sequence coverage of the listed protein from sequenced tryptic peptides.
Fold Change: positive values are indicative of a fold change for transgenic CSF, negative values are fold changes for control CSF, and ↓ indicates the protein was only detected in control CSF.
dSpC: distributive spectral counts which represent the average spectral counts observed per run analysis on the mass spectrometer and corrected using distributive analysis for peptides shared by more than one protein.
Previously Identified Proteins with Expression Changes
Previously, three proteins have been described as increased in CSF from individual(s) suffering from AxD. In one study, a single patient’s CSF was found to have elevated levels of αβ-crystallin and HSP27.44 In a second study, three patients were reported to have elevated levels of GFAP, with concentrations from 4760 ng/L to 30,000 ng/L (compared to <175 ng/L for controls).11 GFAP was detected in our current study; however, the other two proteins were not detected. One possibility is that αB-crystallin and Hsp-27 levels in mouse CSF are too low for detection by MS analysis. In addition, while the transgenic mice display the hallmark pathological feature of AxD in the form of Rosenthal fibers, they do not have any evident leukodystrophy and thus may not display the full range of changes in CSF as might be found in human patients.
Creatine Kinase M
Creatine kinase M (M-CK) is a 43 kDa protein whose role is to reversibly catalyze phosphate transfer between ATP and energy storage compounds. M-CK has been primarily found in muscle tissue, and for humans, creatine kinase M is diagnostically analyzed in the blood for myocardial infarction (heart attack). In addition, M-CK is also found in Purkinje neurons of the cerebellum.45, 46 A related protein, creatine kinase B (B-CK), also exhibited an apparent 2.1 fold increase in transgenic CSF over control, but this difference was not statistically different. B-CK concentration is known to be elevated in CSF following head trauma47 or cerebral infarction,48 but decreased in astrocytes in individuals affected by multiple sclerosis.49
Cathepsin
The data showed multiple cathepsins were up regulated in the CSF of transgenic mice when compared to control mice. The up regulated cathepsins were S, L1, and B isoforms which are all cysteine proteases. Cathepsin S (CS) was never observed in control samples, but observed with an average of 7.3 spectral counts per analysis. Cathepsin L1 (CL1) was up regulated by 9.4 times in transgenic mice. These two cathepsins exhibited significant changes using dNSAF and t-test statistics as shown in Table 1, and Cathepsin B (CB) showed a 4.2 fold increase in transgenic CSF as shown in Table 2.
Cathepsins regulate apoptosis in cells50 which is the major mechanism for elimination of cells deemed by the organism to be dangerous, damaged, or expendable. CL and CB are redundant in the system as thiol proteases and inhibition of CB by CA-047 causes a diminished apoptosis response in multiple cell lines.51 Intriguingly, increased levels of CB or CL are correlated with poor prognosis for cancer patients and shorter disease-free intervals. It is believed that these proteases degrade the extracellular membrane which allows tumor cells to invade adjacent tissue and metastasize.52 With regards to AxD, the up regulation of these cathepsins may be indicative of the body’s natural defense and response to Rosenthal fibers. Thus, stimulation of these cathepsins may provide a further protective stress response, but the positive correlation between these proteases and cancer highlights the multiple roles of these proteins in pathological response. Alternatively, it has been shown that increased CB is involved with the tumor necrosis factor α (TNFα) induced apoptosis cascade.53 The activation of the TNFα could produce oligodendrocyte toxicity54, with the expression of TNFα being elevated in tissue samples from mouse models and AxD patients.55 The potential for a positive or a negative effect in increasing or decreasing cathepsins warrant future research with cathepsins and AxD.
Contactin-1
Contactin-1 (Cntn1) is a 133 kDa cell surface protein that is highly glycosylated and belongs to a family of immunoglobulin domain-containing cell adhesion molecues.56 Table 2 shows that Cntn1 is down regulated in transgenic mice since spectral counts were only observed in control mice. Cntn1 is expressed in neurons and oligodendrocytes and higher levels were observed during brain development.57 In addition, Cntn1 leads to activation of Notch1 which mediates differentiation and maturation of oligodendrocyte precursor cells (OPC). Although the mechanism leading to a reduction of Cntn1 in CSF is not known, it may reflect a change in astrocyte interactions with either neurons or oligodendrocytes to alter their expression of this protein.
Validation of putative biomarkers and MS proteomics data using ELISA and RNA microarray data
To further validate the relative protein expression data obtained via MS-based spectral counting techniques, orthogonal immunological and molecular biological approaches have been examined. As GFAP was shown to be elevated in the CSF of transgenic animals, we used a well-defined GFAP ELISA protocol to test its CSF concentration. CSF from 8 week old male mice was collected from both transgenic and control animals. Five samples of transgenic CSF was prepared by pooling four to eight animals for the GFAP ELISA. In the case of controls, each sample represents a single animal. GFAP concentrations observed by both the MS and ELISA showed significant increases of GFAP in the CSF when comparing transgenic to control animals.
Another validation of MS spectral counts is observed in a microarray analysis performed on transgenic mouse olfactory bulb tissue 55. In this paper, nine of the proteins found by MS showed similar gene expression in the microarray (Table 3). It is not surprising that all the genes observed in the microarray are not the same as the proteins observed by MS analysis. Gene expression and protein synthesis and expression are not always correlated, but the similarities and overlapping trends observed with these two assays are encouraging. As shown in Table 3, gene expression analysis also revealed the up regulation of several cathepsin proteins, GFAP, and down regulation of contactin-1 in CSF collected from transgenic mice, corroborating the MS-based proteomics results.
Table 3.
Validation of changes in proteins revealed by MS-based spectral counting consistent with previously published microarray data
| Microarray fold change (transgenic vs wild-type) in 4 month old mice | |||
|---|---|---|---|
| ↑ regulated in transgenic | ↓ regulated in transgenic | ||
| Cathepsin S | 8.4 ± 0.6 | Contactin-1 | −1.6 ± 0.1 |
| Cathepsin B | 2.0 ± 0.2 | Carboxypeptidase E | −1.4 ± 0.1 |
| Cathepsin L1 | 2.1 ± 0.1 | ||
| Peroxiredoxin-6 | 2.5 ± 0.1 | ||
| Complement C4-B | 6.9 ± 0.9 | ||
| Glial fibrillary acidic protein | 4.0 ± 0.6 | ||
| Serine protease inhibitor A3N | 5.4 ± 0.4 | ||
Fold change ± standard deviation
Note: Validation of putative biomarkers from the current proteomics dataset by previously published RNA microarray data.55 Both up and down regulated proteins were consistent with the RNA microarray data.
CONCLUSIONS
In this study we have produced a panel of proteins with significant up or down regulation in the CSF of transgenic GFAP overexpressor mice. This mouse model for AxD was consistent with the previous studies showing elevation of GFAP in CSF. The development of a modified IgY-14 immunodepletion technique for low amounts of CSF was presented. This improved protocol is useful for future investigations to deal with the unique challenges of mouse CSF analysis. Modified proteomics protocols were employed to profile mouse CSF with biological and technical triplicates addressing the variability and providing quantitation with dNSAF spectral counting. Validation of the MS-based proteomics data were performed using both ELISA and RNA microarray data to provide further confidence in the changes in the putative protein biomarkers. This study presents three classes of interesting targets for future study in AxD, including CK-M/CK-B, cathepsin S, L1, and B isoforms, and contactin-1.
Supplementary Material
Compilation list of proteins identified from all the control and transgenic biological replicates.
Distributive spectral counting calculations performed for proteins observed to share identified peptides.
Proteins with dNSAF spectral counting performed, including t-test analysis for a comparison between transgenic and control CSF.
Figure 6.

Validation of MS spectral counts using a GFAP ELISA. GFAP content (ng/L) measured within mouse CSF from both transgenic and control animals. The data represents the average with standard deviation (n=5, each sample represents pooling of 1 to 8 animals). The statistics are performed using a student t-test, p<0.0001.
Acknowledgments
This work was primarily supported by grants from the NIH (NS060120 and HD03352). Support for this work was also provided by the University of Wisconsin Institute for Clinical and Translational Research (Clinical and Type 1 Translational Pilot Grant) funded by grant 1UL1 RR025011 from the Clinical and Translational Science Award (CTSA) program of the National Center for Research Resources, National Institutes of Health. The authors wish to thank the Analytical Instrument Center at the UW School of Pharmacy for the access to the amaZon ETD ion trap mass spectrometer.
References
- 1.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27(1):117–20. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 2.Herndon RM, Rubinstein LJ, Freeman JM, Mathieson G. Light and electron microscopic observations on Rosenthal fibers in Alexander’s disease and in multiple sclerosis. J Neuropathol Exp Neurol. 1970;29(4):524–51. doi: 10.1097/00005072-197010000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72(3):373–81. doi: 10.1093/brain/72.3.373. 3 pl. [DOI] [PubMed] [Google Scholar]
- 4.Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell. 1989;57(1):71–8. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- 5.Head MW, Goldman JE. Small heat shock proteins, the cytoskeleton, and inclusion body formation. Neuropathol Appl Neurobiol. 2000;26(4):304–12. doi: 10.1046/j.1365-2990.2000.00269.x. [DOI] [PubMed] [Google Scholar]
- 6.Messing A, Daniels CM, Hagemann TL. Strategies for treatment in alexander disease. Neurotherapeutics. 7(4):507–15. doi: 10.1016/j.nurt.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17(11):1540–55. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum Mol Genet. 2009;18(7):1190–9. doi: 10.1093/hmg/ddp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zougman A, Pilch B, Podtelejnikov A, Kiehntopf M, Schnabel C, Kumar C, Mann M. Integrated analysis of the cerebrospinal fluid peptidome and proteome. J Proteome Res. 2008;7(1):386–99. doi: 10.1021/pr070501k. [DOI] [PubMed] [Google Scholar]
- 10.Sjodin MO, Bergquist J, Wetterhall M. Mining ventricular cerebrospinal fluid from patients with traumatic brain injury using hexapeptide ligand libraries to search for trauma biomarkers. J Chromatogr B Analyt Technol Biomed Life Sci. 878(22):2003–12. doi: 10.1016/j.jchromb.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 11.Kyllerman M, Rosengren L, Wiklund LM, Holmberg E. Increased levels of GFAP in the cerebrospinal fluid in three subtypes of genetically confirmed Alexander disease. Neuropediatrics. 2005;36(5):319–23. doi: 10.1055/s-2005-872876. [DOI] [PubMed] [Google Scholar]
- 12.DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem. 2002;81(2):229–36. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- 13.Wong M, Schlaggar BL, Buller RS, Storch GA, Landt M. Cerebrospinal fluid protein concentration in pediatric patients: defining clinically relevant reference values. Arch Pediatr Adolesc Med. 2000;154(8):827–31. doi: 10.1001/archpedi.154.8.827. [DOI] [PubMed] [Google Scholar]
- 14.Roche S, Gabelle A, Lehmann S. Clinical proteomics of the cerebrospinal fluid: Towards the discovery of new biomarkers. PROTEOMICS – Clinical Applications. 2008;2(3):428–436. doi: 10.1002/prca.200780040. [DOI] [PubMed] [Google Scholar]
- 15.Li C, Lee KH. Affinity depletion of albumin from human cerebrospinal fluid using Cibacron-blue-3G-A-derivatized photopatterned copolymer in a microfluidic device. Anal Biochem. 2004;333(2):381–8. doi: 10.1016/j.ab.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Maccarrone G, Milfay D, Birg I, Rosenhagen M, Holsboer F, Grimm R, Bailey J, Zolotarjova N, Turck CW. Mining the human cerebrospinal fluid proteome by immunodepletion and shotgun mass spectrometry. ELECTROPHORESIS. 2004;25(14):2402–2412. doi: 10.1002/elps.200305909. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Harvie G, Feitelson JS, Gramatikoff K, Herold DA, Allen DL, Amunngama R, Hagler RA, Pisano MR, Zhang WW, Fang X. Immunoaffinity separation of plasma proteins by IgY microbeads: meeting the needs of proteomic sample preparation and analysis. Proteomics. 2005;5(13):3314–28. doi: 10.1002/pmic.200401277. [DOI] [PubMed] [Google Scholar]
- 18.Rajic A, Stehmann C, Autelitano DJ, Vrkic AK, Hosking CG, Rice GE, Ilag LL. Protein depletion using IgY from chickens immunised with human protein cocktails. Prep Biochem Biotechnol. 2009;39(3):221–47. doi: 10.1080/10826060902952915. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Fang X. Immunoaffinity fractionation of plasma proteins by chicken IgY antibodies. Methods Mol Biol. 2008;425:41–51. doi: 10.1007/978-1-60327-210-0_4. [DOI] [PubMed] [Google Scholar]
- 20.Greunke K, Braren I, Alpers I, Blank S, Sodenkamp J, Bredehorst R, Spillner E. Recombinant IgY for improvement of immunoglobulin-based analytical applications. Clin Biochem. 2008;41(14–15):1237–44. doi: 10.1016/j.clinbiochem.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Y, Gao X, Taratula O, Treado S, Urbas A, Holbrook RD, Cavicchi RE, Avedisian CT, Mitra S, Savla R, Wagner PD, Srivastava S, He H. Anti-HER2 IgY antibody-functionalized single-walled carbon nanotubes for detection and selective destruction of breast cancer cells. BMC Cancer. 2009;9:351. doi: 10.1186/1471-2407-9-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Qian WJ, Mottaz HM, Gritsenko MA, Norbeck AD, Moore RJ, Purvine SO, Camp DG, 2nd, Smith RD. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol Cell Proteomics. 2006;5(11):2167–74. doi: 10.1074/mcp.T600039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinerfeld D, Innamorati D, Pirro J, Tam SW. Serum/Plasma depletion with chicken immunoglobulin Y antibodies for proteomic analysis from multiple Mammalian species. J Biomol Tech. 2004;15(3):184–90. [PMC free article] [PubMed] [Google Scholar]
- 24.Metz TO, Qian WJ, Jacobs JM, Gritsenko MA, Moore RJ, Polpitiya AD, Monroe ME, Camp DG, 2nd, Mueller PW, Smith RD. Application of proteomics in the discovery of candidate protein biomarkers in a diabetes autoantibody standardization program sample subset. J Proteome Res. 2008;7(2):698–707. doi: 10.1021/pr700606w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ru QC, Zhu LA, Silberman J, Shriver CD. Label-free semiquantitative peptide feature profiling of human breast cancer and breast disease sera via two-dimensional liquid chromatography-mass spectrometry. Mol Cell Proteomics. 2006;5(6):1095–104. doi: 10.1074/mcp.M500387-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Brechlin P, Jahn O, Steinacker P, Cepek L, Kratzin H, Lehnert S, Jesse S, Mollenhauer B, Kretzschmar HA, Wiltfang J, Otto M. Cerebrospinal fluid-optimized two-dimensional difference gel electrophoresis (2-D DIGE) facilitates the differential diagnosis of Creutzfeldt-Jakob disease. Proteomics. 2008;8(20):4357–66. doi: 10.1002/pmic.200800375. [DOI] [PubMed] [Google Scholar]
- 27.Rao PV, Reddy AP, Lu X, Dasari S, Krishnaprasad A, Biggs E, Roberts CT, Nagalla SR. Proteomic identification of salivary biomarkers of type-2 diabetes. J Proteome Res. 2009;8(1):239–45. doi: 10.1021/pr8003776. [DOI] [PubMed] [Google Scholar]
- 28.Yu KH, Barry CG, Austin D, Busch CM, Sangar V, Rustgi AK, Blair IA. Stable isotope dilution multidimensional liquid chromatography-tandem mass spectrometry for pancreatic cancer serum biomarker discovery. J Proteome Res. 2009;8(3):1565–76. doi: 10.1021/pr800904z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 30.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem. 2003;75(24):6843–52. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 33.Xiang F, Ye H, Chen R, Fu Q, Li L. N,N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal Chem. 82(7):2817–25. doi: 10.1021/ac902778d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Zybailov B, Coleman MK, Florens L, Washburn MP. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem. 2005;77(19):6218–24. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]
- 36.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4(10):1487–502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5(9):2339–47. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 38.Mosley AL, Florens L, Wen Z, Washburn MP. A label free quantitative proteomic analysis of the Saccharomyces cerevisiae nucleus. J Proteomics. 2009;72(1):110–20. doi: 10.1016/j.jprot.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Wen Z, Washburn MP, Florens L. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 82(6):2272–81. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- 40.Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152(2):391–8. [PMC free article] [PubMed] [Google Scholar]
- 41.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 43.You JS, Gelfanova V, Knierman MD, Witzmann FA, Wang M, Hale JE. The impact of blood contamination on the proteome of cerebrospinal fluid. Proteomics. 2005;5(1):290–6. doi: 10.1002/pmic.200400889. [DOI] [PubMed] [Google Scholar]
- 44.Takanashi J, Sugita K, Tanabe Y, Niimi H. Adolescent case of Alexander disease: MR imaging and MR spectroscopy. Pediatr Neurol. 1998;18(1):67–70. doi: 10.1016/s0887-8994(97)00128-8. [DOI] [PubMed] [Google Scholar]
- 45.Hemmer W, Wallimann T. Functional Aspects of Creatine Kinase in Brain. Developmental Neuroscience. 1993;15(3–5):249–260. doi: 10.1159/000111342. [DOI] [PubMed] [Google Scholar]
- 46.Hemmer W, Zanolla E, Furter-Graves EM, Eppenberger HM, Wallimann T. Creatine kinase isoenzymes in chicken cerebellum: specific localization of brain-type creatine kinase in Bergmann glial cells and muscle-type creatine kinase in Purkinje neurons. Eur J Neurosci. 1994;6(4):538–49. doi: 10.1111/j.1460-9568.1994.tb00298.x. [DOI] [PubMed] [Google Scholar]
- 47.Nordby HK, Tveit B, Ruud I. Creatine kinase and lactate dehydrogenase in the cerebrospinal fluid in patients with head injuries. Acta Neurochirurgica. 1975;32(3):209–217. doi: 10.1007/BF01405454. [DOI] [PubMed] [Google Scholar]
- 48.Bell RD, Alexander GM, Nguyen T, Albin MS. Quantification of cerebral infarct size by creatine kinase BB isoenzyme. Stroke. 1986;17(2):254–60. doi: 10.1161/01.str.17.2.254. [DOI] [PubMed] [Google Scholar]
- 49.Steen C, Wilczak N, Hoogduin JM, Koch M, De Keyser J. Reduced Creatine Kinase B Activity in Multiple Sclerosis Normal Appearing White Matter. PLoS ONE. 5(5):e10811. doi: 10.1371/journal.pone.0010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chwieralski C, Welte T, Bühling F. Cathepsin-regulated apoptosis. Apoptosis. 2006;11(2):143–149. doi: 10.1007/s10495-006-3486-y. [DOI] [PubMed] [Google Scholar]
- 51.Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, Salvesen GS, Stoka V, Turk V, Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283(27):19140–50. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 52.Duffy MJ. Proteases as prognostic markers in cancer. Clin Cancer Res. 1996;2(4):613–8. [PubMed] [Google Scholar]
- 53.Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106(9):1127–37. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butt AM, Jenkins HG. Morphological changes in oligodendrocytes in the intact mouse optic nerve following intravitreal injection of tumour necrosis factor. J Neuroimmunol. 1994;51(1):27–33. doi: 10.1016/0165-5728(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 55.Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14(16):2443–58. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- 56.Falk J, Bonnon C, Girault JA, Faivre-Sarrailh C. F3/contactin, a neuronal cell adhesion molecule implicated in axogenesis and myelination. Biol Cell. 2002;94(6):327–34. doi: 10.1016/s0248-4900(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 57.Eckerich C, Zapf S, Ulbricht U, Müller S, Fillbrandt R, Westphal M, Lamszus K. Contactin is expressed in human astrocytic gliomas and mediates repulsive effects. Glia. 2006;53(1):1–12. doi: 10.1002/glia.20254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Compilation list of proteins identified from all the control and transgenic biological replicates.
Distributive spectral counting calculations performed for proteins observed to share identified peptides.
Proteins with dNSAF spectral counting performed, including t-test analysis for a comparison between transgenic and control CSF.