

Abstract
Gastrointestinal complications are common in patients with Ehlers-Danlos Syndrome, affecting up to 50% of individuals depending on the subtype. The spectrum of gastrointestinal manifestations is broad and ranges from life threatening spontaneous perforation of the visceral organs to a more benign functional symptoms. Here we describe the clinical and radiographic manifestations of visceroptosis of the bowel, a rare complication of Ehlers-Danlos Syndrome that is characterized by prolapse of abdominal organs below their natural position. We further review the literature on gastrointestinal complications in the different forms of Ehlers-Danlos Syndrome.
Keywords: Ehlers-Danlos syndrome, Visceroptosis of the bowel, Gastrointestinal complications, Hypermobility
Introduction
The Ehlers-Danlos syndrome (EDS) is a heterogeneous group of heritable disorders characterized by generalized fragility of connective tissues. Although some of the joint and skin manifestations are similar among different forms of EDS, it is important to correctly diagnose the specific subtype since the natural history, therapy, and outcome differ. The latest classification, published in 1998, recognizes six EDS subtypes, based on clinical characteristics, mode of inheritance, and biochemical and molecular findings. For each subtype, major and minor clinical diagnostic criteria were defined [1]. An additional group of miscellaneous forms has been proposed; this includes the periodontitis [2], progeroid [3], and other unspecified forms. The most common subtype is the hypermobility type (type III) which comprises ~90% of all diagnosed EDS patients. In five EDS subtypes, single gene defects in collagen proteins or collagen modifying enzymes have been identified. Examples are mutations in PLOD1, the gene encoding the collagen modifying enzyme lysyl hydroxylase in the kyphoscoliotic type of EDS [type VI], mutations in the type III collagen gene (COL3A1) in vascular EDS (type IV), mutations in the genes for type V collagen (COL5A1 & COL5A2)in classic EDS (types I–II), and mutations in the ADAMTS2 gene in the dermatosparaxis type of EDS (type VIIC), illustrating the molecular heterogeneity in the EDS [4]. This classification is now showing its age as several additional forms have been recently clinically and molecularly delineated. These include the dermatan 4-O-sulfotransferase 1 deficient EDS (D4ST1-deficient EDS); [5], and the spondylocheiro dysplastic EDS (caused by mutation in SLC39A13 gene); [6]. Yet, for a substantial proportion of EDS patients the molecular basis remains unknown. The prevalence of EDS has been estimated at 1: 5000, however, it may be higher as it is likely that some individuals with milder clinical manifestations do not seek to medical attention and thus go undiagnosed.
Descriptions of cohorts of patients with EDS report an association with various gastrointestinal manifestations [7,8]. However, the precise incidence of gastrointestinal involvement is unknown, as abdominal symptoms are common and not all affected individuals have undergone the appropriate examination and imaging studies. Here we present a patient that came to medical attention because of a rare and severe gastrointestinal manifestation – visceroptosis of the bowel - that ultimately led to a diagnosis of hypermobile EDS.
Clinical report
The patient is a 28-year-old woman who presented with complaints of bloating and abdominal distention for 4 years that are debilitating and are not allowing her to carry out her normal daily activities of living. She notes that she has been able to deal with her symptoms for 3 years, continued going to school and working, and was able to alleviate her symptoms by fasting occasionally or taking pain medications as needed. Over the past year there was dramatic increase in the amount of distention and the frequency of symptoms causing her to take a medical leave of absence from work. She has been at home for the last 7 months, bedridden due to pain. Her abdomen become so distended at times that people believe she is pregnant, and she started to shop in the maternity section despite being 48 kg (106 pounds) at 167cm (5 feet 6 inches; BMI 17.1). She had a celiac workup and an endoscopy when her symptoms begun, which were normal and biopsies showed minimal nonspecific chronic inflammation and negative Helicobacter pylori.
Imaging studies
Upper gastrointestinal series with small-bowel follow-through, demonstrated that the small bowel loops were tightly bunched together deep in the bony pelvis and colonic abnormality was suggested because solid stool was in the right colon (Fig. 1A). Therefore, a lower gastrointestinal series was completed and showed ptosis of the transverse colon into the pelvis creating an angulation at the splenic junction preventing he stool from clearing the right colon (Fig. 1B). The rectum, sigmoid and descending colons were normal. The patient was subsequently referred for surgical assessment and laparoscopic exploration. A medical genetics consult was requested for evaluation of a possible underlying heritable connective tissue disorder prior to the pending surgery.
Figure 1. Imaging studies of the gastrointestinal tract.

A - Upper gastrointestinal series in upright standing position one hour after administration of oral contrast material. Note that the majority of contrast agent is within small bowel loops located deep in the pelvis, predominantly in the ileum (asterisk). Thus, despite upright position and contrast filling, the radiographic aspect suggests an abnormal downward displacement of small bowel into the pelvis. Notably, there is a large amount of stool in the right colon (arrow) causing a lack of contrast filling of the large bowel.
B - Barium enema in supine patient position after evacuation. Note an abnormal downward displacement and elongated appearance of the transverse colon into the pelvis even in supine position (arrows). The positioning of the ascending, descending and sigmoid colon appears to be normal. Due to evacuation the large bowel demonstrates variable distension and contrast pattern.
Medical Genetics evaluation
The patient reported a history of joint hypermobility and kyphoscoliosis diagnosed at high school. She has been able to sublux her hips as long as she can remember and she recently spontaneously dislocated her left shoulder twice while lying in bed. She also reported a recent thumb dislocation following minor trauma. She had a normal motor development and she hit all milestones. She has high myopia; a recent echocardiogram demonstrated normal aortic root with no valvular disease. The patient is an only child; she reports that her parents do not have any medical problems although an expert has not examined them. A Rheumatology evaluation was normal. On physical examination she has a normal but very thin body habitus, arachnodactyly and small joint laxity [Fig. 2A–B]. She can pop her femur out of the hip sockets bilaterally. Her skin is soft, bruises easily but not significantly hyperextensible, and striae were noted on the thighs and buttocks. Scars were normal. She had loss of subcutaneous fat and muscle mass and a mild scoliosis was noted [Fig. 2C–D]. Based on the joint and skin findings, the patient was given a diagnosis of EDS hypermobility type. Histo-pathological examination of skin revealed minimal alterations in collagen fibers and unremarkable elastin fibers. Subsequently, the patient underwent laparoscopic subtotal colectomy with transverse colon to distal sigmoid colon anastomosis. During surgery, all bowel loops noted to be freely mobile and could be manipulated out of the pelvis without adhesive tethering and seemed to have normal peristalsis, hence, supporting the radiographic findings of bowel ptosis. Postoperatively, the patient had multiple bowel movements and most of her symptoms resolved prior to discharge. Follow up examination one-year post operation revealed well-healed scars (with one scar that is slightly hypertrophic), yet, her bloating symptoms recur and she has difficulty eating. She has lost some weight and is 44.5kg (98 pounds). Her treatment regimen includes total parenteral nutrition with supplemental nutrition shakes. She was refereed for imaging and additional investigative studies that are pending.
Figure 2. Physical findings in a patient with Ehlers-Danlos syndrome and visceroptosis of the bowel.
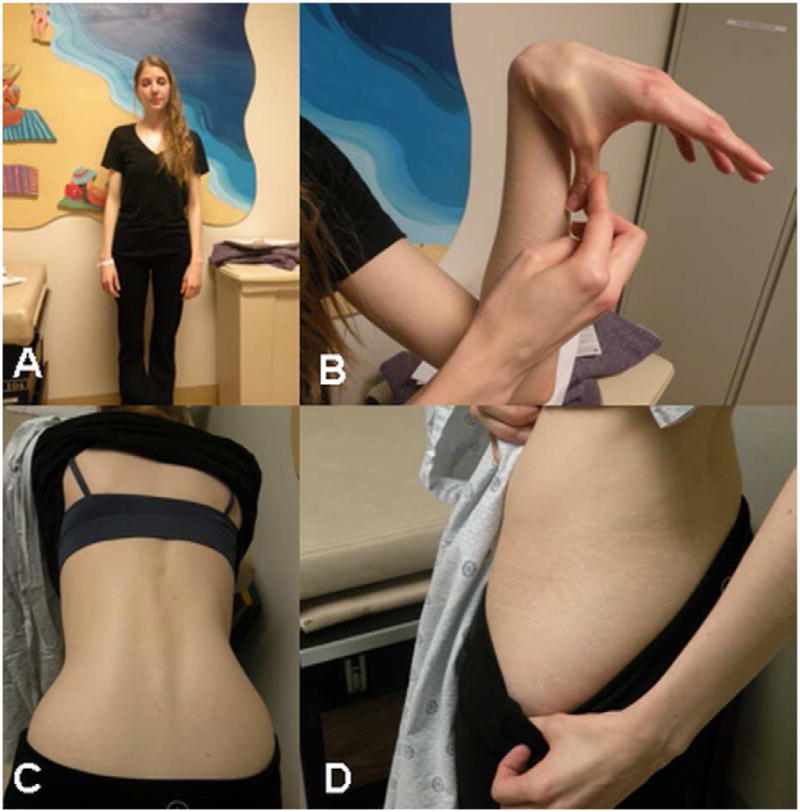
A - Slim body habitus
B - Small joint hypermobility
C - Mild scoliosis
D - Skin striae
Discussion
Visceroptosis is a prolapse or sinking of abdominal organs below their natural position. It can be associated with various gastrointestinal symptoms depending on the severity of the condition and secondary to compression of other internal organs. Visceroptosis can cause kinking of thin-walled abdominal structures such as blood vessels, nerves and ducts. The etiology of visceroptosis is largely unknown although several causes have been suggested, including relaxation of the ligaments that hold the abdominal viscera in place. To the best of our knowledge, there is only one reported case of visceroptosis of the bowel in a patient with EDS from 1941, cited in Beighton et al [8,9]; however clinical information on this patient is not available.
Early reports on cohorts of patients with EDS illustrate a strong association with gastrointestinal complications, suggesting they are common manifestations of this group of disorders. In a large heterogeneous cohort of 125 patients with EDS, Beighton et al. described a wide spectrum of gastrointestinal manifestations ranging from life threatening spontaneous perforation of the intestine and massive gastrointestinal bleeding to a more benign involvement such as hernias, intestinal diverticula, and rectal prolapse during childhood [8]. In recent years, multiple other reports confirmed and extended these earlier observations by demonstrating diverse gastrointestinal manifestations, depending on the subtype of EDS. The most significant complication has been associated with vascular EDS since type III collagen (encoded by COL3A1, the mutated gene in this form) is a major component of the intestinal wall. Rupture of the gastrointestinal tract, most commonly at the level of the sigmoid colon, occurs in up to 25% of patients with vascular EDS harboring missense mutations. Bowel rupture is lethal in only 3% of cases [10], and recurrences proximal to the first sigmoid tear are common. Spontaneous ruptures of the spleen and the liver have also been described in vascular EDS [11–13].
Gastrointestinal involvement in hypermobility EDS is very common, affecting ~ 50% of individuals with this form [14]. However, the manifestations are less severe and most commonly involve functional bowel disorders including gastroesophageal reflux disease, gastritis, and irritable bowel syndrome. A recent study of 31 patients with hypermobility EDS, suggested that in addition to the prevalent functional bowel disorders, celiac disease is also more common in those patients as compared with the general population [15]. No pathogenic single gene mutation can be found in the most common hypermobility type of EDS, with the exception of very rare TNXB mutations in a small subset of patients.
Rectal prolapse has been reported in young patients with classic EDS [8,16,17], as well as megacolon and megacystis [18]. Spontaneous diaphragmatic rupture has been described in a few patients with EDS although the specific subtypes are not clear from these reports [19–21].
Multi-system connective tissue fragility and craniofacial features characterize d4ST1-deficient EDS, a recently delineated form. Several gastrointestinal manifestations have been reported in this form including constipation, abdominal pain, absent gastrocolic omentum and spontaneous volvulus of small intestine, gastric ulcer, and duodenum obstruction due to malrotation [5]. It should be noted however that most of these complication have been observed in single cases, thus it is not clear what their exact prevalence is.
Although characterized by significant connective tissue fragility, significant gastrointestinal complications have not been reported in association with the arthrochalasia type of EDS [22]. This may be related in part to the rarity of the condition and the limited number of reported patients.
In the absence of a known molecular basis, the diagnosis of EDS type III is based entirely on clinical criteria that include musculoskeletal [joint hypermobility and dislocations] and skin findings, but the absence of significant fragility of other connective tissues. Family history appears to be negative in the majority of patients with EDS hypermobility type as in the case presented here. A diagnostic consideration was also given to the previously recognized EDS type II, as there is some phenotypic overlap between the two forms. In conclusion, although gastrointestinal manifestations are common in EDS, their precise incidence is unknown, as abdominal complaints are common and only a minority of affected individuals has undergone appropriate examination. Although the patient presented here reported joint manifestations dating back to her childhood, it is the severe gastrointestinal symptoms that brought her to medical attention and to diagnosis of EDS. The underlying connective tissue disorder can explain, at least in part, the ligamentous laxity of her bowel, although ptosis of large and small bowel has not been characterized in the literature in association with EDS.
Table 1.
Gastrointestinal manifestations in patients with Ehlers-Danlos syndrome
| EDS subtype (historic nomenclature) | Genetic defect | Gastrointestinal manifestations |
|---|---|---|
| Classic (types I+II) | COL5A1, COL5A2 | Rectal prolapse, megacolon, megacystis |
| Hypermobility (type III)* | Functional bowel disorders, gastro-esophageal reflux disease, gastritis, irritable bowel syndrome, celiac disease | |
| Hypermobility, TNXB-related | TNXB | Gastrointestinal bleeding, Rectal prolapse, diverticulitis, bowel perforation |
| Vascular (type IV) | COL3A1 | Bowel, spleen and liver rupture |
| Kyphoscoliotic (type VIA)** | PLOD1 | Gastro-esophageal reflux |
| Arthrochalasia (type VIIB)*** | COL1A1, COL1A2 | |
| Dermatosparaxis (type VIIC)*** | ADAMTS2 | |
| Spondylocheiro dysplastic*** | SLC39A13 | |
| D4ST1-deficient EDS** | CHST14 | constipation, abdominal pain, absent gastrocolic omentum, spontaneous volvulus of small intestine, gastric ulcer, duodenal malrotation |
In most patients with the hypermobility type pathogenic mutations have not been identified
Described in single patients
Significant gastrointestinal complications have not been reported
Acknowledgments
Dr. Rimoin appreciates support from the Steven Spielberg Pediatric Research Center, the NIH/NICHD Program Project Grant (HD36657), the Medical Genetics NIH/NIGMS Training Program Grant (5-T32-GM08243), and the Cedars-Sinai General Clinical Research Center Grant (M01-RR00425).
Footnotes
None of the authors of the manuscript has declared any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers–Danlos Syndromes: Revised Nosology, Villefranche, 1997. Ehlers–Danlos National Foundation [USA] and Ehlers–Danlos SupportGroup [UK] Am J Med Genet. 1998;77:31–7. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Reinstein E, Wang RY, Zhan L, Rimoin DL, Wilcox WR. Ehlers-Danlos type VIII, periodontitis-type: further delineation of the syndrome in a four-generation pedigree. Am J Med Genet A. 2011;155:742–7. doi: 10.1002/ajmg.a.33914. [DOI] [PubMed] [Google Scholar]
- 3.Hernández A, Aguirre-Negrete MG, González-Flores S, Reynoso-Luna MC, Fragoso R, Nazará Z, Tapia-Arizmendi G, Cantú JM. Ehlers-Danlos features with progeroid facies and mild mental retardation. Further delineation of the syndrome. Clin Genet. 1986;30:456–61. doi: 10.1111/j.1399-0004.1986.tb01910.x. [DOI] [PubMed] [Google Scholar]
- 4.Byers PH, Schwarze U. Ehlers–Danlos syndrome. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin’s principles and practice of medical genetics. 5. New York: Elsevier, Churchill-Livingstone; 2007. pp. 3625–3646. [Google Scholar]
- 5.Shimizu K, Okamoto N, Miyake N, Taira K, Sato Y, Matsuda K, Akimaru N, Ohashi H, Wakui K, Fukushima Y, Matsumoto N, Kosho T. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am J Med Genet A. 2011;155A:1949–58. doi: 10.1002/ajmg.a.34115. [DOI] [PubMed] [Google Scholar]
- 6.Giunta C, Elçioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, Steinmann B. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome-an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet. 2008;82:1290–305. doi: 10.1016/j.ajhg.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solomon JA, Abrams L, Lichtenstein GR. GI manifestations of Ehlers-Danlos syndrome. Am J Gastroenterol. 1996;91:2282–8. [PubMed] [Google Scholar]
- 8.Beighton PH, Murdoch JL, Votteler T. Gastrointestinal complications of the Ehlers-Danlos syndrome. Gut. 1969;10:1004–8. doi: 10.1136/gut.10.12.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ota M, Yasuda T. Erster Fall von “Syndrome d’Ehlers-Danlos” in Japan. Zbl Haut Geschl –kr. 1941;60:120. [Google Scholar]
- 10.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–80. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 11.Harris SC, Slater DN, Austin CA. Fatal splenic rupture in Ehlers-Danlos syndrome. Postgrad Med J. 1985;61:259–60. doi: 10.1136/pgmj.61.713.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng SC, Muiesan P. Spontaneous liver rupture in Ehlers-Danlos syndrome type IV. J R Soc Med. 2005;98:320–322. doi: 10.1258/jrsm.98.7.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gelbmann CM, Köllinger M, Gmeinwieser J, Leser HG, Holstege A, Schölmerich J. Spontaneous rupture of liver in a patient with Ehlers Danlos disease type IV. Dig Dis Sci. 1997;42:1724–1730. doi: 10.1023/a:1018869617076. [DOI] [PubMed] [Google Scholar]
- 14.Levy HP. Ehlers-Danlos Syndrome, Hypermobility type. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle [WA]: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1279/ [Google Scholar]
- 15.Danese C, Castori M, Celletti C, Amato S, Lo Russo C, Grammatico P, Camerota F. Screening for celiac disease in the joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. Am J Med Genet A. 2011;155A:2314–2316. doi: 10.1002/ajmg.a.34134. [DOI] [PubMed] [Google Scholar]
- 16.Chen CW, Jao SW. Images in clinical medicine. Ehlers-Danlos syndrome. N Engl J Med. 2007;357:e12. doi: 10.1056/NEJMicm066971. [DOI] [PubMed] [Google Scholar]
- 17.Douglas BS, Douglas HM. Rectal prolapse in the Ehlers-Danlos syndrome. Aust Paediatr J. 1973;9:109–110. doi: 10.1111/j.1440-1754.1973.tb01862.x. [DOI] [PubMed] [Google Scholar]
- 18.Sato T, Ito H, Miyazaki S, Komine S, Hayashida Y. Megacystis and megacolon in an infant with Ehlers-Danlos syndrome. Acta Paediatr Jpn. 1993;35:358–360. doi: 10.1111/j.1442-200x.1993.tb03071.x. [DOI] [PubMed] [Google Scholar]
- 19.Iglesias JL, Renard T. Diaphragmatic hernia in an 8-year old with Ehlers-Danlos syndrome. Pediatr Surg Int. 1998;13:553–555. doi: 10.1007/s003830050401. [DOI] [PubMed] [Google Scholar]
- 20.Ratani RS, Yang DC, Kalani J, Winer-Muram HT, Okadigwe C, Siddalingappa M, Steiner RM. An intrathoracic wandering spleen in a patient with Ehlers-Danlos syndrome and diaphragmatic hernia. Clin Nucl Med. 2000;25:738–739. doi: 10.1097/00003072-200009000-00025. [DOI] [PubMed] [Google Scholar]
- 21.Levine M, Adler J. Acute diaphragmatic rupture in a patient with Ehlers Danlos syndrome. J Emerg Med. 2011;41:366–368. doi: 10.1016/j.jemermed.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Klaassens M, Reinstein E, Hilhorst-Hofstee Y, Schrander J, Malfait F, Staal H, Ten Have L, Blaauw J, Roggeveen H, Krakow D, De Paepe A, van Steensel M, et al. Ehlers-Danlos arthrochalasia type [VIIA-B] - expanding the phenotype: from prenatal life through adulthood. Clin Genet. 2011 doi: 10.1111/j.1399–0004.2011.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]