

Abstract
Allophanate hydrolase (AH) catalyzes the hydrolysis of allophanate, an intermediate in atrazine degradation and urea catabolism pathways, to NH3 and CO2. AH belongs to the amidase signature family, which is characterized by a conserved block of 130 amino acids rich in Gly and Ser and a Ser-cisSer-Lys catalytic triad. In the present study, the first structures of AH were solved from Granulibacter bethesdensis, with and without the substrate analog malonate, to 2.2 Å and 2.8 Å, respectively. The structures confirm the identity of the catalytic triad residues and reveal an altered dimerization interface that is not conserved in the amidase signature family. The structures also provide insights into previously unrecognized substrate specificity determinants in AH. Two residues, Tyr299 and Arg307, are within hydrogen bonding distance to a carboxylate moiety of malonate. Both Tyr299 and Arg307 were mutated and the resulting modified enzymes revealed greater than three orders of magnitude reductions in both catalytic efficiency and substrate stringency. It is proposed that Tyr299 and Arg307 serve to anchor and orient the substrate for attack by the catalytic nucleophile, Ser172. The structure further suggests the presence of a unique C-terminal domain in AH. While this domain is conserved, it does not contribute to catalysis or to the structural integrity of the core domain, suggesting that it may play a role in mediating transient and specific interactions with the urea carboxylase component of urea amidolyase. Analysis of the AH active site architecture offers new insights into common determinants of catalysis and specificity among divergent members of the amidase signature family.
Allophanate hydrolase (AH, EC 3.5.1.54) catalyzes the hydrolysis of allophanate to ammonia and carbon dioxide (Scheme 1). It is a member of the amidase signature (AS) family, characterized by a highly conserved stretch of up to 130 amino acids rich in serine and glycine (1, 2). AS family enzymes are currently known to include over 200 different enzymes in organisms ranging from bacteria to plants and mammals (3). They participate in diverse biological activities, including the catabolism of neuromodulatory fatty acids in mammals by fatty acid amide hydrolase (4), formation of Gln-tRNAGln by GatCAB in a subset of bacteria (5), formation of the plant development regulator, indole-3-acetic acid, by nitrile hydratase (6) and the hydrolysis of malonamate by malonamidase in nitrogen transport from the bacteroid to the plant during nitrogen fixation (7). In addition, AS family enzymes have been implicated in a variety of industrial applications, such as the degradation of nylon polymers (8, 9) and the amidation of synthetic peptides (10). AS family enzymes utilize a conserved Ser-cisSer-Lys triad to catalyze amide bond hydrolysis, where the underlined serine residue serves as the catalytic nucleophile (11). Nucleophilic attack by serine results in a covalent tetrahedral intermediate that is stabilized by an oxyanion hole. After displacement of ammonia, the covalent intermediate is hydrolyzed to release the product. Although the catalytic triad is conserved, AS family enzymes exhibit a wide variation in residues that contribute to substrate specificity. Given the broad cross-section of substrates acted upon by AS family enzymes, this family serves as an intriguing paradigm for relating molecular architecture to substrate specificity.
Scheme 1.
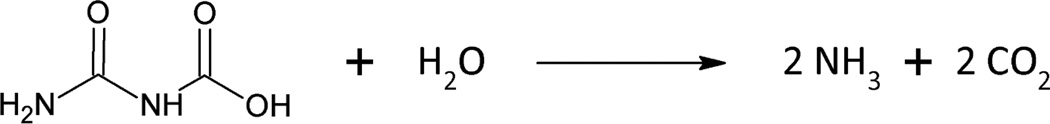
AH was first characterized as one domain of the multi-functional fungal enzyme, urea amidolyase. Urea amidolyase is a biotin-dependent enzyme comprised of two separate enzymatic activities, urea carboxylase and AH, that hydrolyze urea to ammonia and carbon dioxide in a two-step process. A carboxyl group is added to urea by urea carboxylase, forming allophanate, which is subsequently hydrolyzed into ammonia and carbon dioxide by AH. Urea amidolyase is the terminal enzyme of urea degradation and arginine catabolism (12, 13). It is a virulence factor in Candida albicans, an opportunistic pathogen which can evade the human immune system and cause lethal infections in immunocompromised patients. The urea carboxylase component of Kluyveromyces lactis urea amidolyase was recently crystallized and characterized (14). However, the structure of the AH domain remains undefined.
Unlike urea amidolyase enzymes in fungi, urea carboxylase and AH exist as separate subunits in bacteria (15, 16). It is not clear whether and to what degree these subunits physically interact to affect bacterial urea amidolyase activity. In addition, Shapir et al. (17) have determined that AH from Pseudomonas sp. strain ADP-1 is involved in cyanuric acid metabolism and that this enzyme has no association with urea carboxylase. In this pathway, AH serves as the terminal enzyme in s-triazine ring catabolism and serves an important role in the bio-degradation of herbicides such as atrazine. Phylogenetic analysis has revealed that AH is broadly distributed in bacteria, where it functions in the metabolism of both cyanuric acid and urea (18).
Here we report the first crystal structures of AH, both with and without a bound substrate analog, malonate, in the active site. The structure of AH from the bacterium Granulibacter bethesdensis confirms the location of the Ser-cisSer-Lys catalytic triad conserved throughout the AS family. Comparison of the AH structure to those from other AS family enzymes offers insights into substrate specificity determinants in AH. Two amino acids, Tyr299 and Arg307, are conserved in AH and are within hydrogen bonding distance to the substrate analog. Steady-state kinetic analysis of Tyr299 and Arg307 site-directed mutants reveal that the specificity constant (kcat/Km) ratio for allophanate compared to the substrate analog, biuret, decreases ~200 fold for Y299F and that the R307A and R307M mutants are nearly inactive. These results suggest a role for these residues in providing substrate and intermediate stabilization in AH. Finally, the structure suggests the presence of an additional C-terminal domain that is unique to AH and that may act as a functional linker between urea carboxylase and AH.
Materials and methods
Materials
IPTG, NADH and kanamycin were purchased from Research Products International Corp (Mount Prospect, IL). Ni2+-Profinity IMAC resin was obtained from Bio-Rad (Hercules, CA). Q-sepharose ion exchange resin was obtained from Amersham Biosciences, GE Healthcare (Pittsburgh, PA). All other materials were obtained from Sigma-Aldrich. Potassium allophanate was prepared by hydrolyzing ethyl allophanate (Acros Organics, Fisher Scientific, Pittsburgh, PA) as described previously (19).
Construction of an expression vector for His-tagged allophanate hydrolase
Genomic DNA from Granulibacter bethesdensis (strain ATCC BAA-1260 / CGDNIH1) was obtained from American Type Culture Collection (Manassas, VA). The allophanate hydrolase gene (GenBank: ABI62642.1) was amplified by PCR using the primers (5’- GAA GCT GCA GAT GAC GCT GAG CAT G -3’) and (5’- CAG ACT CGA GCT TAC TGC GCC AAA TA -3’). Primers were synthesized by Integrated DNA Technologies (Coralville, IA). The PCR amplified fragment was digested with PstI/XhoI and ligated into the PstI/XhoI digested pET28a-(HIS)8-TEV vector, downstream of the T7 promoter, N-terminal (His)8 tag and rTEV protease recognition sequence (20). The Tyr299 and Arg307 single and double mutations were generated for the present study by QuikChange site-directed mutagenesis (La Jolla, CA 92037) using Pfu Turbo polymerase. The complete sequence of all cloned and mutagenized genes was verified by Functional Biosciences (Madison, WI).
Overexpression and Purification of Protein
E. coli HMS174 (DE3) cells were transformed with the pET28a-(His)8-TEV plasmid encoding wild-type or mutagenized GbAH and were grown in LB media containing 25 µg/mL kanamycin. LB media (1 L) was inoculated with a single colony transformant and incubated overnight at 37°C. The overnight culture was used to inoculate 12 L of LB media which was grown at 37°C to an OD600 = 0.8. The cultures were chilled in an ice water bath prior to induction with IPTG to a final concentration of 1 mM. The cultures were transferred to a 16°C shaking incubator for 24 h prior to cell harvesting.
Both wild-type and mutant GbAH were purified using Ni2+- affinity and ion-exchange chromatography. The harvested cells were disrupted by sonication in buffer containing 50 mM HEPES (pH 8.0), 300 mM NaCl, 0.1 mM EGTA and centrifuged at 12000 rpm, 4°C prior to loading onto a column containing 10 mL Ni2+-Profinity IMAC resin. The column was washed using buffer containing 20 mM imidazole and the protein was eluted from the column using a gradient from 20 mM to 300 mM imidazole, with GbAH eluting between 100–250 mM. The purified protein was subsequently pooled and dialyzed overnight at 4°C against buffer containing 20 mM HEPES (pH 8.0), 50 mM NaCl, 1 mM EGTA and 2 mM DTT prior to loading onto a 10 mL Q-Sepharose ion-exchange column. The column was washed using buffer containing 50 mM NaCl and the protein was eluted using a gradient from 100 mM to 750 mM NaCl. The purified protein fractions were pooled, concentrated to 17 mg/ml, and then dialyzed overnight at 4°C against storage buffer containing 10 mM HEPES (pH 8.0), 50 mM NaCl, 1 mM DTT, and drop frozen in liquid nitrogen for storage at −80°C. The protein concentration was determined spectrophotometrically using the calculated molar extinction coefficient for G. bethesdensis AH of 61,810 M−1cm−1 at 280 nm (21). Typical yields were ~10 mg/L. The purity and subunit molecular weight were estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The purity was >99% and the molecular weight was determined to be ~65 kDa. Analytical gel filtration chromatography was performed on Superose 6 HR 10/30 (GE Healthcare) in buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM DTT.
Protein Crystallization
Crystals of GbAH, both with and without malonate, were grown at room temperature by the hanging-drop vapor diffusion method. Crystals of GbAH with malonate were obtained from a well solution of 100 mM PIPES (pH 6.5) and 1.04 M sodium malonate. The crystals of apo GbAH were obtained from a well solution of 100 mM HEPES (pH 7.5), 24% PEG 4K and 50 mM MgCl2. In both cases, the protein solution (4 mg/ml) and reservoir solution were mixed in a 1:1 ratio to a final volume of 5 µL. The initial GbAH crystals grew spontaneously from a well solution of 100 mM PIPES (pH 6.5), ans 1.04 M sodium malonate after 2 months. Subsequently, all drops were microseeded with pulverized GbAH crystals 24 h after mixing. The resulting diamond shaped GbAH crystals grew within 5–7 days of seeding. After 10–15 days, the crystals of GbAH with malonate were transferred to a cryoprotectant solution consisting of 6 M sodium malonate and 100 mM PIPES (pH 6.5). The crystals of apo GbAH were transferred to a cryoprotectant solution consisting of 100 mM HEPES (pH 7.5), 35% PEG 4K and 50mM MgCl2. All crystals were flash-cooled in a nitrogen gas stream at 100 K.
The crystals belonged to the space group P61, with two subunits in the asymmetric unit and the unit cell parameters were a = 78 Å, b = 78 Å, c = 395 Å, α= β = 90°, γ = 120°.
Data Collection, Structure Determination and Refinement
X-ray diffraction data were collected at Sector 21 (LS-CAT) of the Advanced Photon Source (APS, Chicago, IL). The diffraction data for GbAH with malonate were collected using beamline 21-ID-D with a MAR 300 CCD detector at a wavelength of 1.033Å. The diffraction data for apo GbAH were collected using beamline 21-ID-F with a MAR 225 CCD detector, at a wavelength of 1.033Å. Diffraction images were processed with HKL2000 (22). The structure of GbAH with malonate was solved by molecular replacement using a putative amidase protein (PDB ID: 2DC0) as the search model using the program AutoMR (23) from the Phenix software suite. The apo GbAH structure was solved by molecular replacement using GbAH with malonate as the search model with AutoMR. The initial models were subsequently built by the program AutoBuild (24), extended by several rounds of manual model building with COOT (25) and refined with Phenix.Refine (26). Water molecules were added to the model in COOT with subsequent manual verification. Data collection and processing statistics are summarized in Table 1. The long unit cell axis of 395 Å resulted in some overlapping reflections. This was particularly problematic for data collected on the MAR 225 CCD detector on LS-CAT-21-ID-F. Consequently, the values for Rmerge and some refinement statistics are slightly elevated but still fall well within the acceptable range.
Table 1.
Data Collection and Refinement Statistics
| GbAH + NaMalonate | GbAH apo | |
|---|---|---|
| PDB code | 4GYS | 4GYR |
| Space group | P61 | P61 |
| Cell dimensions | ||
| a, b, c (Å) | 78, 78, 395 | 78, 78, 398 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Resolution range (Å) | 50 - 2.2 | 50 - 2.8 |
| Redundancy | 9.2(6.6)a | 9.0(4.6) a |
| Completeness(%) | 97.2(96.2) | 99.9(100) |
| Rmerge (%) | 11.5(33.2) | 11.4(17.1) |
| Average I/sigma | 21.2(4.9) | 20.8(18.1) |
| Rwork | 0.174(0.218) | 0.256(0.296) |
| Rfree | 0.202(0.275) | 0.307(0.363) |
| No. reflections | 66210 | 33600 |
| No. atoms | 7294 | 6930 |
| Protein | 6840 | 6814 |
| Others | 28 | 0 |
| Water | 426 | 116 |
| Wilson B factor | 31.5 | 40.3 |
| Average B factors (Å) | ||
| Protein | 32.4 | 40.0 |
| Ligand | 36.3 | -- |
| Solvent | 35.7 | 35.7 |
| Ramachandran (%) | ||
| Favored | 89.0 % | 85.5 % |
| Allowed | 11.0 % | 14.3 % |
| Generous | 0 % | 0.3 % |
| Disallowed | 0 % | 0 % |
| R.m.s.deviations | ||
| Bond lengths(Å) | 0.011 | 0.005 |
| Bond angles(°) | 1.28 | 0.91 |
Values in parentheses are for the highest resolution bin.
Enzyme assays
The activity of wild-type GbAH and Tyr299 mutants with allophanate was assayed using a glutamate-dehydrogenase coupled assay at 340 nm (27) in 50 mM HEPES (pH 7.4), containing 50 mM NaCl and 1 mM EGTA in the presence of 10 mM allophanate, 40 U glutamate-dehydrogenase (GDH), 20 mM α-ketoglutarate, and 0.5 mM NADH. The activity of GbAH with substrate analogs and the activity of the GbAH Arg307 mutant with allophanate was assayed using a fixed-time phenol-hypochlorite assay in 50 mM HEPES (pH 7.4), 150 mM NaCl, and 1 mM DTT (28). A background rate was observed in all GDH-coupled reactions but it was subtracted from the AH-catalyzed rate. The reaction with substrate analogs was slow and in some cases was monitored for up to 10 hours in order to estimate rate constants. Despite the slow reaction rates, enzyme-catalyzed reactions (typically 1000 min−1) could easily be measured over the very slow background rate of spontaneous hydrolysis (~ 20 min−1). One enzyme unit is defined as the amount of enzyme required to hydrolyze 1 mole of substrate per min at 25°C.
Enzyme kinetic constants were estimated by nonlinear curve-fitting using GraphPad Prism 3.02 software (GraphPad Software Inc., San Diego, CA). The apparent Km and kcat values were determined by fitting to the Michaelis-Menten equation for single-enzyme systems.
RESULTS
Overall structure of AH
Allophanate hydrolase (AH) was cloned from the genomic DNA of a phylogenetically diverse group of bacteria including Granulibacter bethesdensis, Pseudomonas syringae, Wolinella succinogenes, and Verminephrobacter eiseniae. Only Granulibacter bethesdensis AH (GbAH) was amenable to crystallization. Granulibacter bethesdensis is an emerging opportunistic pathogen, first isolated in 2006 from patients with chronic granulomatous disease (29). The X-ray crystal structure of GbAH with malonate was determined by molecular replacement using a putative amidase (PDB ID: 2DC0, 32% sequence identity with GbAH) as a search model and was refined to 2.2 Å resolution. The apo GbAH structure was determined to 2.8 Å (table 1). A large number of overlapping reflections in the data set collected for the apo GbAH structure resulted in acceptable but higher than optimal values for Rmerge, R and Rfree. For this reason, descriptions of the apo GbAH structure are limited to comparative analyses of global and active site structure and the figures and conclusions presented herein are based entirely on the high quality structure of GbAH+malonate.
In both the apo and ligand-bound structures, the GbAH crystal lattice is composed of a homodimer in the asymmetric unit (Figure 1A). In malonate-bound GbAH, each subunit includes 461 residues (residues 3–463) while in the apo GbAH structure, each subunit includes 462 residues (residues 2–463). In all cases, the subunits consist of a globular amidase domain comprised of 15 α-helices and 11 β-strands, consistent with structures from other AS family enzymes (11). The N-terminal poly-His tag and the C-terminal residues 463–592 were not defined in the electron density of either the apo or malonate-bound structure and are not included in the models. SDS-PAGE of isolated GbAH crystals grown in the presence of malonate demonstrated that the complete 65kDa protein was intact in the crystals (data not shown). Therefore, the undefined C-terminus results from a lack of direct phasing information combined with intrinsic disorder in this region. Structural alignments of GbAH with other AS family enzymes show conservation of the overall structure despite low sequence similarities, ranging from 9 – 32% (Figure 1B–D). Analysis of the AH structure using the DALI server (30) confirms that AH is homologous with several AS family member enzymes (Supplementary Table S1). The Glutamyl-tRNA amidotransferase subunit A (GatA) is the most closely related in structure, as measured both by z-score and root mean squared deviation.
Figure 1. Structure of GbAH and structural comparison with other AS family enzymes.

(A) Overall structure of AH. The overall structure is shown in cartoon representation. Malonate binding at the active sites is shown in CPK-colored spheres. The three subdomains in each subunit are individually colored: N-terminal subdomain (blue), the amidase signature sequence (Phe71~Leu182) subdomain (hotpink), and the C-terminal subdomain (yellow). The corresponding subdomains in the second subunit are colored in light blue, light pink and light yellow. (B~D) Structural superimposition of the GbAH monomer (cyan) with other AS family enzymes. (B) Alignment of AH and Staphylococcus aureus GatA (PDB ID: 2F2A, salmon). (C) Alignment of AH and Bradyrhizobium japonicum malonamidase (MAE2, PDB ID: 1OCL) subunit A (white). (D) Alignment of AH and rat fatty acid amide hydrolase (FAAH, PDB ID: 1MT5) subunit A (magenta). (E~F) Structural superimposition of the GbAH dimer (cyan) with other AS family enzyme dimers: (E) MAE2 (white); (F) FAAH (magenta).
Analytical gel filtration analysis of GbAH revealed a holoenzyme with a molecular weight of ~130kDa, while SDS-PAGE revealed a ~65kDa monomer (data not shown), confirming that the biological unit is a dimer, consistent with characterization of other AH enzymes (15, 17). The dimerization interface was analyzed using the PDBe PISA server (31). Three dimerization interfaces were identified as having a large buried surface area and favorable ΔG (Supplementary Figure S1A). The biological dimer was determined to be that with the largest buried surface area and the most favorable ΔG (Supplementary Figure S1B). Of the three possible interfaces, this dimerization interface is most similar, though not identical, to the dimerization interface of the AS family enzymes fatty acid amide hydrolyase and malonamidase (Figure 1E–F, Supplementary Figure S1B).
GbAH active site architecture
AH shares a common catalytic mechanism with other AS family members, using a conserved Ser-cisSer-Lys triad to catalyze amidolysis of allophanate (17, 32). Similar to other AS family members, three conserved residues, Ser172, cis-Ser148 and Lys74, form the expected catalytic triad in GbAH with an oxyanion hole formed by the backbone amide nitrogens of Asp168, Thr169, Ala170 and Gly171. When GbAH was crystallized in sodium malonate (1.04 M), one molecule of malonate was bound at the active site and a second molecule was bound at the protein surface. In crystals grown in the absence of malonate, no electron density corresponding to a bound ligand was observed. The binding of malonate in the active site did not result in any structural rearrangements compared with the apo enzyme (Figure 2A, B). Based on the proposed mechanism of homologous AS family enzymes and analysis of inactivated and mutated AH enzymes, Ser172 acts as the catalytic nucleophile to attack the amide carbon on allophanate, while Ser148 acts as a general acid catalyst to stabilize the enzyme-substrate intermediate (11, 17, 32). The deprotonated form of Ser148 is maintained by Lys74 through a hydrogen bond and the tetrahedral intermediate is stabilized by the oxyanion hole. The catalytic triad facilitates the subsequent hydrolysis of the terminal amide bond to yield a Ser172-dicarboxylated amide and one molecule of NH3. It is not clear whether the dicarboxylated amide further decomposes while tethered to Ser172 or is hydrolyzed intact from the enzyme and subsequently decomposes in solution (32). The nucleophilic Ser172 points away from malonate in the bound structure, but is oriented toward the active site cavity in the apo enzyme (Figure 2B, C)
Figure 2. Structural superposition of GbAH apo with the malonate-bound structure.

(A) Superimposition of the GbAH monomer with (purple) and without (cyan) bound malonate reveals no significant conformational changes accompanying substrate binding. The active-site-bound malonate is shown in CPK-colored spheres. (B) A stereo figure showing the structural superimposition of the active sites of apo GbAH (cyan) and the malonate-bound GbAH (purple). A simulated annealing omit map for malonate, contoured at 3.0 σ, is shown in green. The 2Fo-Fc map, contoured at 1.0 σ, is shown in grey. For clarity, only side chains are displayed for Tyr299, Arg307, Ser148, Ser172 and Lys74. Only mainchain atoms are displayed for the oxyanion hole. (C) A stereo figure showing the active site of malonate-bound GbAH (purple). The distances (in Å) are indicated. The nucleophilic Ser172 adopts a conformation that is oriented away from malonate in the malonate-bound GbAH structure. However, this residue can also adopt an orientation that is oriented toward the substrate’s electrophilic center, as is observed in the overlaid side-chain of Ser172 from the apo GbAH structure (pink) (D) Sequence alignment of AH, UAL and other AS family enzymes shows conservation of Tyr299 and Arg307 among AH enzymes. GbAH (Granulibacter bethesdensis allophanate hydrolase); VeAH (Verminephrobacter eiseniae allophanate hydrolase); PsAH (Pseudomonas syringae allophanate hydrolase); ScUAL (Saccharomyces cerevisiae urea amidolyase); CaUAL (Candida albicans urea amidolyase); GatA (Thermotoga maritima Glutamyl-tRNA(Gln) amidotransferase subunit A); MAE2 (Bradyrhizobium japonicum malonamidase E2); FAAH (rat fatty acid amide hydrolase).
The crystal structures of GbAH with bound malonate also reveal two residues, Tyr299 and Arg307, within interacting distance (2.6 and 2.8 Å, respectively) of one of the malonate carboxylate moieties (Figure 2C). Sequence alignments reveal that both Tyr299 and Arg307 are conserved in all AH enzymes but that they are not conserved across the AS family (Figure 2D). This suggests a role for these residues in facilitating substrate binding in the enzyme active site.
The size of the active site cavity is relatively small compared to other AS family enzymes. Measurement of the active site cavity using VOIDOO (33) reveals that the cavity size is closest to GatA, which uses glutamine or asparagine as a substrate. The cavity size comparison shows a pronounced difference between AH and malonamidase, an AS family enzyme which hydrolyzes a very similar substrate, malonamate.
GbAH enzyme activity
The kcat for wild-type GbAH is 18.2 s−1, the Km is 0.10 mM and kcat/Km is 1.8×105 s−1 M−1 (Table 2). This falls in a range of kcat/Km values previously determined for AH from Oleomonas sagaranensis (2.7×106 s−1 M−1) (15), AtzF from Pseudomonas sp. strain ADP (1.1×104 s−1 M−1) (17) and TrzF from Enterobacter cloacae strain 99 (8.8×103 s−1 M−1) (32).
Table 2.
Kinetic data of GbAH wild-type and site-directed mutants with allophanate.
| kcat(s−1)* | Km (mM) * |
kcat/Km (s−1 M−1) |
|
|---|---|---|---|
| wt | 18 ± 1 | 0.10 ± 0.002 | (1.8 ± 0.03)×105 |
| C_del | 20 ± 1 | 0.30 ± 0.004 | (6.9 ± 0.2) ×104 |
| Y299F | (4.2 ± 0.1)×10−2 | 1.2 ± 0.1 | 35 ± 3 |
| Y299A | (7.1 ± 0.2)×10−2 | 3.0 ± 0.4 | 24 ± 3 |
| R307A | - | - | (1.7 ± 0.2) ×10−3 |
| R307M | - | - | (3.6 ± 0.8) ×10−3 |
| Y299A/R307M | - | - | (1.9 ± 0.1) ×10−2 |
| Y299F/R307M | - | - | (7.1 ± 0.6) ×10−3 |
| Y299A/R307A | - | - | (2.7 ± 0.2) ×10−2 |
mean ± standard deviation (n=3).
kcat/Km estimated from slope of vi vs [S] at [S]<<Km; mean ± standard deviation (n=3).
Substrate specificity
AH has been shown to exhibit high specificity for allophanate (15, 32). To assess the substrate specificity of GbAH, the rate of ammonia release was measured for a series of allophanate analogs, including biuret, malonamide, acetyl-urea, hydantoic acid, L-glutamine and L-asparagine (Supplementary Figure S2). Among this group of substrate analogues, activity was only detected with biuret and malonamide, consistent with previous reports (32). The measured kinetic constants for GbAH with allophanate and the substrate analogs biuret and malonamide are provided in Table 3 with representative data provided in Supplementary Figure S3.
Table 3.
Kinetic data for GbAH wild-type and mutants with different substrate analogs
| Biuret kcat/Km* (s−1 M−1) |
Ratio to wt |
Malonamide kcat/Km* (s−1 M−1) |
Ratio to wt | |||||
|---|---|---|---|---|---|---|---|---|
| wt | 0.30 ± 0.01 | (1.7 ± 0.1) ×10−6 | 1 | 0.38 ± 0.01 | (2.1 ± 0.2) ×10−6 | 1 | ||
| C_del | 0.59 ± 0.02 | (8.6 ± 0.4) ×10−6 | 5.2 | 0.35 ± 0.01 | (5.1 ± 0.2) ×10−6 | 2.4 | ||
| Y299F | 0.35 ± 0.02 | (1.0 ± 0.1) ×10−2 | 6.1×103 | 0.12 ± 0.01 | (3.5 ± 0.1) ×10−3 | 1.6×103 | ||
| Y299A | 1.0 ± 0.02 | (4.3 ± 0.6) ×10−2 | 2.6×104 | 0.36 ± 0.02 | (1.5 ± 0.2) ×10−2 | 7.2×103 |
kcat/Km estimated from slope of vi vs [S] at [S]<<Km; mean ± standard deviation (n=3).
Tyr299 and Arg307 are within hydrogen bonding distance (2.6 and 2.8 Å, respectively) of one carboxylate moiety of malonate (Figure 2C). Both Tyr299 and Arg307 were mutated in GbAH to investigate the role of these two residues in catalysis. A series of single mutants (Y299F, Y299A, R307M, R307A), and double mutants (Y299A/R307M, Y299F/R307M, Y299A/R307A, and Y299F/R307A) were assayed for catalytic activity. While the dimerization interface is located in close proximity to the active sites, analytical gel-filtration confirmed that mutations in the active site residues located near the interface (ie. Ser172, Tyr299, Arg307) did not alter the oligomerization state of the enzyme (Supplementary Figure S4). The kinetic constants measured with allophanate revealed a ~400 fold and ~250 fold reduction in kcat for Y299F and Y299A, respectively, and a ~5000 fold and ~7500 fold reduction in kcat/Km for Y299F and Y299A, respectively (Table 2). In addition, mutations in Tyr299 altered the enzyme’s discrimination between substrates, increasing the specificity constants for biuret and malonamide relative to allophanate. The ratio of kcat/Km for biuret compared to allophanate increased by three orders of magnitude in Y299F and four orders of magnitude in Y299A (Table 3). By comparison, mutations at Arg307 had a much more dramatic effect on the rate of catalysis. The kcat/Km is estimated at 2–4×10−3 s−1M−1 for Arg307 mutations, resulting in a decrease in kcat/Km of eight orders of magnitude. No activity could be observed for Arg307 mutants with biuret or malonamide.
Function of the C-terminal domain
While the structures in the presence and absence of malonate defined the 462 amino acid globular amidase domain, the C-terminal 126 amino acids were disordered in both structures. This suggests that an additional, less structured domain of unknown function resides at the C terminus of AH. This domain is conserved in all AH enzymes, but is missing in other AS family enzymes (Figure 3). To determine whether the C-terminal domain influences the overall amidase activity, the C-terminal 126 amino acids were truncated by introducing two stop codons immediately following the last ordered residue from the crystal structure, Thr462. The C-terminally truncated enzyme was over-expressed and purified and the molecular weight was confirmed to be ~50 kDa by SDS-PAGE, compared to the full-length ~65 kDa wild-type enzyme. The truncated enzyme maintained its ability to dimerize, as measured by analytical gel filtration (Supplementary Figure S4). Furthermore, truncation of the C-terminus had very little effect on the kinetic constants. The kcat and Km values measured for the C-terminal truncation were very similar to those measured for the wild-type enzyme, both with the substrate and with substrate analogs (Tables 2 and 3), indicating that the C-terminal domain does not significantly influence amidase activity or substrate specificity.
Figure 3. The C-terminal domain of GbAH.

(A) Structure of amidase domain of AH with a cartoon representation showing the missing C terminal domain, depicted as blue ovals encircled by a dotted line. (B) Schematic representation of the domain arrangement of AH, urea carboxylase, urea amidolyase and other AS family enzymes.
DISCUSSION
Acyclamide amidohydrolase enzymes (E.C. classification group 3.5.1) catalyze the hydrolysis of linear amide bonds in a wide range of substrates. Several enzyme superfamilies with unrelated sequence and structure have converged on this common enzymatic function, including the amidohydrolase, nitrilase and amidase signature (AS) superfamilies. The AS superfamily catalyzes amidolysis without the assistance of a metal ion, instead using a nucleophilic serine and oxyanion hole to mediate the formation and stabilization of the tetrahedral transition state. The superfamily was originally identified and characterized by a signature block of 130 amino acids rich in conserved Gly and Ser residues (2). Subsequent structural and kinetic studies of AS family enzymes revealed a conserved Ser-cisSer-Lys catalytic triad that is essential for catalysis (11, 34, 35, 36). The structures for a few well characterized members of the AS family have been reported in recent years, including GatA (37), malonamidase (11), fatty acid amide hydrolase (38), and peptide amidase (39). These structures have confirmed a common catalytic core and have revealed a diverse active site architecture that accommodates both stringent and promiscuous activities over a wide range of substrates.
AH is a typical AS family enzyme using a Ser-cisSer-Lys catalytic triad to hydrolyze allophanate to ammonia and carbon dioxide. In some bacterial strains, AH works in conjunction with cyanuric acid amidohyrolase (EC 3.5.2.15) and biuret amidohydrolase (EC 3.5.1.84) to catalyze the final steps of s-triazide degradation (17, 32, 40). In certain fungi and prokaryotes, AH also functions in concert with urea carboxylase to degrade urea (16, 41). Prior to this investigation, no structure of AH had been reported. Several studies described a high degree of substrate specificity in AH (15, 17, 32) but, in the absence of structural insights, the molecular basis for this specificity has not been defined.
General Features of the AH Structure
Both the overall fold and the position of the catalytic triad in GbAH superimpose well with other members of the AS family. However, comparison of the AH dimer with homologous structures reveals that the superfamily exhibits a surprising plasticity at the dimerization interface. A structural alignment of GbAH with dimeric superfamily members shows that the dimerization interface varies between enzymes, leading to a wide variation in the distance between active sites, ranging from 33 to 60 Å (Figure 1E–F). The absence of a conserved dimerization interface in the AS family suggests that dimerization does not play an essential role in catalysis and that the individual active sites most likely function independently. Indeed, some AS family members do not form homodimers: GatA, the glutamine amidohydrolase component of GatCAB, makes up a heterotrimer with GatB and GatC subunits (5) and in vivo and in vitro studies of Arabidopsis amidase-1 have concluded that there is no interaction between the amidase monomers (42). Nevertheless, the relationship between dimerization and catalytic activity in AH warrants further investigation considering that the essential active site residues Tyr299 and Arg307 (vide infra) are positioned near the dimerization interface. A connection between dimerization and substrate specificity has been proposed for the nitrile amidase from Rhodococcus sp. N-771, where a modified dimerization interface reduces the width of the substrate binding tunnel, thereby limiting the access of larger substrates (6).
Comparison of the apo and malonate bound structures of GbAH reveals no structural rearrangements accompanying ligand binding in the active site (Figure 2B), consistent with other AS family enzymes (8, 11). Thus, from X-ray structures, it appears that the active sites of AS enzymes are rigid assemblies and that substrate discrimination arises by limiting substrate access to the active site and by the design of specific architectures that are complementary to the substrate(s). This apparent rigidity of AS active sites makes the superfamily particularly amenable to functional predictions by in silico ligand docking, as described in a recent review (43).
Substrate Specificity Determinants in AH
Previous studies on AH cloned from Enterobacter cloacae (TrzF) and Pseudomonas sp. Strain ADP (AtzF) confirmed the identity of the Ser-cisSer-Lys catalytic triad and documented a detectable promiscuous activity with the alternate substrates biuret, malonamide and malonamate (17, 32). TrzF has a specific activity of 0.5 nmol min−1mg−1 with biuret and 1.5 nmol min−1mg−1 with malonamide (32). GbAH has a similar activity of 8.3 nmol min−1mg−1 with biuret and 2.3 nmol min−1mg−1 with malonamide. Conversely, AH from Oleomonas sagaranensis had no detectable activity with biuret or malonamide substrates (15). Whereas the identity of the catalytic triad and the low tolerance for analogous substrates has been documented in AH, the active site residues contributing to substrate recognition have not been described owing to a lack of structural information. In the current study, the structure of the GbAH-malonate complex combined with kinetic analysis of GbAH modified by site-directed mutagenesis has revealed an important role for two conserved residues: Tyr299 and Arg307. The side chains for these amino acids are positioned at a distance of 2.6–2.8 Å from the carboxylate moiety of the substrate analogue, malonate (Figure 2C). Mutations at Arg307 result in a nearly complete loss of catalytic activity, while mutations at Tyr299 substantially decrease kcat and kcat/Km for allophanate and reduce the substrate preference of AH for allophanate compared with biuret and malonamide (Tables 2 and 3).
While malonate is not an ideal isosteric mimic of allophanate, the GbAH structure with bound malonate can be combined with kinetic analysis of GbAH site-directed mutations to propose a role for Tyr299 and Arg307 in AH catalysis. The side chain of Tyr299 is 2.6 Å from the carboxylate oxygen of malonate. This residue is proposed to donate a hydrogen bond to the deprotonated carboxylate of allophanate. Hydrogen bond donation from Tyr299 would facilitate discrimination between the carboxylate moiety of the bona fide substrate, allophanate, and the amide substituent of the substrate analogues, biuret and malonamide. The reduced discrimination between allophanate and malonamide/biuret in Tyr299 mutations is consistent with this proposal. In addition, the side chain of Arg307 is 2.8 Å from the other carboxylate oxygen of malonate, suggesting that Arg307 interacts with the substrate carboxylate via a salt bridge in a manner analogous to interactions with substrate carboxylate moieties in malonamidase and GatA (vide infra). The nearly complete loss of catalytic activity with allophanate observed in site-directed mutations at Arg307 is consistent with this proposal. Formation of a salt bridge between Arg307 and the substrate would serve both to bind and orient the substrate in proximity to the catalytic triad and also to assist in stabilizing developing negative charge in the transition state. The inability of wild-type AH to form a salt bridge with the alternate substrates biuret and malonamide may further account for the poor reactivity of these substrates. Substrate discrimination in AH, therefore, may originate from anchoring the carboxylate moiety of allophanate in the active site opposite the catalytic triad and, thus, properly positioning and orienting the amide portion of the substrate for nucleophilic attack. It is conceivable that substrate analogues such as biuret, malonamide and asparagine are anchored improperly and, consequently, the terminal amide of these substrates is improperly positioned for attack by the Ser172 nucleophile of the catalytic triad. Confirmation of this proposal awaits structures of GbAH bound with alternate substrates.
Malonamate is a structural analogue of allophanate and the substrate of the AS family enzyme, malonamidase. Given the similarity between the substrates, these two enzymes might be expected to share similar active site architectures. However, structural and phylogenetic analysis (17) reveals that malonamidase and AH are distantly related AS superfamily members. Instead, AH is most closely related to GatA, the bacterial glutamyl-tRNA amidotransferase subunit A. Interestingly, all three of these enzymes position an Arg side-chain for salt-bridge formation with the substrate carboxylate moiety. In malonamidase, this residue (Arg158) is located on the opposite side of the active site compared with AH and GatA, resulting in a completely different substrate binding orientation (Figure 3A). This arginine is replaced by a valine in AH and a glutamine in GatA. In addition, an active site loop (amino acids 119–125 in GbAH) present in most AS family enzymes adopts a variety of conformations and contributes to substrate specificity by dictating the orientation of the substrate in the active site. In malonamidase, this loop adopts an atypical conformation, forcing the substrate into a different binding orientation (Figure 4A and Supplementary Figure S5).
Figure 4. Comparison of substrate binding residues at the active sites of GbAH with other AS family enzymes.

(A) Alignment of malonate-bound GbAH (cyan) and malonate-bound Bradyrhizobium japonicum malonamidase (PDB ID 1OCL; white). (B) Alignment of malonate-bound GbAH (cyan) and Staphylococcus aureus GatA with covalently bound glutamine (PDB ID 2F2A; salmon). (C) Alignment of malonate-bound GbAH (cyan) and benzamide-bound Rhodococcus sp. N-771 amidase (PDB ID 3A1I; yellow). (D) Alignment of malonate-bound GbAH (cyan) with rat fatty acid amide hydrolase (PDB ID 1MT5; pink) with the bound inactivator methoxy arachidonyl fluorophosphonate (MAY). Residues and substrates are shown as sticks, labeled and colored according to their structure.
The substrate binding orientation and the position and identity of the side-chains responsible for substrate binding are very similar between GatA and AH (Figure 4B). In the GatA structure, an alanine replaces the Arg307 of GbAH. However, GatA positions a different arginine side-chain at a similar location in the active site, allowing the guanidinium side chain to form a salt bridge with the substrate carboxylate. This guanidinium group is 7 Å further removed from the catalytic triad in GatA relative to GbAH, allowing the longer substrates, asparagine and glutamine, to be properly positioned in the triad of GatA. It is notable that AH does not catalyze the hydrolysis of either glutamine or asparagine. The difference in the arginine side-chain position may explain this discrimination, accounting for the specificity of GatA for longer substrates and AH for shorter substrates. In the heterotrimeric GatCAB complex, an ammonia channel runs from the GatA glutaminase active site to the transamidase catalytic pocket in GatB (5, 37). This same channel appears to be present in GbAH and may serve as an exit channel for the release of the ammonia (Supplementary Figure S6).
A structurally independent C-terminal domain in AH
The structures of GbAH reveal the presence of a unique C-terminal domain which is not present in other AS family enzymes but which is conserved in AH and urea amidolyase. Deletion of this domain did not significantly influence the dimerization or the catalytic activity of GbAH. The functional role of this domain, therefore, cannot yet be unambiguously assigned. However, given that AH activity is typically coupled to the reaction of urea carboxylase and that they comprise separate domains of urea amidolyase in yeast (Figure 3B), it is tempting to speculate that the C-terminal domain of AH facilitates the interaction and coupling between AH and urea carboxylase enzymes or between the individual catalytic domains in urea amidolyase. In the heterotrimeric GatCAB, a small GatC subunit links GatA and GatB and is essential for maintaining GatCAB structure and activity (37, 44, 45). In bacteria, the genes encoding urea carboxylase and AH are located in close proximity, suggesting that they may coordinate their activities (16). In G. bethesdensis, the gene encoding AH is located 2 nucleotides immediately downstream of the gene encoding urea carboxylase and two additional genes of unknown function (GenBank Accession Numbers: YP_745567 and YP_745568) are also tightly clustered into what likely constitutes a single operon. However, studies examining the interaction between urea carboxylase and AH indicate that AH and urea carboxylase do not form a stable complex (15), consistent with studies on complex formation between AH and urea carboxylase from Pseudomonas syringae (Supplementary Figure S7). The absence of a stable complex between AH and urea carboxylase does not, however, preclude the possibility of a transient interaction between the two enzymes that may serve to facilitate the efficient transfer of intermediates (46, 47). Furthermore, additional proteins may be required to maintain a stable complex between AH and urea carboxylase and it is notable that two additional putative cytoplasmic proteins of unknown function are located in close proximity to the genes for AH and urea carboxylase in many bacteria. Structural and kinetic studies are currently underway to clarify whether additional proteins mediate multi-enzyme complex formation via the C-terminal domain of AH and to determine the extent of substrate channeling that exists between urea carboxylase and AH.
Supplementary Material
Acknowledgements
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
ABBREVIATIONS
- AH
allophanate hydrolase
- APS
Advanced Photon Source
- AS family
amidase signature family
- DTT
dithiothreitol
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- GatA
Glutamyl-tRNA amidotransferase subunit A
- GatCAB
heterotrimeric glutamyl-tRNA amidotransferase CAB
- GbAH
Granulibacter bethesdensis allophanate hydrolase
- GDH
glutamate-dehydrogenase
- Gln-tRNAGln
glutaminyl- transfer RNAGln
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- NADH
reduced nicotinamide adenine dinucleotide
- TEV
tobacco etch virus
- PEG
poly(ethylene glycol)
- PIPES
piperazine-N,N′-bis(2-ethanesulfonic acid)
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Footnotes
This work is supported by the National Institute of General Medical Sciences of the National Institute of Health under Award Number R15GM097724.
Supporting information. Supplementary Methods, Table S1 and Supplementary Figures S1 through S7 are provided in the supporting information. This material may be accessed free of charge online at http://pubs.acs.org.
Accession codes. The atomic coordinates for apo GbAH and GbAH+malonate have been deposited in the Protein Data Bank as entry 4GYR and 4GYS, respectively.
REFERENCES
- 1.Koo HM, Choi SO, Kim HM, Kim YS. Identification of Active-Site Residues in Bradyrhizobium japonicum Malonamidase E2. Biochem. J. 2000;349:501–507. doi: 10.1042/0264-6021:3490501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chebrou H, Bigey F, Arnaud A, Galzy P. Study of the Amidase Signature Group. Biochim. Biophys. Acta. 1996;1298:285–293. doi: 10.1016/s0167-4838(96)00145-8. [DOI] [PubMed] [Google Scholar]
- 3.Shin S, Yun YS, Koo HM, Kim YS, Choi KY, Oh BH. Characterization of a Novel Ser-cisSer-Lys Catalytic Triad in Comparison with the Classical Ser-His-Asp Triad. J. Biol. Chem. 2003;278:24937–24943. doi: 10.1074/jbc.M302156200. [DOI] [PubMed] [Google Scholar]
- 4.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Fatty Acid Amide Hydrolase Substrate Specificity. Bioorg. Med. Chem. Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Bu W, Sheppard K, Kitabatake M, Kwon ST, Soll D, Smith JL. Insights into tRNA-Dependent Amidotransferase Evolution and Catalysis from the Structure of the Aquifex aeolicus Enzyme. J. Mol. Biol. 2009;391:703–716. doi: 10.1016/j.jmb.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohtaki A, Murata K, Sato Y, Noguchi K, Miyatake H, Dohmae N, Yamada K, Yohda M, Odaka M. Structure and Characterization of Amidase from Rhodococcus Sp. N-771: Insight into the Molecular Mechanism of Substrate Recognition. Biochim. Biophys. Acta. 2010;1804:184–192. doi: 10.1016/j.bbapap.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Kim YS, Kang SW. Novel Malonamidases in Bradyrhizobium japonicum. Purification, Characterization, and Immunological Comparison. J. Biol. Chem. 1994;269:8014–8021. [PubMed] [Google Scholar]
- 8.Yasuhira K, Shibata N, Mongami G, Uedo Y, Atsumi Y, Kawashima Y, Hibino A, Tanaka Y, Lee YH, Kato D, Takeo M, Higuchi Y, Negoro S. X-Ray Crystallographic Analysis of the 6-Aminohexanoate Cyclic Dimer Hydrolase: Catalytic Mechanism and Evolution of an Enzyme Responsible for Nylon-6 Byproduct Degradation. J. Biol. Chem. 2010;285:1239–1248. doi: 10.1074/jbc.M109.041285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heumann S, Eberl A, Fischer-Colbrie G, Pobeheim H, Kaufmann F, Ribitsch D, Cavaco-Paulo A, Guebitz GM. A Novel Aryl Acylamidase from Nocardia farcinica Hydrolyses Polyamide. Biotechnol. Bioeng. 2009;102:1003–1011. doi: 10.1002/bit.22139. [DOI] [PubMed] [Google Scholar]
- 10.Cerovsky V, Kula MR. Studies on Peptide Amidase-Catalysed C-Terminal Peptide Amidation in Organic Media with Respect to its Substrate Specificity. Biotechnol. Appl. Biochem. 2001;33:183–187. doi: 10.1042/ba20000082. [DOI] [PubMed] [Google Scholar]
- 11.Shin S, Lee TH, Ha NC, Koo HM, Kim SY, Lee HS, Kim YS, Oh BH. Structure of Malonamidase E2 Reveals a Novel Ser-cisSer-Lys Catalytic Triad in a New Serine Hydrolase Fold that is Prevalent in Nature. EMBO J. 2002;21:2509–2516. doi: 10.1093/emboj/21.11.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roon RJ, Hampshire J, Levenberg B. Urea Amidolyase. the Involvement of Biotin in Urea Cleavage. J. Biol. Chem. 1972;247:7539–7545. [PubMed] [Google Scholar]
- 13.Ghosh S, Navarathna DH, Roberts DD, Cooper JT, Atkin AL, Petro TM, Nickerson KW. Arginine-Induced Germ Tube Formation in Candida albicans is Essential for Escape from Murine Macrophage Line RAW 264.7. Infect. Immun. 2009;77:1596–1605. doi: 10.1128/IAI.01452-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan C, Chou CY, Tong L, Xiang S. Crystal Structure of Urea Carboxylase Provides Insights into the Carboxyltransfer Reaction. J. Biol. Chem. 2012;287:9389–9398. doi: 10.1074/jbc.M111.319475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanamori T, Kanou N, Kusakabe S, Atomi H, Imanaka T. Allophanate Hydrolase of Oleomonas sagaranensis Involved in an ATP-Dependent Degradation Pathway Specific to Urea. FEMS Microbiol. Lett. 2005;245:61–65. doi: 10.1016/j.femsle.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 16.Strope PK, Nickerson KW, Harris SD, Moriyama EN. Molecular Evolution of Urea Amidolyase and Urea Carboxylase in Fungi. BMC Evol. Biol. 2011;11:80. doi: 10.1186/1471-2148-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shapir N, Sadowsky MJ, Wackett LP. Purification and Characterization of Allophanate Hydrolase (AtzF) from Pseudomonas Sp. Strain ADP. J. Bacteriol. 2005;187:3731–3738. doi: 10.1128/JB.187.11.3731-3738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng G, Shapir N, Sadowsky MJ, Wackett LP. Allophanate Hydrolase, Not Urease, Functions in Bacterial Cyanuric Acid Metabolism. Appl. Environ. Microbiol. 2005;71:4437–4445. doi: 10.1128/AEM.71.8.4437-4445.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitney PA, Cooper TG. Urea Carboxylase and Allophanate Hydrolase. Two Components of Adenosine Triphosphate:Urea Amido-Lyase in Saccharomyces cerevisiae. J. Biol. Chem. 1972;247:1349–1353. [PubMed] [Google Scholar]
- 20.Lietzan AD, Menefee AL, Zeczycki TN, Kumar S, Attwood PV, Wallace JC, Cleland WW, St Maurice M. Interaction between the Biotin Carboxyl Carrier Domain and the Biotin Carboxylase Domain in Pyruvate Carboxylase from Rhizobium etli. Biochemistry. 2011;50:9708–9723. doi: 10.1021/bi201277j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zbyszek Otwinowski WM. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative Model Building, Structure Refinement and Density Modification with the PHENIX Auto-Build Wizard. Acta Crystallogr. D Biol. Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Afonine PV, Grosse-Kunstleve RW, Adams PD. The Phenix Refinement Framework. CCP4 Newsletter. 2005 Jul [Google Scholar]
- 27.Kimura S, Yamanishi H, Iyama S, Yamaguchi Y, Kanakura Y. Enzymatic Assay for Determination of Bicarbonate Ion in Plasma using Urea Amidolyase. Clin. Chim. Acta. 2003;328:179–184. doi: 10.1016/s0009-8981(02)00430-8. [DOI] [PubMed] [Google Scholar]
- 28.Weatherburn MW. Phenol-Hypochloride Reaction for Determination of Ammonia. Anal. Chem. 1967;39:971–974. [Google Scholar]
- 29.Greenberg DE, Porcella SF, Stock F, Wong A, Conville PS, Murray PR, Holland SM, Zelazny AM. Granulibacter Bethesdensis Gen. Nov., Sp. Nov., a Distinctive Pathogenic Acetic Acid Bacterium in the Family Acetobacteraceae. Int. J. Syst. Evol. Microbiol. 2006;56:2609–2616. doi: 10.1099/ijs.0.64412-0. [DOI] [PubMed] [Google Scholar]
- 30.Holm L, Rosenstrom P. Dali Server: Conservation Mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Shapir N, Cheng G, Sadowsky MJ, Wackett LP. Purification and Characterization of TrzF: Biuret Hydrolysis by Allophanate Hydrolase Supports Growth. Appl. Environ. Microbiol. 2006;72:2491–2495. doi: 10.1128/AEM.72.4.2491-2495.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleywegt GJ, Jones TA. Detection, Delineation, Measurement and Display of Cavities in Macromolecular Structures. Acta Crystallogr. D Biol. Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 34.Valina AL, Mazumder-Shivakumar D, Bruice TC. Probing the Ser-Ser-Lys Catalytic Triad Mechanism of Peptide Amidase: Computational Studies of the Ground State, Transition State, and Intermediate. Biochemistry. 2004;43:15657–15672. doi: 10.1021/bi049025r. [DOI] [PubMed] [Google Scholar]
- 35.McKinney MK, Cravatt BF. Evidence for Distinct Roles in Catalysis for Residues of the Serine-Serine-Lysine Catalytic Triad of Fatty Acid Amide Hydrolase. J. Biol. Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- 36.Shin S, Lee TH, Koo HM, Kim SY, Lee HS, Kim YS, Oh BH. Crystallization and Preliminary X-Ray Crystallographic Analysis of Malonamidase E2, an Amidase Signature Family Member. Acta Crystallogr. D Biol. Crystallogr. 2002;58:562–563. doi: 10.1107/s0907444902001415. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura A, Yao M, Chimnaronk S, Sakai N, Tanaka I. Ammonia Channel Couples Glutaminase with Transamidase Reactions in GatCAB. Science. 2006;312:1954–1958. doi: 10.1126/science.1127156. [DOI] [PubMed] [Google Scholar]
- 38.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural Adaptations in a Membrane Enzyme that Terminates Endocannabinoid Signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 39.Labahn J, Neumann S, Buldt G, Kula MR, Granzin J. An Alternative Mechanism for Amidase Signature Enzymes. J. Mol. Biol. 2002;322:1053–1064. doi: 10.1016/s0022-2836(02)00886-0. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Gonzalez V, Govantes F, Porrua O, Santero E. Regulation of the Pseudomonas Sp. Strain ADP Cyanuric Acid Degradation Operon. J. Bacteriol. 2005;187:155–167. doi: 10.1128/JB.187.1.155-167.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lombard J, Moreira D. Early Evolution of the Biotin-Dependent Carboxylase Family. BMC Evol. Biol. 2011;11:232. doi: 10.1186/1471-2148-11-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pollmann S, Neu D, Weiler EW. Molecular Cloning and Characterization of an Amidase from Arabidopsis thaliana Capable of Converting Indole-3-Acetamide into the Plant Growth Hormone, Indole-3-Acetic Acid. Phytochemistry. 2003;62:293–300. doi: 10.1016/s0031-9422(02)00563-0. [DOI] [PubMed] [Google Scholar]
- 43.Gerlt JA, Babbitt PC, Jacobson MP, Almo SC. Divergent Evolution in Enolase Superfamily: Strategies for Assigning Functions. J. Biol. Chem. 2012;287:29–34. doi: 10.1074/jbc.R111.240945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagao A, Suzuki T, Katoh T, Sakaguchi Y, Suzuki T. Biogenesis of Glutaminyl-Mt tRNAGln in Human Mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16209–16214. doi: 10.1073/pnas.0907602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheppard K, Soll D. On the Evolution of the tRNA-Dependent Amidotransferases, GatCAB and GatDE. J. Mol. Biol. 2008;377:831–844. doi: 10.1016/j.jmb.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geck MK, Kirsch JF. A Novel, Definitive Test for Substrate Channeling Illustrated with the Aspartate aminotransferase/malate Dehydrogenase System. Biochemistry. 1999;38:8032–8037. doi: 10.1021/bi983029c. [DOI] [PubMed] [Google Scholar]
- 47.James CL, Viola RE. Production and Characterization of Bifunctional Enzymes. Substrate Channeling in the Aspartate Pathway. Biochemistry. 2002;41:3726–3731. doi: 10.1021/bi0159074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.