

Abstract
Site-specific labeling of biomolecules in vitro with gold clusters can enhance the information content of electron cryomicroscopy experiments. This chapter provides a practical overview of well-established techniques for forming biomolecule/gold cluster conjugates. Three bioconjugation chemistries are covered: Linker-mediated bioconjugation, direct gold–biomolecule bonding, and coordination-mediated bonding of nickel(II) nitrilotriacetic acid (NTA)-derivatized gold clusters to polyhistidine (His)-tagged proteins.
1. Introduction
Electron cryomicroscopy comprises a set of techniques brimming with promises and possibilities as varied as atomic resolution protein structure on noncrystalline biomolecules (Cong et al., 2010) and visual proteomics (Nickell et al., 2006). The suite of electron cryomicroscopy techniques fails to achieve its theoretical limits in large part because of various contrast problems: poor signal-to-noise ratio (SNR) constitutes a substantial practical limitation for all applications of electron cryomicroscopy to “unstained” biological materials.
Site-specific labeling with gold cluster tags has been used to skirt the SNR limitations inherent to electron cryomicroscopy techniques. Gold clusters have amplitude contrast in addition to phase contrast in the electron microscope, and so are easily distinguished in images and in image processing from biological molecules which have only phase contrast. While in principle, clusters or nanoparticles comprised of any heavy metal could be used as an electron contrast marker, gold is adopted most widely because it is comparatively inert, insensitive to molecular oxygen, forms nanoparticles comparatively easily because of the low surface energy of Au (Alvarez et al., 1997), and because the chemistries for surface functionalization of gold nanoparticles are comparatively well understood and controlled in aqueous solution.
This chapter focuses on methods for site-specific gold cluster labeling of biomolecules. Here, we define “biomolecules” as including proteins, nucleic acids, lipids, and biologically active small molecules. Gold clusters are here defined as a cluster of gold atoms containing multiple gold–gold bonds which are rendered kinetically metastable by a protecting or stabilizing ligand layer. They are differentiated from colloidal gold in that they have molecule-like properties and often well-defined molecular formulae, and sometimes, crystal structure. This is in contrast to colloidal gold where “monodisperse” is usually defined as ±10% or lower coefficient of variation in core size, and the gold cores are generally comprised of multiply twinned crystallites with many different crystal structures present in the same preparation. Figure 9.1 shows a selection of well-established gold clusters, with structures shown if presently determined. A representation of 5 nm colloidal gold is included for size comparison.
Figure 9.1.
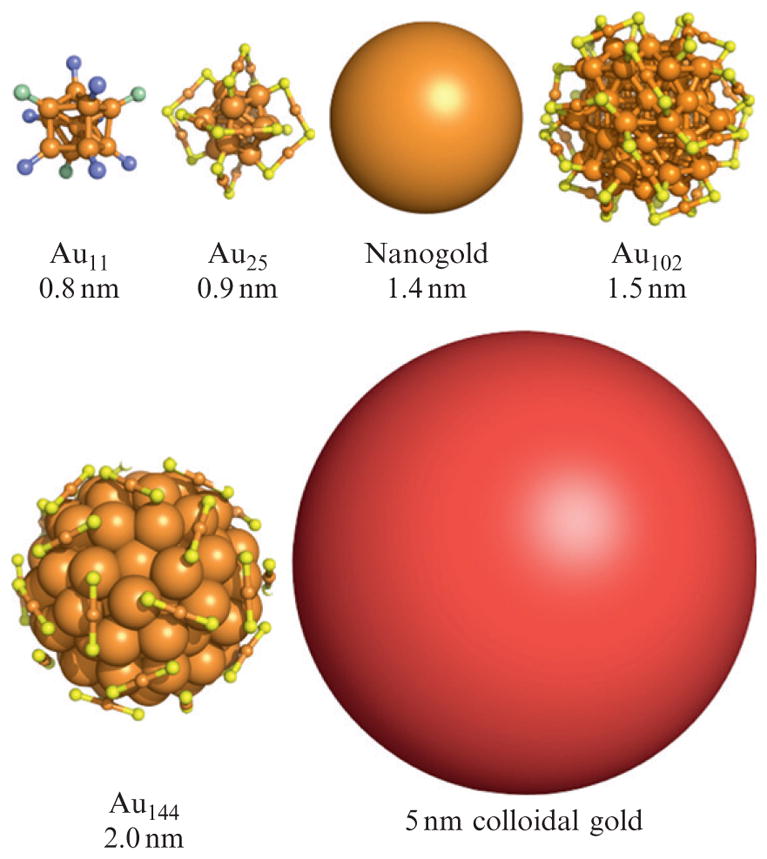
Representative inorganic gold clusters. The organic components of the ligand layer of each cluster are not shown. Orange, yellow, purple, and green represent gold, sulfur, phosphorous, and chlorine, respectively. The images of Au11, Au25, and Au102 are created from X-ray crystallographic coordinates. The image of Au144 is from a density functional theory model (Lopez-Acevedo et al., 2009). A 5-nm diameter colloidal gold particle is shown for comparison.
Just as there are many different modalities of cryomicroscopy, there are multiple approaches to making gold cluster/biomolecule conjugates for electron cryomicroscopy (Table 9.1; Fig. 9.2). Generally, the methods can be categorized according to the nature of the interface between biomolecule and gold cluster. There are three commonly used nanoparticle/biomolecule interfacial chemistries: Electrostatic, linker-mediated bonding, and direct bonding (Fig. 9.2).
Table 9.1.
Properties of selected gold cluster labels
| Gold cluster | Bioconjugate reactive group(s) of cluster | Labeled biomolecule moiety | Nominal diameter |
|---|---|---|---|
| Undecagold | Monoamino-, maleimido, Sulfo-NHS−, | –SH, –NH2 | 0.8 nm |
| Nanogold | Amino-, maleimido, Sulfo-NHS, | –SH, –NH2 | 1.4 nm |
| Ni-NTA–Nanogold | Ni(II) nitrilotriacetic acid (NTA) | Polyhistidine, particularly His6 | 1.8, 5 nm |
| MPCs: Au25, Au38, Au68, Au102, Au144 | Au | –SH | 0.9, 1.1, 1.3, 1.5, 2.0 nm |
Figure 9.2.

Schematic of the bioconjugation chemistries presented in this chapter, which are summarized in Table 9.1. In the upper left corner is shown a “direct” biomolecule/gold cluster conjugate where a thiol moiety such as a cysteine residue bonds directly to the gold cluster core of Au102(p-mercaptobenzoic acid)44 through a sulfur–gold bond. The crystallographically determined ligand layer of the cluster is rendered as a partially transparent surface in that image, illustrating the steric constraint of this linkage. Shown in upper and lower right panels are the bioconjugates formed by NHS and malimido derivatives of tris (aryl) ligands which protect clusters such as Nanogold and undecagold. Shown at lower left is an Ni2+-mediated interaction between a polyhistidine tag and a nitroloacetic acid (NTA)-derivatized gold cluster. Note that all stable water-soluble gold clusters and colloids have a full organic ligand shell like that shown for Au102 in this diagram, but these ligand layers are sometimes not as well characterized as the cluster core.
This chapter focuses on the linker-mediated and direct bioconjugation strategies that make stable covalent bonds between well-defined gold cluster compounds and biomolecules. This introduction provides an overview of the bioconjugate chemistries shown in Fig. 9.2. These various clusters can be used to form gold cluster-cysteine, -amine, or Ni(II)-His6 cross-links to biomolecules (Fig. 9.2 and Table 9.1).
Linker-mediated bioconjugate chemistries, as depicted in the upper right and lower right panels of Fig. 9.2, have been used for decades to reliably make gold cluster bioconjugates that may be molecularly defined. Given the widespread and easy use of these clusters, their use will be discussed in detail. The best known examples of gold nanoparticles used for linker-mediated biological labeling are undecagold (Bartlett et al., 1978; Cariati and Naldini, 1971; Safer et al., 1982) and the commercial product Nanogold (Hainfeld and Furuya, 1992).
Nanogold and undecagold are available in nominally monofunctional forms (Nanoprobes, Inc., Yaphank, NY). The monomaleimido and mono-Sulfo-NHS forms of Nanogold and undecagold have been used widely for site-specific labeling of cysteine and primary amine-containing residues respectively within proteins. This bioconjugate chemistry has been used to label antibodies and their fragments (Hainfeld and Furuya, 1992; Hainfeld and Powell, 1997; Ribrioux et al., 1996), other proteins (Rappas et al., 2005; Yonekura et al., 2006), peptides (Segond von Banchet et al., 1999), oligonucleotides (Alivisatos et al., 1996; Hamad-Schifferli et al., 2002; Park et al., 2005; Xiao et al., 2002), and lipids that assemble into liposomes (Hainfeld et al., 1999a). The biological (Hainfeld and Powell, 1997, 2000) and nanobiotechnological (Hainfeld et al., 2004) aspects of covalent gold labeling with Nanogold and undecagold have been reviewed previously.
Metal cluster labels can also be targeted to genetically encoded tags. A 1.8-nm Nanogold cluster label, stabilized and solubilized using a different ligand shell, has been functionalized with a derivative of nitrilotriacetic acid (NTA). Upon incubation with nickel(II) salts, the Nanogold cluster forms a nickel(II) complex with two vacant adjacent binding sites, which binds strongly to histidine residues (Hainfeld et al., 1999b). Consequently, this structure binds very strongly to polyhistidine (His) tags such as hexahistidine (His6), which are commonly used as affinity purification tags. A 5-nm analog has recently been introduced (Dubendorff et al., 2010; Reddy et al., 2005).
It is also possible to form bonds between cysteine residues within proteins and gold clusters, on the basis of gold–thiol bonding. This strategy eliminates the organic linker between the protein and gold core, which is advantageous for highest resolution localization of cluster labels on proteins. This labeling strategy also takes advantage of a set of molecularly defined, and in some cases, structurally defined (Heaven et al., 2008; Jadzinsky et al., 2007) gold cluster compounds whose gold atom nuclearities include Au25, Au38, Au68, Au102, and Au144. This strategy has been used to form gold cluster conjugates with proteins (Ackerson et al., 2006, 2010; Aubin-Tam and Hamad-Schifferli, 2005; Aubin-Tam et al., 2009), peptides (Krpetic et al., 2009; Levy et al., 2004), oligonucleotides (Ackerson et al., 2005), and bioactive small molecules (Bowman et al., 2008). This labeling strategy is more complicated than the linker-mediated strategies, because the site of label and chemical composition of the surface layer of the gold cluster influences both the yield of the labeling reaction and the biological function of the cluster/protein conjugate (Aubin-Tam et al., 2009). Fortunately, guidelines for choosing sites for gold cluster labeling exist to guide the design of these experiments.
The bioconjugation strategies presented in this chapter are chosen because they are robust and have been repeated in many laboratories. Other in vitro labeling means have been used to make nanoparticle/protein bioconjugates. Tetrairidium clusters have been used to derivatize virus capsids for high-resolution EM and single particle analysis (Cheng et al., 1999). Although it is too small to be directly visualized in a standard TEM, and hence is suited mainly to single particle analysis, this cluster provides very high resolution and minimal structural perturbation. Larger platinum clusters 0.8–2.5 nm in diameter, stabilized by 1,10-phenanthroline derivatives, have been used as covalent Fab′ labels (Powell et al., 1999). However, covalent gold labels are preferred because they afford better synthetic control of particle size distribution, solubility, and stability, and more specific biomolecule conjugation.
Colloidal gold nanoparticles, defined here as particulate gold with diameters between 5 and 250 nm, have found widespread use in biological electron microscopy. Most commonly, they are used as fiducial markers in electron tomography and in the gold colloid/antibody conjugates widely used in the immunohistochemistry (Handley, 1989a,b). In contrast to the discrete molecular particle sizes that have recently emerged for gold nano-particles approximately 3 nm in diameter or smaller, these colloidal particles can be prepared with average core diameters that comprise a more or less continuous distribution. From a physicochemical standpoint, when compared to the molecular or molecule-like gold clusters that are the focus of this chapter, these particles are generally larger, comprised of multiply twinned crystallites, and defined as monodisperse when the coefficient of variation of particle diameter within a preparation is 10% of the average diameter or less. From a bioconjugate chemical standpoint, gold colloids generally bind to biomolecules through electrostatic, rather than covalent, interactions.
A consequence of the electrostatic nature of the colloidal gold–protein bond is that site-specific biomolecule labeling with gold colloids is not routine. For instance, colloidal gold antibody conjugates are usually defined in terms of “average number of antibodies per colloid” for large colloids or “average number of colloids per antibody” for small colloids, as opposed to a specific labeled residue within a protein as is the case for the conjugates that are the focus of this chapter. Other consequences of the electrostatic interaction typical in gold colloid/biomolecule conjugates include dissociation of the colloid which can result in competition between labeled and unlabeled probe giving lower labeling (Kramarcy and Sealock, 1990), and the residual affinity of colloidal gold for proteins which sometimes results in nonspecific binding (Behnke et al., 1986). To avoid some of these problems, additional macromolecules are sometimes used for stabilization, such as carbowax (polyethylene glycol) or bovine serum albumin. These, however, can add to probe size and hinder and target access. Thus, while colloidal gold conjugates have greatly enhanced the microscopy of fixed and plastic-embedded biological samples, their utility in the higher resolution experiments typical in electron cryomicroscopy is more limited.
There are also emerging strategies, and substantial excitement about these strategies, for producing electron contrast in a clonable manner, with the goal of producing a “Green Fluorescent Protein” for electron microscopy. Many efforts have focused on adapting metallothionein, a member of the metal binding class of phytochelatin proteins (Diestra et al., 2009; Mercogliano and DeRosier, 2007). Metallothioneins function within cells as metal scavengers and are highly expressed when cultured cells are exposed to toxic metal ions. Mercogliano and DeRosier showed that recombinant concatenated metallothioneins can sequester a sufficient number of gold atoms to observe metal–nanoparticle-like densities in in vitro electron cryomicroscopy experiments. Subsequent in vivo experiments by other research groups have demonstrated some promise for such strategies.
Proteins with unusual electron density have also been used to localize areas of proteins (Alcid and Jurica, 2008). These strategies also show promise, but are not yet widely adopted.
2. General Considerations
Of the site-specific gold cluster bioconjugation strategies discussed here, the bioconjugate chemistry chosen will depend on the goal of the cryomicroscopy experiment. For intermediate resolution goals, the linker-mediated bioconjugate strategies incorporated in Nanogold and undecagold (Bartlett et al., 1978; Cariati and Naldini, 1971; Safer et al., 1982) deserve the highest consideration because of their commercial availability and ease of use. For the highest resolution goals, direct bonding strategies should be considered, because gold clusters of varying sizes with the exactly known molecular formulae of Au25, Au38, Au68, Au102, and Au144, and in some cases, exactly known structure, can be affixed directly and rigidly to a specific site within the target biomolecule.
This chapter contains discussions and detailed protocols for labeling and purification of gold cluster bioconjugates with the direct, NHS-ester, malimido-, and Ni(II)-mediated bioconjugate chemistries shown in Fig. 9.2. While the reaction conditions for formation of the bioconjugates presented are specific for the particular bioconjugate chemistry, the assays of bioconjugate product, for instance, by gel electrophoresis, UV/VIS spectroscopy, or electron microscopy and purification conditions may be considered more general, applicable to any of the gold cluster bioconjugates presented in this chapter with minor adaptations.
With regard to purification in particular, colloidal gold conjugates are usually separated by “pelleting” in a centrifuge or ultracentrifuge, followed by resuspension. However, most of the gold cluster labels discussed in this chapter are not amenable to such a simple purification procedure. For instance, Nanogold is sufficiently small that it does not pellet at 100,000×g. Although centrifugation is inefficient for purification of gold cluster bioconjugates, a number of general strategies have proved successful for purification of bioconjugates, and several specific separation strategies are discussed in the context of purification of specific bioconjugates. The purification strategies presented for particular bioconjugate strategies, however, may work well for other gold cluster bioconjugates. For instance, we include a detailed discussion on gel filtration chromatography strategies for Nanogold conjugate purification, including recommended reaction stoichiometries, to make the purification steps easier. These strategies are certainly also adaptable for purification of monolayer protected cluster (MPC) bioconjugates. Thus, when working through the purification of a novel gold cluster bioconjugate, a “mix and match” approach with the purification strategies presented in each subsection may be considered.
2.1. Stoichiometric labeling
In some applications, 100% labeling is desired, for example, to count the number of target sites on a protein or complex. Although this is possible, there are several factors that may interfere with the desired result: (a) it should be appreciated that these are chemical reactions that may not yield 100% efficiency; (b) reactions may generate some cross-reactivity with other chemical functional groups; (c) the gold particle may bind “nonspecifically” to the protein being studied; and (d) a particular gold cluster may have more than one reactive group. Thus, in any labeling experiment where subsequent electron cryomicroscopy might require stoichiometric labeling, appropriate controls must always be included. These include, for instance, competing or blocking agents, removal of the target site (e.g., cleavage of His tag), and use of nonfunctionalized gold particles or (for labeling at thiol sites obtained by disulfide reduction) nonreduced biomolecules.
3. Monolayer Protected Cluster Labeling of Biomolecules
Beginning in the 1990s was a great increase in research in “nanotechnology,” resulting in a great number of discoveries relating to nanoparticles in general and gold nanoparticles in particular. Just as an example, there were, according to Web of Science, 1060, 1299, and 1390 original research papers on the topic of “gold nanoparticle” in the years 2007, 2008, and 2009. Some of this research has involved developing, characterizing, and applying novel gold cluster/biomolecule conjugates that may be of direct interest for application in electron cryomicroscopy.
Among the research that contributes to improvements in gold nanoparticle/protein conjugates includes the work of several groups investigating the interactions of nanoparticle preparations with biological molecules (Bowman et al., 2008; Tkachenko et al., 2003; Verma et al., 2004). Some research groups have put forth strategies for monofunctionalization of gold nanoparticles (Huo and Worden, 2007; Liu et al., 2006; Worden et al., 2004a,b). Another group has investigated the direct coupling of His6 tags to gold nanoparticle surfaces, presenting spectroscopic evidence for His–gold bonds and also slow denaturation of the protein conjugated to the gold surface (Kogot et al., 2008, 2009). Multidentate strategies for bioconjugation have also been put forth (Krpetic et al., 2009). Of greatest interest to electron cryomicroscopy is the work of the Kornberg and Hamad-Schifferli groups, where gold–protein bonds through cysteine thiolate functionality have been formed. These results will form the basis for the methods that follow.
The gold nanoparticles that are amenable to the protein labeling strategies that are described herein come from a series of now well-defined thiolate MPCs that have been investigated for the past ~15 years (Sardar et al., 2009). MPCs are composed of a compact crystalline gold core in which every gold surface atom is bonded to an organothiolate ligand (Fig. 9.3) and may generally be formulated as Aun(SR)m. These compounds are now understood to occur in a “magic number series” resulting from the complete filling of superatomic electron orbitals (Walter et al., 2008). This means that MPCs of the generic formulae Au25(SR)18, Au38(SR)22, Au68(SR)34, Au102(SR)44, and Au144(SR)60 are understood to have special stability, analogous to the stability of a noble gas atom.
Figure 9.3.
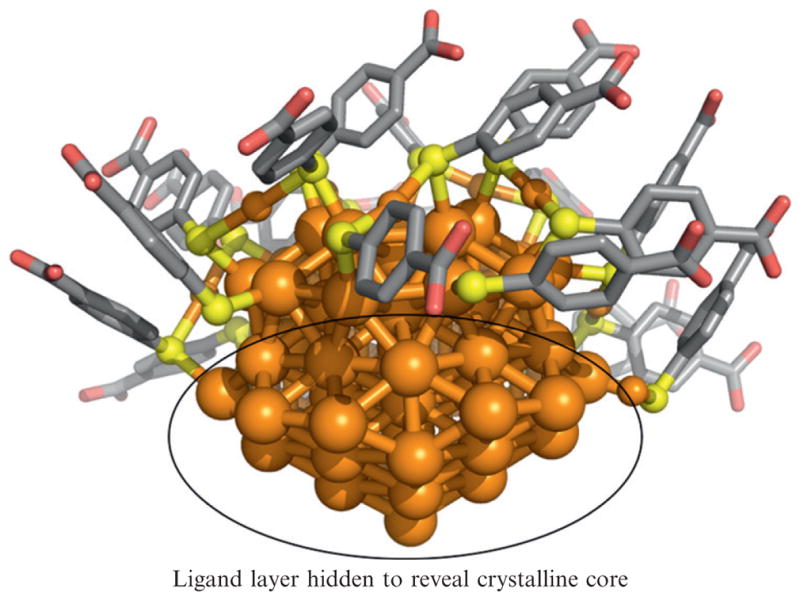
An image generated from Au102(p-mercaptobenzoic acid)44 ligand protected cluster X-ray crystal coordinates. Some of the ligand layer is hidden to reveal the crystalline core which is fcc (bulk) gold. Orange is gold, yellow is sulfur, gray is carbon, and red is oxygen.
MPCs may be functionalized in well-known ligand exchange reactions (Scheme 9.1; Hostetler et al., 1999; Song and Murray, 2002; Templeton et al., 1998, 2000). The kinetics, rate, and extent of a ligand exchange reaction depend upon the nature of the incoming and outgoing ligands, as well as the charge on the cluster core. The incoming ligand can be any molecule presenting a thiol, including modified nucleic acids, small molecule drugs, and cysteine-containing proteins and peptides, allowing the formation of biomolecule/MPC conjugates.
Scheme 9.1.
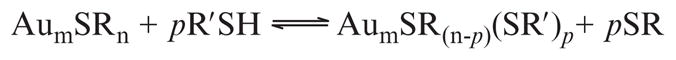
This protein–gold bonding can lead to a remarkably rigid interface between the protein and the metal cluster. For instance, Au144(p-mercaptobenzoic acid)60 clusters can be conjugated to a protein such that the majority of Au144 clusters have a positional displacement (mobility) of less than 3 Å relative to the center of mass of the protein (Sexton and Ackerson, 2010). This means that the in vitro conjugated cluster/protein conjugate is defined both with respect to metal cluster formula and position of the metal relative to the protein with a “precision” that approaches that of naturally occurring metalloproteins.
The primary caution when making directly bonded gold cluster/protein conjugates is that forming such conjugates can perturb the structure and function of the labeled protein (Aubin-Tam and Hamad-Schifferli, 2005, 2008; Aubin-Tam et al., 2009; Kogot et al., 2009). The nature of this perturbation depends upon the ligand layer used to protect the clusters and the location of the cysteine within the protein that is labeled. Because diminishment or destruction of protein structure will complicate or make impossible any electron cryomicroscopy analysis, the methods section contains a detailed discussion of design of the conjugates. A secondary consideration is labeling yield, which also depends upon the ligand layer of the cluster and the site of label. Generally, when labeling yield is good, protein structure is retained, but this is not always true.
3.1. Design considerations
The careful design of an MPC/protein conjugation experiment can help insure a resulting MPC/protein conjugate that behaves in the expected manner. There are three essential parameters to consider when designing the bioconjugation process. These are the position of the labeled cysteine residue within the larger protein, the choice of size of the gold cluster core, and the choice of ligand protecting the gold cluster core. Because of the anticipated audience of this chapter, we will limit the discussion to choosing the position of the labeled cysteine residue, as this single parameter has the greatest effect on the structure and function of the resulting gold cluster bioconjugate.
When the cysteine position is chosen well, the Au144(p-mercaptobenzoic acid)60 does not diminish protein function and will label in high yield. The synthesis of this cluster is also reasonably straightforward in a laboratory equipped for standard molecular biology procedures. Other MPCs (Table 9.1) may also be adapted in the design and bioconjugation process, depending upon the desired result. These compounds are generally harder to synthesize as discrete products in good yield, and while some have been well investigated for protein labeling, they are less well established.
The “direct” labeling of a protein with a gold cluster as shown in Fig. 9.2, upper right panel, takes advantage of the spontaneous bonding that can occur by a ligand exchange reaction (Scheme 9.1) between a gold cluster and the thiol functional group of cysteine. Many, if not most, cysteine residues within proteins, however, are not amenable to direct labeling with MPCs. In our experience, only solvent exposed surface cysteine residues can be labeled, and in some cases, for instance, if the cysteine residue sits in a concave region of protein surface, not even solvent exposed residues can be labeled (Ackerson et al., 2010). Cysteine residues that are disulfide bonded or buried within the interior of proteins cannot be labeled.
If the X-ray structure of the target protein is known, then it is possible to examine the structure for the presence of any unpaired cysteine residues. If they are present, it is quite easy to test empirically if the target protein is label-able and whether the resulting structure is perturbed, which can be accomplished by taking a circular dichroism (CD) spectrum of the labeled protein. If the experimental goal can be accomplished with an adventitious naturally occurring cysteine residue, then more careful design is unnecessary.
If the structure of the target protein is unknown, but cysteine residues are present, in the primary sequence, then it is also recommended that a test should be done empirically (vide infra) for “label-ability” and maintenance of structure.
In most cases, adventitious cysteine residues will not exist. Standard molecular cloning protocols can be used to make serine for cysteine mutations for native cysteine residues that form undesired conjugates, and a single cysteine residue may be inserted into a position that allows the binding of a gold cluster to the region of the protein optimal for the goal of the broader experiment.
The goal of the broader experiment can only provide an approximate placement of the cysteine residue. Two other factors must be taken into account.
The first factor is that the cysteine residue that is labeled will become an integral component into the monolayer of the cluster which is labeled, and thus it must protrude from the protein a sufficient distance to penetrate the monolayer of the MPC. Solvent-accessible cysteine residues that are in recessions or cavities in a protein surface will usually not form conjugates. The second factor is that some cysteine residue positions, when labeled, will cause the protein to partially or completely denature, while other labeled cysteine positions leave protein structure intact. The studies of the Hamad-Schifferli group on labeling different cysteine positions of cytochrome c provide the greatest insight in choosing positions for labeled cysteine residues within proteins (Aubin-Tam et al., 2009). Hamad-Schifferli concludes that to preserve protein structure, flexible and loosely folded motifs should be labeled. Labeling of nucleation centers of protein folding should be avoided. We have not done the same systematic study that Hamad-Schifferli has done, but we do note that some residue positions, when labeled, appear to diminish or abolish the affinity of a labeled antibody for its target.
In our own work, we have labeled cysteine residues that are both naturally occurring and engineered into proteins. We have the most experience with labeling cysteine residues appended to an engineered C-terminal extension of the NC10 single chain Fv antibody fragment (Ackerson et al., 2010). In the constructs that we made, the labeled cysteine residue is separated from the normal C-terminus of the protein by genetically engineered linkers of 0–7 amino acid residues. These mutants were originally generated when the gold nanoparticles that we made did not label cysteine residues engineered elsewhere in the scFv and labeled cysteine residues on C-terminal linkers with never more than 50% conjugate yield (Ackerson et al., 2006). We have overcome the yield and position limitations with subsequently described conjugation chemistries (Ackerson et al., 2010).
With a sufficiently long linker, we observe no diminishment of practical protein function as measured by ELISA assay, and we also observe that with a three amino acid residue extension to the C-terminus of the protein, we observe a remarkably rigid interface between protein and cluster (Sexton and Ackerson, 2010). Thus, the diminishment of protein structure and presumably, function that sometimes occurs when labeling cysteine residues can certainly be avoided by labeling a C- or N-terminal extension, if the C- or N-terminal of the target protein is appropriately accessible. We have noticed that sometimes expression of proteins in recombinant over-expression systems (i.e., Escherichia coli) can be complicated by a C-terminal cysteine. Thus, we generally include a Glycine residue after the Cysteine residue so that the Cysteine is not the terminal residue. With a sufficiently long linker, the penetration of ligand layer problems is obviated as well. The primary drawback to C- or N-terminal labeling is that the label may not be positioned at a site of particular interest within the protein. For identifying subunits within a multicomponent protein complex, however, C- or N-terminal labeling may work very well and still produce a tight interface between gold cluster and protein.
In summary, designing a protein with a single label-able cysteine residue requires consideration of multiple factors. If it is compatible with the goal of the experiment, then the most facile position of a cysteine may be on a short N or C-terminal extension of the protein. To make a site-directed gold label within the “framework” of the protein, it is best to locate the labeled cysteine on a protruding and loosely folded motif.
3.2. Protocols
The protocol presented will describe how to site-specifically label a designed cysteine-containing protein with an Au144 cluster. Because synthesis of Au144 clusters is straightforward, the protocol will be divided into two sections, one describing particle synthesis, and a second describing particle bioconjugation.
3.3. Synthesis of Au144( pMBA)60
Generally, when gold clusters are synthesized, the result is a mixture containing a wide distribution of particle sizes, with particularly stable clusters (i.e., Magic Number clusters, vide supra) being more abundant, but generally within a large range of cluster core sizes. The Au144(p-mercaptobenzoic acid)44 synthesis that we have discovered is rare in that it produces in a single synthetic step a uniform cluster. Other compounds in the “magic number series” such as Au102 or Au68 are synthetically more elusive and are thus less appropriate for synthetic discussion in this chapter.
Reagents
We have found that the vendor of 4-mercaptobenzoic acid (pMBA) matters in the synthesis, but have sourced the other components in the synthesis from many different vendors without detriment. Reagents required for this synthesis include 4-mercaptobenzoic acid (90%; pMBA), sourced from TCI America, hydrogen tetracholoroaurate trihydrate (HAuCl4), sodium borohydride(NaBH4), reagent grade methanol, nano-pure or comparable water, and ammonium acetate.
Dissolve 0.002 mol of HAuCl4 in 100 mL of methanol.
Dissolve 0.0068 mol of pMBA in 48 mL of water, to this add 3.2 mL of 10 M NaOH. Check that pH is above 13, and if so, add water to a final volume of 80 mL. If pH is not above 13, then add NaOH until pH is above 13, and then add water to a final volume of 80 mL.
Mix solutions made in steps 1 and 2, and stir at least overnight in a sealed vessel. This can be allowed to stir for many days if desired.
This makes a gold(I)-p-mercaptobenzoic acid compound, and the solution should appear clear at this point.
Dilute the gold(I)-pMBA made above to a final [Au] of ~500 μM and a final water/methanol ratio of 27% water, 73% methanol. For instance, in a ~1 L scale, take 50 mL of the gold/pMBA solution above, and add it to 260 mL of MeOH and 740 mL of water.
To this mixture add a ~4.5-fold molar excess of NaBH4:Au(I). For instance, in the 1.05-L scale referenced earlier, add 10 mL of 0.25 M NaBH4 (dissolved freshly in water). This is the hardest step to reproduce, because it is difficult to know the potency of a particular stock of NaBH4. This is because in normal storage, NaBH4 will slowly absorb water from the atmosphere and convert to sodium borate, releasing H2 gas. Thus, it is advised to initially attempt this reaction on a small scale (scale down to ~1.0 mL) and run five reactions with different equivalents of NaBH4, going from a 1.5× excess to a 10× excess. Assay these small-scale reactions and then use the optimized of NaBH4 for scaling up.
Seal the reaction vessel and let it stir for 18 h at room temperature. The extended reduction time seems important for getting monodisperse product.
Once the reaction is complete, the product can be precipitated by addition of excess methanol. For instance, in the 1.05-L scale, addition of an additional 1.0-L of methanol should completely precipitate the product.
The precipitated product may be collected by centrifugation or filtration over a medium frit filter. If it is by centrifugation, limit the g-force that the particles are exposed to no more than 8000×g. While 8000×g may be tolerated for hours, it is generally not necessary to centrifuge for more than 3 or 4 min, as the precipitated metal clusters will rapidly collect at the bottom of the centrifuge tube. Higher centrifugal forces can cause the particles to sinter.
After collection by filtration or centrifugation, the particles may be redissolved in 10 mM ammonium acetate, pH 8.0. For the 1.05-L volume referenced earlier, 50 mL is an appropriate volume of buffer to use. The product should then be subjected to two additional methanol precipitation and redissolve rounds to remove any unreacted starting materials. For the 50 mL redissolved volume, it is appropriate to add 50 mL methanol to provoke precipitation.
3.4. Assay of Au144(pMBA)60 synthesis
The most straightforward assay is to dry the product on a glow-discharged continuous carbon TEM grid and examine the product in the electron microscope. The apparent diameter of the product should be 2.0 nm, and it should appear very uniform, sometimes forming large ordered crystalline arrays. If the product appears polydisperse and larger than 2.0 nm, the culprit is probably too large of an excess of NaBH4 in the synthetic step. If the product appears sufficient for the goals of the experiment at this stage, then it may not be necessary to do further analysis. If it is important to confirm molecular or near molecular dispersity of the product, then forming crystals and analyzing by mass spectroscopy can be convenient methods for this.
Product quality may be assessed by attempting to form crystals. Hexagonal plate crystals measuring between 100 and 300 μm in maximum dimension should form from a solution of the product placed in a 250-μM NaCl solution that is allowed to slowly evaporate. A microscope suitable for crystallography will be required to examine these crystals.
The product may also be amenable to assessment by MALDI-mass spectroscopy, although depending upon the instrument often data is not acquired. MALDI matrices of 2,5-dihydroxybenzoic acid and DCTB have been successful in our lab and others for generating good spectra (Chaki et al., 2008; Dass, 2009). The product may be quantitated using its weak absorption peak at 510 nm for which we have calculated a Beer’s law extinction coefficient of ε510 = 4.34 × 105 M−1cm−1.
3.5. Conjugation of Au144(pMBA)60 clusters
To conjugate the Au144 clusters described, it is first necessary to prepare the protein by reducing any oxidized or disulfide cysteine residues to thiol form. Thiol-based reductants, commonly used in protein handling, such as β-mercaptoethanol, mercaptoethylamine, and dithiothreitol (DTT), can participate in ligand exchange, altering or even destroying the cluster core in an “etching” process (Schaaff and Whetten, 1999) when they are used in amounts commonly used in protein chemistry (i.e., 1 mM). If these reductants are used, they must be completely removed, for instance, by gel filtration chromatography. We have found, however, that the phosphine-based cysteine reductant Tris-carboxyethyl phosphine (TCEP) can be used prior to the gold labeling reaction without the need for removal.
Reagents
Protein to be labeled, with cysteine residue as described (vide supra), 1.0 mg or more in solution at a concentration of 1.0 mg/mL or greater.
TCEP
pMBA-protected Au144 clusters from previous protocol.
Add TCEP to the protein to a final concentration of 300 μM.
Add a fivefold molar excess of Au144 to the protein/TCEP mixture. Incubate at 37 °C for 1 h. Labeling may be complete in much shorter time periods (as short as 5 min) if the protein being used will not tolerate 37 °C for 1 h.
Prior to purifying the protein from excess Au144, check the yield of the labeling reaction by running an SDS-PAGE gel. Note that reducing agents such as β-mercaptoethanol should not be included in these gels, as such reagents will disrupt the protein–gold bond. A coffee-colored band corresponding to the labeled protein should run gel-shifted about 8 kDa relative to the protein, as shown in Fig. 9.4. A successful labeling reaction will appear quantitative in this assay. The intensity of gold-labeled bands may be enhanced with an autometallographic reaction, as described in the Nanogold labeling section of this chapter.
Figure 9.4.
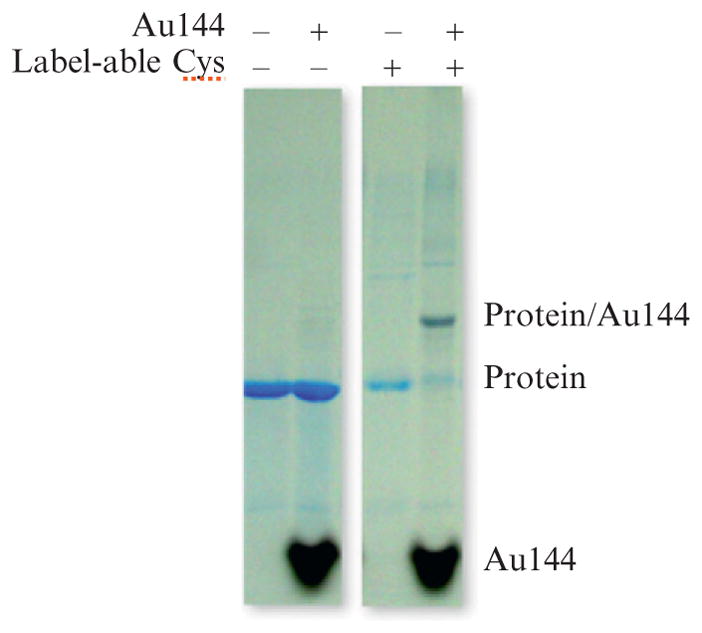
Coomassie-stained SDS–polyacrylamide gels showing the gel shift and color change in a protein band that results from gold cluster labeling, in the far right lane. The third lane from the right shows no gel-shifted band for a protein without label-able cysteine residues.
3.6. Separation of Au144(pMBA)60 conjugates
Purification of the conjugate from free Au144 depends upon the nature of the labeled protein. Purification generally aims to accomplish removal of three undesired components of the labeling reaction: Excess Au144, unlabeled protein, and Au144 conjugated to more than one target protein.
Purification can be achieved in a single step by native gel electrophoresis and glycerol gradient centrifugation for which standard protocols easily found elsewhere will work well. In each case, free Au144 is very well separated from Au144 conjugated to protein, and the protein that is conjugated to Au144 retains the coffee-like color of Au144, making identifying the labeled protein in a gel or gradient possible by simple visual inspection. These methods provide single-step purification, but do not scale well, although this is often not problematic given the small material requirements of electron cryomicroscopy.
We have had success with other methods of purification. If the protein has an affinity tag, the protein can usually be purified away from excess gold by affinity chromatography. We have used FLAG and TAP tags successfully for this purpose. While this will remove free Au144, there may be unlabeled protein retained and also Au144 with two or more proteins bound retained. We have found that ion exchange chromatography with a Q-column will generally produce excellent separation between unlabeled and labeled protein because of the high negative charge of the mercaptobenzoic acid ligand layer. We have used gel filtration chromatography to separate Au144 with two or more proteins from Au144 with only one protein.
In some instances, Au144 may become flocculent inside of chromatography columns, discoloring the column and possibly fouling it. This can be minimized by adding glycerol to the column running buffer to a concentration of 5%. If a column becomes discolored or fouled, the Au144 clusters may be effectively dissolved and removed with an injection of 200 mM glutathione, which will etch any flocculant gold nanoparticles on the column and solubilize them.
4. Nanogold Labeling of Proteins
The use of metal cluster compounds functionalized using synthetic organic ligands, which can be tailored to impart desired solubility properties or functionalized for selective cross-linking to specific functional groups on biomolecules, provides gold clusters with an extensive conjugation chemistry. The 0.8-nm undecagold cluster, containing 11 gold atoms, and the 1.4-nm Nanogold cluster label, are stabilized with organic ligands terminated with water-soluble functional groups. These compounds are typically prepared with a small number of amines, then chromatographed over an anionic resin to isolate the monoamino forms. These are then converted to maleimido-derivatives, which may be used to label thiol groups (cysteine residues) or Sulfo-NHS-derivatives, which may be used to label primary aliphatic amino groups (lysine residues or N-terminal amines). Both NHS-and malimido-bioconjugate chemistries are shown schematically in Fig. 9.2. In addition, the monoamino-functionalized clusters may be activated and cross-linked to a variety of biomolecules or other types of probes using homo- and heterobifunctional cross-linkers.
These reagents have been used to label a variety of proteins (Rappas et al., 2005; Yonekura et al., 2006) and peptides (Kelly and Taylor, 2005; Segond von Banchet et al., 1999) for electron cryomicroscopy, enabling the preparation of probes that have been used for the molecular localization of protein subunits (Opalka et al., 2003) and the reconstruction of a variety of protein complexes ( Jeon and Shipley, 2000a,b; Volkmann et al., 2001). Using maleimido-Nanogold, antibody IgG molecules and Fab′ fragments may be labeled site-specifically at hinge thiol sites, generated by the selective reduction of IgG molecules or F(ab′)2 fragments with a mild reducing agent such as DTT or mercaptoethylamine hydrochloride (MEA); this positioning minimizes interference with target binding (Hainfeld and Furuya, 1992; Hainfeld and Powell, 1997). Even smaller Fv fragments may be labeled at naturally occurring lysine residues using Sulfo-NHS-Nanogold (Ribrioux et al., 1996).
4.1. Conjugation of the Nanogold label
Nanogold and undecagold are supplied in with a choice of three different reactive functional groups, intended for different labeling applications (Fig. 9.2); each contains close to one reactive functional group. The starting material used to prepare all three is a “Monoamino” form of each cluster, which is separated from a statistical mixture of gold clusters containing different numbers of amines using ion exchange chromatography (Hainfeld, 1989). This is then converted to the maleimido-form using an excess of a bifunctional reagent that introduces a maleimide group. The activated undecagold or Nanogold reagents are separated from excess cross-linker by gel filtration over a desalting gel (GH25, Chisso Corporation). The reagents are eluted with the same buffer in which labeling is usually conducted: 0.02 M sodium phosphate, pH 6.5 with 150 mM sodium chloride, and 1 mM disodium dihydrogen EDTA to chelate transition metal ions, which might otherwise catalyze oxidation of the thiol-labeling sites. The highest selectivity of maleimides for thiols is between pH 6.0 and 7.0; at higher pH, competing hydrolysis and reactions with amines decrease specificity. Immediately after separation, the reagents are diluted to 30 nmol/mL (Nanogold) or 50 nmol/mL (undecagold) in the same buffer, then flash-frozen and lyophilized before being packaged for use. Sulfo-N-hydroxysuccinimido (NHS)-Nanogold is similarly activated and separated, but using a cross-linking reagent that introduces a Sulfo-NHS group; the activated Nanogold or undecagold is separated by gel filtration, but in this case is eluted with 0.02 M sodium phosphate at pH 7.5 with 150 mM sodium chloride. This pH is within the optimum range (7.5–8.2) for reaction of the Sulfo-NHS group with primary aliphatic amines.
This means that the only preparation required for use is reconstitution in degassed, deionized water to give a solution in a buffer that is optimized for labeling. When labeling thiol-containing compounds, the amenability of the thiol to maleimide labeling should be checked carefully before reaction. In many proteins, such as antibody IgG molecules, the target thiols are in the form of disulfides. If so, these should be carefully reduced before use. Thiol-based reducing agents such as DTT or MEA at concentrations between 10 and 50 mM are usually appropriate for selective reduction of antibody hinge thiols leaving intrachain thiols intact; generally, we have found that lower concentrations (20 mM) work well for goat antibodies, while mouse and, in particular, rabbit antibodies may require higher concentrations of reducing agent. Reduction is typically carried out for 1 h at room temperature in 0.1 M sodium phosphate buffer at pH 6.0. The reduced protein is then separated from excess reducing agent by gel filtration over a desalting gel (GH25, Chisso) eluted with the buffer in which the labeling reaction is to be conducted, usually 0.02 M sodium phosphate, pH 6.5, with 150 mM sodium chloride and 1 mM disodium dihydrogen EDTA. Because the reducing agent is used in relatively large excess, even residual traces can react with the maleimido-Nanogold, and it is therefore critical that it be completely separated from the reduced protein. Dialysis does not provide an acceptable level of purification: gel filtration is required to ensure complete removal.
A second consideration for labeling is accessibility of the target functionality to the relatively large Nanogold cluster. Nanogold has a molecular weight (MW) close to 15,000, and its diameter, including its coordinated ligands, is 2.5–2.7 nm; this may hinder conjugation if the target group is located in a deep cleft. Therefore, it is usually worth checking the structure of the conjugate biomolecule before labeling. In addition, searching for parallel labeling reactions (such as fluorescent or enzymatic labeling) is highly recommended since the conditions used for successful labeling can provide insights into the conditions that may work well with Nanogold. Suitability for labeling may be assessed using 14C labeling or colorimetrically (Grassetti and Murray, 1967). Buffers should be thoroughly degassed and plastic implements used to handle and transfer specimens, since transition metal ions, especially in the presence of atmospheric oxygen, can mediate the oxidation or reoxidation of thiols to disulfides.
Maleimido Nanogold and Sulfo-NHS-Nanogold, as sold, will react in a monovalent (1:1) manner with most biomolecules. While accurate analytical methods to “prove” monofunctionality are not readily available, monovalent reactivity has been demonstrated for a reasonably wide range of conjugates. To maximize the yield of 1:1 conjugate, and also to provide an easy separation, the relative sizes of the molecule to be labeled and the Nanogold particle should be compared before the reaction is carried out. In most labeling reactions, the products will be separated by gel filtration or size exclusion chromatography. Therefore, a reaction stoichiometry should be used that gives the greatest size difference between the conjugate and the reagent used in excess. For labeling proteins or other macromolecules that are larger than Nanogold, an excess of Nanogold reagent should be used; this can range from 2:1 up to 5:1, with the smaller ratios used where the reagents are similar in size. For small proteins or peptides that are similar in size to Nanogold or smaller, a stoichiometric excess of the protein or peptide should be used; a twofold excess is recommended for proteins close to Nanogold in mass, and a larger excess for those that are smaller, up to 10-fold for small peptides or small molecules. This strategy is to enable size separation, for example, by gel filtration column chromatography. If the protein is large, it can be easily separated from unreacted free Nanogold due to the size difference. For peptides smaller than Nanogold, the Nanogold can be separated from the peptide, but labeled Nanogold may not separate well from free Nanogold. By using an excess of the peptide, most of the Nanogold will be labeled, and it is then just necessary to separate excess peptide. A typical labeling procedure is as follows:
Dissolve protein in labeling buffer, at a concentration of 1.0 mg/mL or higher if possible.
Reconstitute the Nanogold reagent in deionized water. Brief vortexing may help dissolve any residual precipitate.
Combine the Nanogold reagent with the protein or biomolecule to be labeled. Gentle agitation for 40 min to 1 h may be used to ensure reaction; otherwise, and afterwards, reaction mixtures should be incubated at 4 °C overnight.
Separate next day.
4.2. Separation of Nanogold conjugates
Gel filtration chromatography usually provides a successful method for purification of Nanogold bioconjugates. For conjugate purification, a gel should be used with an MW separation range that extends from below the smaller of the two reagents (Nanogold or conjugate biomolecule) to significantly above that of the conjugate, in order to allow the separation of multimers or small aggregates from monomeric 1:1 conjugates. For example, when labeling antibody Fab′ fragments, Superose-12 or Superdex-75 (GE Healthcare), which have MW separation ranges of 10,000–300,000 and 500–70,000 respectively, are appropriate. An example of the separation that can be obtained is shown in Fig. 9.5: note that the conjugate product is separated from both larger and smaller impurities.
Figure 9.5.

Chromatogram showing the separation of Nanogold-labeled Fab′ from larger species, excess Nanogold, and smaller species by gel filtration over Superose-12 gel filtration media. Column volume = 16 mL (50 cm length × 0.67 cm diameter).
Larger proteins and protein complexes may be separated using gels with higher MW ranges such as Superose-6 (GE Healthcare) or A-5m (Bio-Rad), which have MW separation ranges of 5000–1,000,000 or 10,000–5,000,000, respectively. Smaller peptide conjugates may be separated using Superdex-75. For separating conjugates prepared with molecules that are significantly smaller than Nanogold, such as small peptides or substrate analogs, a gel such as Superdex Peptide/PG30, which has a separation range from 100 to 7000 with an exclusion limit of 10,000, often works well. It should be noted that because the majority of the mass of Nanogold is contributed by heavy gold atoms, its hydrodynamic radius is smaller than that of a protein of comparable MW: on many media, it will elute in a position similar to a protein of about 8000 MW. Before injection onto the column, the reaction mixture should be concentrated to a volume of 5% of the column or less using a membrane centrifuge filter with an MW cutoff below the MW of the conjugate: this will ensure maximum chromatographic resolution.
In some situations, gel filtration may not be the most appropriate method for separation. Alternative chromatographic methods may provide better separation. Hydrophobic interaction chromatography may be used to separate labeled from unlabeled proteins with similar MWs in situations where this is required (Hainfeld, 1989). Reverse-phase chromatography may be used to separate labeled oligonucleotides: a butyl column, eluted with a gradient of 0–70% acetonitrile in 0.1 M triethylammonium acetate buffer at pH 7, was found to separate labeled and unlabeled oligonucleotides from unconjugated Nanogold.
Caution should be exercised when using other separation methods, as the behavior of Nanogold or its conjugates is not always predictable. Gel electrophoresis may be used to separate Nanogold conjugates in a manner similar to that shown in Fig. 9.5; however, as discussed for MPC conjugates, nonreducing gels should be used, and the shift arising from the Nanogold has been found to be significantly less than that predicted from its mass (Hainfeld and Furuya, 1995). When using gel electrophoresis, it is recommended that the product is run in duplicate gel lanes so that one may be developed using a protein-specific colorimetric stain such as Coomassie blue to visualize protein-containing bands, and the other with an autometallographic reagent such as LI Silver, which visualizes the bands containing Nanogold (Note that silver stain reagents for proteins, which use a different chemistry, are not appropriate for developing gels containing gold cluster bioconjugates.) A gold cluster/protein bioconjugate is indicated by a band that develops with both types of stains. Dialysis may also be used, but the membrane should be tested before use to ensure that it does not bind to Nanogold.
4.3. Assay of labeling and activity
The extent of labeling may be calculated from the UV/visible absorption spectrum of the conjugate. Gold labeling of most proteins is best calculated using the extinction coefficients at 280 nm, where proteins usually absorb strongly and their extinction coefficients are known, and at 420 nm, where Nanogold absorbs strongly but most proteins do not. If your protein does not absorb at 420 nm, use the following method to calculate labeling:
- Since the absorption at 420 nm arises solely from the Nanogold, use the extinction coefficient of Nanogold at 420 nm (110,000) to calculate the concentration of Nanogold:
- Use the absorption at 420 nm and the extinction coefficients of Nanogold at 280 and 420 nm to calculate the absorption at 280 nm due to the Nanogold:
- Subtract this from the measured absorption at 280 nm. The difference is the absorption due to protein:
-
Use the absorption arising from protein to calculate the protein concentration:where E is the extinction coefficient of the protein at 280 nm. Convert optical densities to extinction coefficients using the MW as follows:
- The number of Nanogold labels per protein is simply the concentration of Nanogold divided by the concentration of protein:
The activity of the labeled conjugate may be checked by dot blots. We have developed an optimized detection procedure that maintains the very high sensitivity these conjugates provide, combined with a greatly reduced background and enhanced signal clarity. Best results are usually obtained using a procedure that incorporates (a) gold enhancement (Powell and Hainfeld, 2002) rather than silver enhancement to develop the signal after application of the Nanogold conjugate; in most cases, this will provide substantially lower background or nonspecific signal; (b) 0.1% Tween 20 (detergent) in the buffers used for blocking, antibody incubation, and washing; this will dramatically reduce background binding; and (c) inclusion of 1% nonfat dried milk (such as the material sold in supermarkets and food stores) as an additive in the incubation buffer in which the Nanogold conjugate is applied to the blot, and 5% nonfat dried milk in the blocking buffer used to block the membrane before application of probes. The suggested procedure is as follows.
Antigen application
Prepare antigen solutions with a series of dilutions (0.01, 0.001, 0.0005, 0.0001, 0.00005, 0.00001, and 0.000005 mg/mL) using PBS, pH 7.4.
Pipet 1 μL of the aforementioned solutions on to a dry nitrocellulose membrane (0.2 μm pore size); prepare two duplicates as a negative control: (a) Negative control 1: No antigen, No antibody; and (b) Negative control 2: No antigen with Nanogold conjugate incubation.
Air-dry for 30 min.
Blocking
Immerse membranes in 8 mL of TBS–Tween 20 (20 mM Tris pH 7.6, with 150 mM NaCl and 0.1% Tween 20) for 5 min.
Block membranes in 8 mL of TBS–Tween 20 containing 5% nonfat dried milk for 30 min at room temperature.
Binding of Nanogold conjugate
Dilute Nanogold conjugate in TBS–Tween 20 containing 1% nonfat dried milk to about 0.05 nmol/mL (about 4 μg/mL for antibodies; 1:20 dilution, 300 μL conjugate + 5.30 mL TBS-gelatin containing 1% nonfat dried milk).
Incubate the membranes in 8 mL of diluted conjugate solution for 30 min at room temperature.
Incubate the control membrane in 8 mL of TBS–Tween 20 containing 1% nonfat dried milk for 30 min at room temperature.
Autometallographic detection
Wash membranes three times for 3 min each in 8 mL of TBS–Tween 20. Wash membranes thoroughly in 8 mL of deionized water (4 × 3 min). Make sure strips are washed separately according to what they are incubated in (strips incubated in one lot of a conjugate are washed in a separate dish from strips that are incubated in TBS–Tween 20 with 1% nonfat dried milk without conjugate; strips incubated in different lots are washed separately).
Perform Gold Enhancement using GoldEnhance EM (Nanoprobes Product No. 2113) according to instructions (mix solutions A and B, wait 5 min, then add C and D).
Record the number of observed spots and time when the spots appear. Record the time at which background appears on the control.
After 15 min, remove the enhancement solution. Rinse membranes with water (3 × 3 min) and air-dry for storage.
4.4. Electron cryomicroscopy with incorporated Nanogold conjugates
Many electron cryomicroscopy experiments require the incorporation of a Nanogold-labeled component into the structure that is being observed, rather than labeling of the assembled structure, and this can require a modified approach to microscopy. Because of the variety of systems and probes that can be used for such experiments, a single best procedure is difficult to identify; however, that described by Kelly and Taylor (2005), who used Nanogold-labeled β1-integrin to identify the binding site on α-actinin, provides a useful starting point and demonstrates the resolution that is possible with this approach. The cytoplasmic domain of β1-integrin is a 41 amino acid peptide. In order to identify its binding site, the authors synthesized the cytoplasmic domain with a histidine tag at its N-terminus. The binding of this peptide to a lipid monolayer containing a chelated nickel (II) atom mimics the native environment at the cytoplasmic leaflet of the plasma membrane. Preliminary results using a binding assay indicated the presence of two binding sites on the β1-integrin: electron cryomicroscopy studies were then pursued in order to localize these. It was synthesized with two different cysteine modifications: in the first, a cysteine was inserted in the middle of the sequence just after T757 (C758), and in the second, a cysteine was added at the C-terminus (C779). Monomaleimido Nanogold was conjugated to the cysteines by mixing the peptides with the target molecule (without reducing agents) for approximately 16 h at room temperature. Conjugates were chromatographically purified by gel filtration using Sephadex G-25 (Sigma), and labeling efficiency was determined from the UV/visible spectrum of the conjugate.
Nickel-modified lipid monolayers were set up in wells 5 mm in diameter and 1 mm deep milled into Teflon blocks. Dilaurylphosphatidylcholine (DLPC) was used as a filler lipid, with different percentages of dimyristoylphosphatidyl cholinesuberimidenitriloacetic acid nickel(II) salt (nickel-chelating lipid), and different amounts of DDMA to facilitate crystallization.
Monolayers were cast upon aqueous buffer containing His-tagged β1-integrin peptide.
α-Actinin was then injected into the aqueous phase. Arrays were obtained using 0.0114 nmol of α-actinin and 0.148 nmol of the β1-integrin peptide, using 20 mM Tris buffer (pH 7.5) with 50 mM NaCl and 1 mM MgCl2. Crystals formed overnight at 4 °C.
Specimens were recovered from the monolayer using hydrophobic reticulated carbon films on 300 Mesh copper grids, and were either negatively stained with 2% aqueous uranyl acetate or plunge frozen without stain in liquid ethane for cryomicroscopy. Low-dose EM data were collected on a Philips CM300-FEG, a 300-kV electron microscope equipped with a field emission gun. Frozen hydrated specimen grids were transferred to a cryoholder and examined at a temperature of −180 °C. Images were recorded at magnifications of 3,040,000 under low-dose conditions.
The 2D arrays of the β1-integrin–α-actinin complex were examined with and without the gold label. Averaged projections were calculated for each specimen along with a difference map to determine the relative position of the gold-labeled β1-integrin peptide. The β1-integrin peptide with gold label at two sites gives difference peaks consistent with binding of the β1-integrin cytoplasmic domain to α-actinin between the first and second of the four 3-helix motifs in the central rod domain (at the R1–R2 junction). The observed differences in binding for the two gold-labeled peptides were attributed to the asymmetrical arrangement of the R1–R4 domain with respect to the lipid monolayer, steric factors because of packing of adjacent molecules within the 2D array, and flexibility of the C-terminal region of the β1-integrin cytoplasmic domain.
A comparison of their results with the model of α-actinin (Liu et al., 2004) is shown in Fig. 9.6. A principle advantage of electron cryomicroscopy is the lack of specimen processing, so the goal is to minimize the use of counterstains and processing steps. However, negative stains are helpful for defining the edges of protein particles, particularly where particle averaging is planned.
Figure 9.6.
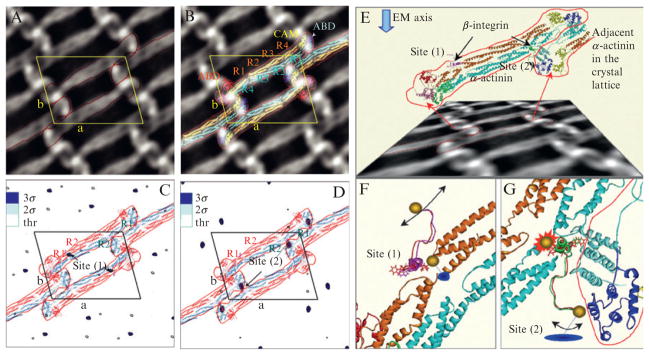
Use of Nanogold-labeled β1-integrin to identify the binding site on α-actinin (Kelly and Taylor, 2005). (Left) (A) 2D projection map of the β1-integrin: α-actinin arrays. α-Actinin dimers are indicated by the red outline; the unit cell is indicated in yellow. (B) Docking of the α-actinin atomic model (Liu et al., 2004) into the EM projection map. P2-symmetry-related dimers were also generated. (C) 2D difference map between unlabeled β1-integrin:α-actinin arrays, and arrays labeled with the Nanogold attached to C758, that is, in the middle of the integrin peptide sequence. The binding in this case occurs at site (1) on α-actinin. The most significant differences are at the level of 3σ (blue). (D) 2D difference map between unlabeled β1-integrin:α-actinin arrays and arrays with Nanogold labeling at C779. Labeling in this case occurs at site (2) on α-actinin. The density levels in both difference maps were truncated at a threshold corresponding to the values of 2σ (two times the standard deviation) for each map, respectively. 2σ peaks are colored sky-blue in (C) and (D). (Right) (E) Model of the unlabeled β1-integrin bound to both potential sites on α-actinin between the first and second spectrin repeats. The view direction chosen to show the atomic model is not perpendicular to the arrays, but rather at an angle that is approximately parallel with the local twofold axis of the R1–R4 domain. The effect is accentuated by the foreshortened appearance of the projection image shown below the model. This direction is maintained in (F) and (G), but because the local twofold symmetry of R1–R4 is only approximate, the integrin peptides are not seen in identical orientations. Arrows in (F) and (G) indicate the general degree of disorder in the gold label when attached to C779. (F) Integrin binding at site (1), while (G) refers to the integrin binding site (2) related by the local twofold of the R1–R4 domain. (F) The Nanogold label at C758 in the integrin peptide (red) fits into the α-actinin crystal lattice in an ordered fashion to give rise to a significant signal seen in the first difference map. The Nanogold label at C779 in the peptide (magenta) is too disordered to detect at a significant level in the second difference map. W755 is shown in stick rendering here and in (G). (G) Integrin cytoplasmic domain binding at site (2). When the Nanogold label is bound to C758 in the peptide (red), the fit is very tight because the inserted amino acid shifts the position of W755 toward the neighboring α-actinin molecule. No difference density is detected for this peptide when bound to site (2) suggesting that the peptide cannot bind under these conditions within the 2D lattice. However, the Nanogold probe positioned at C779 (green) of the integrin peptide fits with less restriction into the α-actinin lattice. The red line in (G) delineates an adjacent α-actinin molecule in the 2D lattice. Blue disks in (F) and (G) indicate the approximate location of the difference peak if the model were projected in a direction perpendicular to the plane of the 2D arrays (gold sphere = Nanogold).
5. Ni(II)-NTA–Nanogold Labeling of His-Tagged Proteins
Studies on single His-tag–NTA-Ni(II) interactions have determined values for the dissociation constant between 7 × 10−8 and 7 × 10−7 M (Kröger et al., 1999; Nieba et al., 1997). Recombinant proteins are often expressed with His tags so that they can be purified over chromatographic resins derivatized with NTA-Ni(II) groups (Hochuli et al., 1988). In the context of chromatographic resins in which multiple NTA-Ni(II) groups can chelate to a single polyhistidine tag, the dissociation constant has been estimated to be 10−13, significantly tighter binding than antibody–antigen (Schmitt et al., 1993). Ni(II)-NTA–Nanogold contains multiple NTA-Ni (II) chelates, and therefore a similar value is reasonable, making Ni(II)-NTA–Nanogold a sensitive and specific probe for His-tagged proteins.
This probe has an important advantage over antibody and protein probes: the targeting entity is much smaller even than the Fab′ antibody fragment. Therefore, the distance from the center of the Nanogold particle to the His-tag site is much shorter, estimated to be about 2.6 nm (Fig. 9.7). This provides significantly higher resolution, which is sufficient to obtain specific structural information on the position and orientation of protein subunits within a protein complex. Its initial use to localize the PsbH subunit in the photosynthetic reaction center Photosystem II (PSII; Buchel et al., 2001; Bumba et al., 2005) provides an excellent illustration of the power of this approach to locating specific proteins within multi-subunit protein complexes. To identify the location of the His-tagged PsbH subunit within PSII, Bumba et al. (2005) used dimeric PSII complexes from cyanobacterium Synechocystis sp. PCC 6803 (obtained by solubilization of thylakoid membranes with 1% dodecyl-maltoside and affinity chromatography over an Ni(II) chelate gel). These were applied to glow-discharged grids, excess buffer was removed using filter paper, and the grid was then incubated upside-down on a droplet of NTA-Ni(II)–Nanogold solution (170 nM in 50 mM MES, pH 6.5) for 10 min at 4 °C. Labeling was terminated by removing the grid from the droplet, rinsing thoroughly with water, and staining with 1% uranyl acetate. A typical EM image showed dispersed particles with uniform size and shape, almost free of contaminants: these are dimeric PSII particles, mostly in top-view projections. All the projections had the same handedness and no mirror images were detected, indicating preferred orientation of the PSII dimers with their stromal side to the carbon support film. Not all the complexes were labeled, and this was attributed to the location of the PsbH His-tag on the stromal side of the complex, which renders the labeling site somewhat inaccessible. The short N-terminus of the Synechocystis PsbH protein enabled precise identification of the location of the His site within the PSII complex. A careful comparison of the location of the gold clusters in Synechocystis PSII with those in Chlamydomonas reinhardtii revealed that gold label in Synechocystis preparation is slightly shifted with respect to the longer edge of the complex. The location of the PsbH subunit is in good agreement with the assignment of the PsbH subunit in the model of Ferreira et al. (2004), and suggests that a single transmembrane helix close to the CP47 subunit corresponds to the PsbH protein.
Figure 9.7.

Ni(II)-NTA–Nanogold, showing binding mechanism (top) and size comparison with Nanogold-labeled Fab′.
This reagent has since become quite widely used for the structural characterization of protein complexes (Adami et al., 2007; Balasingham et al., 2007) and a viral capsid (Chatterji et al., 2005) by high-resolution electron cryomicroscopy. Ni(II)-NTA–Nanogold has been used in the structural characterization of protein complexes by single particle analysis (Collins et al., 2006; Young et al., 2008), with electron tomography and single particle analysis to determine the structure of the Saccharomyces cerevisiae gamma-tubulin small complex (gamma-TuSC) at 25-Å resolution (Kollman et al., 2008), and even as a heavy atom derivative in the assignment of the 3D structure of Cav3.1 (Walsh et al., 2009).
Larger Ni-NTA-derivatized gold nanoparticles have been prepared and used for derivatization of His-tagged proteins, and afford similar resolution combined with higher EM visibility. A 3.9-nm analog was used to assemble Mycobacterium tuberculosis 20S proteasomes tagged with 6×-histidine into 2D arrays, thus improving electron cryomicroscopic resolution by 6–8 Å compared to analysis of single particles (Hu et al., 2008), and a 4.4-nm Ni-NTA–gold label was used to label adenovirus serotype 12 (Ad12) knob protein for STEM observation (Hu et al., 2007; Briñas et al., 2008). A 5-nm NTA-Ni(II)–gold probe is now available commercially (Dubendorff et al., 2010; Reddy et al., 2005).
Ni(II)-NTA–Nanogold is usually used to tag subunits or components of a specimen, which are then assembled or modified to give the functional unit under investigation; this is then examined by electron cryomicroscopy. An important consideration in the preparation of Ni(II)-NTA–Nanogold-labeled conjugates is that each particle contains multiple NTA-Ni(II) groups, and therefore care should be taken to avoid cross-linking and formation of multimers. This can usually be achieved by control of binding stoichiometry: the use of a moderate excess of Ni(II)-NTA–Nanogold (5–10-fold) to label a protein with a single His tag will usually ensure single labeling with minimal cross-linking. For proteins with multiple His tags, larger excesses may be appropriate. A protocol for this procedure usually includes the following elements:
Mix the His-tagged protein or peptide with Ni(II)-NTA–Nanogold in a binding buffer of 20 mM Tris, 0.15 M NaCl, pH 7.6 with 0.1% (w/v) Tween 20 and 1% (w/v) nonfat dried milk. Including Tween 20 and nonfat dried milk helps reduce the nonspecific binding of Ni-NTA–Nanogold to the proteins. The binding buffer must be free from thiols such as β-mercaptoethanol or DTT, or chelators such as EDTA or citrate which can remove Ni(II) ions and produce low binding, However, 5–20 mM imidazole may be added to reduce non-specific binding.
Incubate for 5–30 min at room temperature. For most applications, 5–10 min incubation is sufficient to obtain positive staining.
The Nanogold-labeled His-tagged protein can be separated chromatographically in the same manner as proteins labeled with maleimido-Nanogold or Sulfo-NHS-Nanogold. However, because the mode of binding is different, different buffers are appropriate for elution and reaction. 50 mM Tris, 300 mM NaCl, 10–200 mM imidazole, and 0.1% Tween 20 adjusted to pH 7.6 are recommended for elution, reaction, and washing.
Mix the Nanogold-labeled His-tagged protein or peptide with the other components of the system under study, and allow to assemble or assume natural functionality.
Process for electron cryomicroscopy according to the usual procedure used for unlabeled specimens.
The wash buffer may be optimized for specific applications by adjusting the imidazole and sodium chloride concentrations. Because Ni(II)-NTA–Nanogold is charged, high salt concentrations (up to 1 M) may be helpful in preventing nonspecific interactions. Increasing the imidazole concentration may also help reduce interactions of the negatively charged NTA-Ni (II) entity with protein components. However, it can also decrease the binding to the target proteins: the optimum concentration may be found by trial and error.
The extent of labeling may be calculated in the same manner as for conjugates labeled with maleimido-Nanogold or Sulfo-NHS-Nanogold, using the extinction coefficients at 280 and 420 nm. However, the values for the extinction coefficients are different to those for maleimido-Nano-gold or Sulfo-NHS-Nanogold, and can vary from lot to lot. The values that should be used for calculation with each lot are given in a specification sheet that is supplied with the product.
6. Conclusions
Site-specific labeling of biomolecules with gold clusters offers a flexible, adaptable approach to enhancing information content in electron cryomicroscopy of biological samples. The development of site-specific labeling methods have the potential to bring to electron microscopy many of the advantages that the new generations of fluorophores developed in the past three decades have brought to light and fluorescence microscopy. These advantages are particularly relevant to cryo-EM where specimens are observed at high resolution with significantly less processing and perturbation than with conventional EM methods, and a wealth of molecular information is available to be harvested.
References
- Ackerson CJ, et al. Defined DNA/nanoparticle conjugates. Proc Natl Acad Sci USA. 2005;102:13383–13385. doi: 10.1073/pnas.0506290102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerson CJ, et al. Rigid, specific, and discrete gold nanoparticle/antibody conjugates. J Am Chem Soc. 2006;128:2635–2640. doi: 10.1021/ja0555668. [DOI] [PubMed] [Google Scholar]
- Ackerson CJ, et al. Synthesis and bioconjugation of 2 and 3 nm-diameter gold nanoparticles. Bioconjug Chem. 2010;21:214–218. doi: 10.1021/bc900135d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adami A, Garcia-Alvarez B, Arias-Palomo E, Barford D, Llorca O. Structure of TOR and its complex with KOG1. Mol Cell. 2007;27:509–516. doi: 10.1016/j.molcel.2007.05.040. [DOI] [PubMed] [Google Scholar]
- Alcid EA, Jurica MS. A protein-based EM label for RNA identifies the location of exons in spliceosomes. Nat Struct Mol Biol. 2008;15:213–215. doi: 10.1038/nsmb.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alivisatos AP, Johnsson KP, Peng X, Wilson TE, Loweth CJ, Bruchez MP, Jr, Schultz PG. Organization of “Nanocrystal Molecules” using DNA. Nature. 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- Alvarez MM, Khoury JT, et al. Critical sizes in the growth of Au clusters. Chem Phys Lett. 1997;266:91–98. [Google Scholar]
- Aubin-Tam ME, Hamad-Schifferli K. Gold nanoparticle cytochrome c complexes: The effect of nanoparticle ligand charge on protein structure. Langmuir. 2005;21:12080–12084. doi: 10.1021/la052102e. [DOI] [PubMed] [Google Scholar]
- Aubin-Tam ME, Hamad-Schifferli K. Structure and function of nanoparticle-protein conjugates. Biomed Mater. 2008;3:034001. doi: 10.1088/1748-6041/3/3/034001. [DOI] [PubMed] [Google Scholar]
- Aubin-Tam ME, et al. Site-directed nanoparticle labeling of cytochrome c. Proc Natl Acad Sci USA. 2009;106:4095–4100. doi: 10.1073/pnas.0807299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasingham SV, Collins RF, Assalkhou R, Homberset H, Frye SA, Derrick JP, Tonjum T. Interactions between the Lipoprotein PilP and the Secretin PilQ in Neisseria meningitidis. J Bacteriol. 2007;189:5716–5727. doi: 10.1128/JB.00060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett P, et al. Synthesis of water-soluble undecagold cluster compounds of potential importance in electron microscopic and other studies of biological systems. J Am Chem Soc. 1978;100:5085–5089. [Google Scholar]
- Behnke O, Ammitzbøll T, Jessen H, Klokker M, Nilausen K, Tranum-Jensen J, Olsson L. Non-specific binding of protein-stabilized gold sols as a source of error in immunocytochemistry. Eur J Cell Biol. 1986;41:326–338. [PubMed] [Google Scholar]
- Bowman MC, et al. Inhibition of HIV fusion with multivalent gold nanoparticles. J Am Chem Soc. 2008;130:6896–6897. doi: 10.1021/ja710321g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briñas RP, Hu M, Qian L, Lymar ES, Hainfeld JF. Gold nanoparticle size controlled by polymeric Au(I) thiolate precursor size. J Am Chem Soc. 2008;130:975–982. doi: 10.1021/ja076333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchel C, Morris E, Orlova E, Barber J. Localisation of the PsbH subunit in photosystem II: A new approach using labelling of His-tags with a Ni(2+)-NTA gold cluster and single particle analysis. J Mol Biol. 2001;312:371–379. doi: 10.1006/jmbi.2001.4951. [DOI] [PubMed] [Google Scholar]
- Bumba L, Tichy M, Dobakova M, Komenda J, Vacha F. Localization of the PsbH subunit in photosystem II from the Synechocystis 6803 using the His-tagged Ni–NTA Nanogold labeling. J Struct Biol. 2005;152:28–35. doi: 10.1016/j.jsb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Cariati F, Naldini L. Trianionoheptakis(triarylphosphine)undecagold cluster compounds. Inorg Chim Acta. 1971;5:172–174. [Google Scholar]
- Chaki NK, et al. Ubiquitous 8 and 29 kDa gold:alkanethiolate cluster compounds: Mass-spectrometric determination of molecular formulas and structural implications. J Am Chem Soc. 2008;130:8608–8610. doi: 10.1021/ja8005379. [DOI] [PubMed] [Google Scholar]
- Chatterji A, Ochoa WF, Ueno T, Lin T, Johnson JE. A virus-based nanoblock with tunable electrostatic properties. Nano Lett. 2005;5:597–602. doi: 10.1021/nl048007s. [DOI] [PubMed] [Google Scholar]
- Cheng N, Conway JF, Watts NR, Hainfeld JF, Joshi V, Powell RD, Stahl SJ, Wingfield PE, Steven AC. Tetrairidium, a 4-atom cluster, is readily visible as a density label in 3D cryo-EM maps of proteins at 10–25 Å resolution. J Struct Biol. 1999;127:169–176. doi: 10.1006/jsbi.1999.4120. [DOI] [PubMed] [Google Scholar]
- Collins RF, Beis K, Clarke BR, Ford RC, Hulley M, Naismith JH, Whitfield C. Periplasmic protein–protein contacts in the inner membrane protein Wzc form a tetrameric complex required for the assembly of Escherichia coli group 1 capsules. J Biol Chem. 2006;281:2144–2150. doi: 10.1074/jbc.M508078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Baker ML, et al. 4.0-A resolution cryo-EM structure of the mammalian chaperonin TRiC/CCT reveals its unique subunit arrangement. Proc Natl Acad Sci USA. 2010;107:4967–4972. doi: 10.1073/pnas.0913774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dass A. Mass spectrometric identification of Au(68)(SR)(34) molecular gold nanoclusters with 34-electron shell closing. J Am Chem Soc. 2009;131:11666–11667. doi: 10.1021/ja904713f. [DOI] [PubMed] [Google Scholar]
- Diestra E, et al. Visualization of proteins in intact cells with a clonable tag for electron microscopy. J Struct Biol. 2009;165:157–168. doi: 10.1016/j.jsb.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Dubendorff JW, Lymar E, Furuya FR, Hainfeld JF. Gold labeling of protein fusion tags for EM. Microsc Microanal. 2010;16(Suppl 2) Proceedings CD866. [Google Scholar]
- Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Grassetti DR, Murray JF., Jr Determination of sulfhydryl groups with 2, 2′-or 4, 4′-dithiodipyridine. Arch Biochem Biophys. 1967;119:41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF. Undecagold-antibody method. In: Hayat MA, editor. Colloidal Gold: Principles, Methods, and Applications. Vol. 2. Academic Press; San Diego: 1989. pp. 413–429. [Google Scholar]
- Hainfeld JF, Furuya FR. A 1.4-nm gold cluster covalently attached to antibodies improves immunolabeling. J Histochem Cytochem. 1992;40:177–184. doi: 10.1177/40.2.1552162. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Furuya FR. Silver-enhancement of Nanogold and undecagold. In: Hayat MA, editor. Immunogold-Silver Staining: Principles, Methods and Applications. CRC Press; Boca Raton, FL: 1995. pp. 71–96. [Google Scholar]
- Hainfeld JF, Powell RD. Nanogold technology: New frontiers in gold labeling. Cell Vis. 1997;4:408–432. [Google Scholar]
- Hainfeld JF, Powell RD. New frontiers in gold labeling. J Histochem Cytochem. 2000;48:471–480. doi: 10.1177/002215540004800404. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Furuya FR, Powell RD. Metallosomes. J Struct Biol. 1999a;127:152–160. doi: 10.1006/jsbi.1999.4145. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Liu W, Halsey CMR, Freimuth P, Powell RD. Ni-NTA–gold clusters target his-tagged proteins. J Struct Biol. 1999b;127:185–198. doi: 10.1006/jsbi.1999.4149. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Powell RD, Hacker GW. Nanoparticle molecular labels. In: Mirkin CA, Niemeyer CM, editors. Nanobiotechnology. Chapter 23 Wiley-VCH; Weinheim, Germany: 2004. pp. 353–386. [Google Scholar]
- Hamad-Schifferli K, Schwartz JJ, Santos AT, Zhang S, Jacobson JM. Remote electronic control of DNA hybridization through inductive coupling to an attached metal nanocrystal antenna. Nature. 2002;415:152–155. doi: 10.1038/415152a. [DOI] [PubMed] [Google Scholar]
- Handley DA. The development and application of colloidal gold as a microscopic probe. In: Hayat MA, editor. Colloidal Gold: Principles, Methods and Applications. Chapter 1 Academic Press; San Diego, CA: 1989a. pp. 1–11. [Google Scholar]
- Handley DA. Methods for synthesis of colloidal gold. In: Hayat MA, editor. Colloidal Gold: Principles, Methods and Applications. Chapter 2 Academic Press; San Diego, CA: 1989b. pp. 12–22. [Google Scholar]
- Heaven M, et al. Crystal structure of the gold nanoparticle [N(C(8)H(17))(4)][Au (25)(SCH(2)CH(2)Ph)(18)] J Am Chem Soc. 2008;130:3754–3755. doi: 10.1021/ja800561b. [DOI] [PubMed] [Google Scholar]
- Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stber D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Bio/Technology. 1988;6:1321–1325. [Google Scholar]
- Hostetler M, et al. Dynamics of place-exchange reactions on monolayer-protected gold cluster molecules. Langmuir. 1999;15:3782–3789. [Google Scholar]
- Hu M, Qian L, Brinas RP, Lymar ES, Hainfeld JF. Assembly of nanoparticle-protein binding complexes: From monomers to ordered arrays. Angew Chem Int Ed Engl. 2007;46:5111–5114. doi: 10.1002/anie.200701180. [DOI] [PubMed] [Google Scholar]
- Hu M, Qian L, Briñas RP, Lymar ES, Kuznetsova L, Hainfeld JF. Gold nanoparticle-protein arrays improve resolution for cryo-electron microscopy. J Struct Biol. 2008;161:83–91. doi: 10.1016/j.jsb.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Q, Worden JG. Monofunctional gold nanoparticles: Synthesis and applications. J Nanopart Res. 2007;9:1013–1025. [Google Scholar]
- Jadzinsky PD, et al. Structure of a thiol monolayer-protected gold nanoparticle at 1.1 angstrom resolution. Science. 2007;318:430–433. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- Jeon H, Shipley GG. Vesicle-reconstituted low density lipoprotein receptor: Visualization by cryoelectron microscopy. J Biol Chem. 2000a;275:30458–30464. doi: 10.1074/jbc.M002583200. [DOI] [PubMed] [Google Scholar]
- Jeon H, Shipley GG. Localization of the N-terminal domain of the low density lipoprotein receptor. J Biol Chem. 2000b;275:30465–30470. doi: 10.1074/jbc.M002582200. [DOI] [PubMed] [Google Scholar]
- Kelly DF, Taylor KA. Identification of the beta1-integrin binding site on alpha-actinin by cryoelectron microscopy. J Struct Biol. 2005;149:290–302. doi: 10.1016/j.jsb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Kogot JM, et al. Single peptide assembly onto a 1.5 nm Au surface via a histidine tag. J Am Chem Soc. 2008;130:16156–16157. doi: 10.1021/ja8064717. [DOI] [PubMed] [Google Scholar]
- Kogot JM, et al. Analysis of the dynamics of assembly and structural impact for a histidine tagged FGF1-1.5 nm Au nanoparticle bioconjugate. Bioconjug Chem. 2009;20:2106–2113. doi: 10.1021/bc900224d. [DOI] [PubMed] [Google Scholar]
- Kollman JM, Zelter A, Muller EG, Fox B, Rice LM, Davis TN, Agard DA. The structure of the gamma-tubulin small complex: Implications of its architecture and flexibility for microtubule nucleation. Mol Biol Cell. 2008;19:207–215. doi: 10.1091/mbc.E07-09-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R. Commercial preparations of colloidal gold-antibody complexes frequently contain free active antibody. J Histochem Cytochem. 1990;39:37–39. doi: 10.1177/39.1.1983872. [DOI] [PubMed] [Google Scholar]
- Kröger D, Liley M, Schiwek W, Skerra A, Vogel H. Immobilization of histidine-tagged proteins on gold surfaces using chelator thioalkanes. Biosens Bioelectron. 1999;14:155–161. doi: 10.1016/s0956-5663(98)00116-x. [DOI] [PubMed] [Google Scholar]
- Krpetic Z, et al. A multidentate peptide for stabilization and facile bioconjugation of gold nanoparticles. Bioconjug Chem. 2009;20:619–624. doi: 10.1021/bc8003028. [DOI] [PubMed] [Google Scholar]
- Levy R, et al. Rational and combinatorial design of peptide capping ligands for gold nanoparticles. J Am Chem Soc. 2004;126:10076–10084. doi: 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Taylor KA. A 3-D reconstruction of smooth muscle -actinin by cryoEM reveals two diVerent conformations at the actin binding region. J Mol Biol. 2004;338:115–125. doi: 10.1016/j.jmb.2004.02.034. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. Monofunctionat gold nanopartictes prepared via a noncovalent-interaction-based solid-phase modification approach. Small. 2006;2:1126–1129. doi: 10.1002/smll.200600162. [DOI] [PubMed] [Google Scholar]
- Lopez-Acevedo O, Akola J, et al. Structure and bonding in the ubiquitous icosahedral metallic gold cluster Au-144(SR)(60) J Phys Chem C. 2009;113:5035–5038. [Google Scholar]
- Mercogliano CP, DeRosier DJ. Concatenated metallothionein as a clonable gold label for electron microscopy. J Struct Biol. 2007;160:70–82. doi: 10.1016/j.jsb.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell S, et al. A visual approach to proteomics. Nat Rev Mol Cell Biol. 2006;7:225–230. doi: 10.1038/nrm1861. [DOI] [PubMed] [Google Scholar]
- Nieba L, Nieba-Axmann SE, Persson A, Hämäläinen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson ÅF, Pluückthun A. BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Anal Biochem. 1997;252:217–228. doi: 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- Opalka N, Beckmann R, Boisset N, Simon MN, Russel M, Darst SA. Structure of the filamentous phage pIV multimer by cryo-electron microscopy. J Mol Biol. 2003;325:461–470. doi: 10.1016/s0022-2836(02)01246-9. [DOI] [PubMed] [Google Scholar]
- Park SH, Yin P, Liu Y, Reif JH, LaBean TH, Yan H. Programmable DNA self-assemblies for nanoscale organization of ligands and proteins. Nano Lett. 2005;5:729–733. doi: 10.1021/nl050175c. [DOI] [PubMed] [Google Scholar]
- Powell RD, Hainfeld JF. Silver- and gold-based autometallography of Nanogold. In: Hacker GW, Gu J, editors. Gold and Silver Staining: Techniques in Molecular Morphology. Chapter 3 CRC Press; Boca Raton, FL: 2002. pp. 29–46. [Google Scholar]
- Powell RD, Halsey CMR, Liu W, Joshi VN, Hainfeld JF. Giant platinum clusters: 2 nm covalent metal cluster labels. J Struct Biol. 1999;127:177–184. doi: 10.1006/jsbi.1999.4153. [DOI] [PubMed] [Google Scholar]
- Rappas M, Schumacher J, Beuron F, Niwa H, Bordes P, Wigneshweraraj S, Keetch CA, Robinson CV, Buck M, Zhang X. Structural insights into the activity of enhancer-binding proteins. Science. 2005;307:1972–1975. doi: 10.1126/science.1105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V, Lymar E, Hu M, Hainfeld JF. 5 nm Gold–Ni-NTA binds His Tags. In: Price R, Kotula P, Marko M, Scott JH, Vander Voort GF, Nanilova E, Mah Lee Ng M, Smith K, Griffin P, Smith P, McKernan S, editors. Microsc Microanal. Suppl 2. Vol. 11. Cambridge University Press; 2005. p. 1118CD. [Google Scholar]
- Ribrioux S, Kleymann G, Haase W, Heitmann K, Ostermeier C, Michel H. Use of Nanogold- and fluorescent-labeled antibody Fv fragments in immunocytochemistry. J Histochem Cytochem. 1996;44:207–213. doi: 10.1177/44.3.8648079. [DOI] [PubMed] [Google Scholar]
- Safer D, et al. Biospecific labeling with undecagold: Visualization of the biotin-binding site on avidin. Science. 1982;218:290–291. doi: 10.1126/science.7123234. [DOI] [PubMed] [Google Scholar]
- Sardar R, Funston A, et al. Gold nanoparticles: Past, present, and future. Langmuir. 2009;25:13840–13851. doi: 10.1021/la9019475. [DOI] [PubMed] [Google Scholar]
- Schaaff T, Whetten R. Controlled etching of Au:SR cluster compounds. J Phys Chem B. 1999;103:9394–9396. [Google Scholar]
- Schmitt J, Hess H, Stunnenberg HG. Affinity purification of histidine-tagged proteins. Mol Biol Rep. 1993;18:223–230. doi: 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- Segond von Banchet G, Schindler M, Hervieu GJ, Beckmann B, Emson PC, Heppelmann B. Distribution of somatostain receptor subtypes in rat lumbar spinal cord examined with gold-labelled somatostatin and anti-receptor antibodies. Brain Res. 1999;816:254–257. doi: 10.1016/s0006-8993(98)01226-8. [DOI] [PubMed] [Google Scholar]
- Sexton JZ, Ackerson CJ. Determination of rigidity of protein bound Au144 clusters by electron cryomicroscopy. J Phys Chem C. 2010 doi: 10.1021/jp101970x. Published on Web July 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Murray R. Dynamics and extent of ligand exchange depend on electronic charge of metal nanoparticles. J Am Chem Soc. 2002;124:7096–7102. doi: 10.1021/ja0174985. [DOI] [PubMed] [Google Scholar]
- Templeton A, et al. Gateway reactions to diverse. Polyfunctional monolayer-protected gold clusters. J Am Chem Soc. 1998;120:4845–4849. [Google Scholar]
- Templeton A, et al. Monolayer-protected cluster molecules. Acc Chem Res. 2000;33:27–36. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- Tkachenko AG, et al. Multifunctional gold nanoparticle-peptide complexes for nuclear targeting. J Am Chem Soc. 2003;125:4700–4701. doi: 10.1021/ja0296935. [DOI] [PubMed] [Google Scholar]
- Verma A, et al. Recognition and stabilization of peptide alpha-helices using templatable nanoparticle receptors. J Am Chem Soc. 2004;126:10806–10807. doi: 10.1021/ja047719h. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Amann KJ, Stoilova-McPhie S, Egile C, Winter DC, Hazelwood L, Heuser JE, Li R, Pollard TD, Hanein D. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 2001;293:2456–2459. doi: 10.1126/science.1063025. [DOI] [PubMed] [Google Scholar]
- Walsh CP, Davies A, Butcher AJ, Dolphin AC, Kitmitto A. Three-dimensional structure of CaV3.1: comparison with the cardiac L-type voltage-gated calcium channel monomer architecture. J Biol Chem. 2009;284:22310–22321. doi: 10.1074/jbc.M109.017152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, et al. A unified view of ligand-protected gold clusters as superatom complexes. Proc Natl Acad Sci USA. 2008;105:9157–9162. doi: 10.1073/pnas.0801001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden JG, et al. Monofunctional group-modified gold nanoparticles from solid phase synthesis approach: Solid support and experimental condition effect. Chem Mater. 2004a;16:3746–3755. [Google Scholar]
- Worden JG, et al. Controlled functionalization of gold nanoparticles through a solid phase synthesis approach. Chem Commun. 2004b:518–519. doi: 10.1039/b312819a. [DOI] [PubMed] [Google Scholar]
- Xiao S, Liu F, Rosen AE, Hainfeld JF, Seeman NC, Musier-Forsyth K, Kiehl RA. Self assembly of metallic nanoparticle arrays by DNA scaffolding. J Nanopart Res. 2002;4:313–317. [Google Scholar]
- Yonekura K, Yakushi T, Atsumi T, Maki-Yonekura S, Homma M, Namba K. Electron cryomicroscopic visualization of PomA/B stator units of the sodium-driven flagellar motor in liposomes. J Mol Biol. 2006;357:73–81. doi: 10.1016/j.jmb.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Young MT, Fisher JA, Fountain SJ, Ford RC, North RA, Khakh BS. Molecular shape, architecture, and size of P2×4 receptors determined using fluorescence resonance energy transfer and electron microscopy. J Biol Chem. 2008;283:26241–26251. doi: 10.1074/jbc.M804458200. [DOI] [PMC free article] [PubMed] [Google Scholar]