

Abstract
Recent advances in far-field microscopy have demonstrated that fluorescence imaging is possible at resolutions well below the long-standing diffraction limit. By exploiting photophysical properties of fluorescent probe molecules, this new class of methods yields a resolving power that is fundamentally diffraction unlimited. Although these methods are becoming more widely used in biological imaging, they must be complemented by suitable data analysis approaches if their potential is to be fully realized. Here we review the basic principles of diffraction-unlimited microscopy and how these principles influence the selection of available algorithms for data analysis. Furthermore, we provide an overview of existing analysis strategies and discuss their application.
Keywords: diffraction, STED microscopy, fluorescence photoactivation localization microscopy, super-resolution, image processing
1. INTRODUCTION
To forward our understanding of cell architecture and function, we need imaging techniques that can observe processes inside living specimens at the molecular level, in two or three dimensions, using highly specific markers. Fluorescence microscopy, one of the most dominant imaging techniques, provides all these criteria except that its resolution is fundamentally limited by diffraction to >200 nm. Recent years have seen the invention of methods that overcome this constraint and provide diffraction-unlimited resolution (1). In this review, we focus on two of the most prominent methods of optical nanoscopy: stimulated emission depletion (STED) microscopy, and single-molecule localization and reconstruction microscopy, or for short, localization microscopy (LM). The application of these methods in biological imaging is becoming increasingly popular, and their principles have been extended to multicolor, three-dimensional (3D), and live-cell imaging (2). However, the true merit of these techniques must be judged by the effectiveness with which new biological insights can be obtained. In this respect, there is a need for analysis tools that extract the desired information and fully exploit the novel kinds of data provided by these new microscopy techniques; an example of these data includes large sets of positions of individual probe molecules. Fortunately, the field has recently seen promising developments in this area. Here, we briefly review the principles behind diffraction-unlimited microscopy and highlight the analysis methods successfully applied to date.
2. PRINCIPLES OF DIFFRACTION-UNLIMITED MICROSCOPY
2.1. The Point-Spread Function and Conventional Resolution
Diffraction prevents focusing a beam of light into a spot substantially smaller than half a wavelength in size. Similarly, diffraction causes the image of a point-like emitter, such as a single fluorescent molecule, to appear as an extended intensity distribution in the image plane. An image of an extended object taken with a fluorescence microscope is equivalent to the superposition of images of point sources composing the object. The image of a point source, the point-spread function (PSF), therefore describes the imaging properties of these linear systems: To form the observed image of an object, every point of the object is blurred to have a shape given by the PSF. The size of the PSF thus describes the resolution of an imaging system. This resolution is commonly defined by the full width at half maximum (FWHM) of the PSF, which is approximated by:
| (1) |
| (2) |
where Δr and Δz are the PSF FWHMs in the lateral and axial directions, respectively; λ is the wavelength of the light; n is the refractive index of the medium; and NA is the numerical aperture of the lens (3). For λ = 500 nm and a high-NA objective lens, Equation 1 reveals that diffraction limits resolution to Δr ≈ 200 nm. The axial value is typically two to three times larger than the lateral value. Such limitations on resolution hinder the study of biological processes that occur on molecular length scales. Fortunately, STED microscopy and LM allow imaging below the resolution limits given by Equations 1 and 2.
2.2. Concept of Stimulated Emission Depletion Microscopy
The principle of STED microscopy (4) relies on the targeted switching of fluorescent molecules at the periphery of the excitation focus. Fluorophores excited in the focus of a laser-scanning microscope can be driven back to the ground state through stimulated emission by an additional photon, typically red-shifted from the excitation light. These photons are provided by a second laser termed the STED beam or depletion beam. The result of this optical transition, which competes against the spontaneous emission of fluorescence, is the quenching of fluorescence in regions where the intensity of the STED beam is sufficiently high. In STED microscopy, the focus of this beam is typically shaped to feature a central intensity zero surrounded by a ring of high intensity and is aligned to the center of the excitation focus. When the intensity of the STED beam saturates the stimulated emission process, an effective PSF is created in which fluorescence is confined to the immediate vicinity of the intensity zero with a size that is not limited by diffraction (Figure 1). Using depletion intensities ≥100 MW cm−2, resolution on the order of a few tens of nanometers can typically be reached (5).
Figure 1.

Concepts of diffraction-unlimited microscopy. (a) Concept of localization microscopy (LM). Sparse subsets of single molecules are identified in a raw frame (red boxes). Each molecule is fitted (cyan curve) to determine its position (x0) with a precision that is inversely proportional to the square root of the number of detected photons (N), as indicated by the green curve. An LM image is generated from the coordinates of molecules localized from many frames. (b) Concept of stimulated emission depletion (STED) microscopy. The conventional excitation focus of a laser-scanning microscope (cyan) is coaligned to the donut-shaped depletion focus, which features an intensity zero at its center (red ). Saturating the depletion efficiency quenches fluorescence emission except at the center of the depletion focus ( gray curve). An effective STED point-spread function (PSF) ( green) is generated with a size that is inversely proportional to the square root of the ratio of maximum depletion intensity to characteristic saturation intensity (Imax/Isat).
The concept of targeted switching in the vicinity of an intensity zero has been generalized to include any reversible, saturable, optical fluorescence transitions (RESOLFT) (6). Although STED has been the most common implementation, using ground state depletion (7, 8) and reversibly switching proteins (9–11) have allowed imaging beyond the diffraction barrier using laser intensities lower than those required for STED.
The lateral FWHM of the effective PSF in STED microscopy (and RESOLFT microscopy in general) is well approximated by an extension of Equation 1:
| (3) |
where Imax is the peak intensity in the focus of the STED beam, and Isat is a characteristic saturation intensity of the fluorophore (12, 13). For arbitrarily large Imax/Isat values, the resulting resolution becomes arbitrarily small and is therefore diffraction-unlimited in principle.
2.3. Concept of Localization Microscopy
Whereas the resolution in conventional light microscopy is limited by diffraction, single-particle tracking has demonstrated that subdiffraction localization is possible (14, 15). Localization refers to the determination of the position of an object using its image. This achievement is based on the fact that the center of a measured (diffraction-limited) intensity distribution can be determined with a precision much better than the width of the distribution. In a background-free scenario with a Gaussian-shaped PSF with standard deviation σPSF, if effects from the finite pixel size within the imaging system are neglected, the localization precision σloc for a given number of detected photons N is
| (4) |
and can therefore be far smaller than the PSF width (16, 17). Particle-tracking microscopy allows one to trace the trajectories of single particles (including single fluorophores), with nanometer precision (18). However, single-particle tracking does not allow the image generation of complex structures with diffraction-unlimited resolution. Generating such images could be improved through spectral separation (19, 20), step-wise photobleaching (21, 22), or blinking of quantum dots (23, 24), all of which have demonstrated localization of a few fluorophores per diffraction-limited area.
A breakthrough in subdiffraction imaging based on localization was achieved by combining localization algorithms with photoswitchable fluorescent probes (25–27). These methods, which collectively fall under the aforementioned LM (also known as FPALM, PALM, and STORM), image numerous sparse subsets of individual fluorescent molecules over time with a camera and then analyze those images to localize visible molecules at concentrations of up to hundreds or thousands of molecules per diffraction-limited area (Figure 1). A super-resolution image is created by rendering sets of molecular coordinates.
Although LM was originally demonstrated with fluorescent probes that can be switched from a dark state (or a state emitting fluorescence outside the detection band) to a bright state (25–27), the intermittency of individual conventional fluorescent molecules interconverting between dark and bright states can also be exploited to obtain super-resolution images (28–32). Probes that become fluorescent upon binding to a target can be utilized in the same manner (33). The key requirements are that only a few molecules are visible at any time and that their images are separable from one another to permit their localization with high spatial precision.
2.4. Conceptual Similarities and Differences Between Stimulated Emission Depletion and Localization Microscopy
At first sight, STED microscopy and LM techniques may seem to have few points in common: STED microscopy is a laser-scanning microscopy technique with two laser beams being focused into the same spot, one of which is ring shaped. In contrast, LM is usually implemented in a widefield geometry in which a large field of view is homogeneously illuminated and repeatedly imaged.
Both approaches, however, utilize the photophysical or chemical properties of fluorescent probes to overcome the diffraction limit. In both cases, switching molecules between detectable and nondetectable states generates the resolution improvement (5). In STED microscopy, the ring-shaped STED beam forces excited molecules back to their ground state through stimulated emission in a switching process targeted at the outer areas of the excitation focus. LM relies on stochastic activation (switching on) of individual probe molecules (triggered either by light or spontaneously) and photobleaching (or deactivation) to control the visible probe density. Both methods exploit the response of the probe to one or more of the illumination wavelengths: STED microscopy by saturation of stimulated emission, and LM by the temporally discontinuous (step-wise) response of individual fluorescent molecules.
Because the detection process in any far-field microscope is fundamentally diffraction limited, fluorescence signals detected simultaneously from neighboring subdiffraction areas overlap and are indistinguishable. Both STED microscopy and LM solve this problem by sequentially imaging these areas. In STED microscopy, the combined excitation and depletion laser beams defining a subdiffraction observation volume are scanned systematically across the sample. Whereas high depletion rates (>109 s−1) in STED microscopy allow rapid scanning, LM typically utilizes photoswitching rates of ~10−1 to 10−4 s−1 per molecule; a standard scan configuration is therefore inefficient compared with parallel recording over the whole (illuminated) field of view. Sequential imaging of sparse, stochastic subsets of emitters assures that molecules closer than the diffraction limit will be imaged at different times and can therefore be distinguished.
The biggest difference between LM and STED microscopy, in the context of this review, is data processing (Figure 2). Resolution improvement in STED microscopy is based on a purely physical effect that reduces the effective volume from which fluorescence is emitted. The diffraction-unlimited image is produced directly by sequentially plotting the recorded signal according to the scan pattern, in a process analogous to confocal microscopy. LM, however, requires that imaged molecules from many camera frames be identified and localized. Images are then generated from the list of determined molecule positions. For STED microscopy, data processing is an option, but for LM, it is mandatory (Figure 2). However, this additional analysis in LM yields useful information not obtained by other methods such as STED and confocal microscopy: LM directly measures the positions, numbers, and intensities of individual probe molecules and can measure the polarization anisotropy (related to the molecular orientation) (34) and emission wavelength properties of each molecule (35, 36). This kind of single-molecule information can be used to identify population heterogeneities inaccessible to ensemble measurements or simply to separate multiple emitters with highly overlapping emission spectra (35, 36).
Figure 2.

Data processing in conventional and diffraction-unlimited microscopy. Abbreviation: STED, stimulated emission depletion.
2.5. Three-Dimensional Imaging
One of the main advantages of (far-field) fluorescence microscopy, especially confocal microscopy, is its capability of recording 3D data sets. As is the case for resolution in the lateral plane, the axial resolution, or depth resolution, is also limited by diffraction (Equation 2). Consequently, improving the axial resolution beyond the diffraction limit is as important as improving the lateral resolution, and 3D extensions of STED microscopy and LM have been developed to achieve this purpose. Both have been combined with total internal reflection excitation (25, 27, 37) to limit imaging to within ~100 nm of the coverslip. Phase patterns for STED microscopy have been developed to quench fluorophores not only laterally but also axially (38). Numerous 3D LM techniques have been developed on the basis of multiplane detection (39), astigmatic PSFs (40), other PSFs that unambiguously change their shapes with depth (41), and detection of the fluorescence emission from two different angles using parallax-like effects (42, 43). In all cases, axial resolution scales with the axial extent of the diffraction-limited PSF. Naturally, the best axial resolutions have therefore been obtained by combining LM or STED microscopy with 4Pi microscopy (44–46), i.e., in geometries utilizing two opposing objective lenses that sandwich the sample (47, 48).
The extension from 2D to 3D creates fundamentally different situations in data processing: The effect of imaging a volume versus a plane has to be considered from the perspective of background levels and particle densities. 3D structures can be dramatically more complex than, for example, membrane-bound geometries, and 3D data sets are often dramatically larger than 2D ones. However, objects that were not decipherable in their 2D projections now become potentially understandable through the availability of the additional imaging dimension.
3. RESOLUTION
3.1. Spatial Resolution
The resolution that may be achieved in STED microscopy or LM is, in principle, unlimited as indicated by Equations 3 and 4. However, in practice, resolution is still limited by experimental factors such as the achievable beam quality and intensity or the signal-to-noise ratio (SNR). Moreover, for better than 20-nm resolution and fluorescent label sizes that can exceed 10 nm, the probe distribution can differ significantly from the targeted structures, potentially leading to misinterpretations. The localization accuracy of probe positions therefore needs to be considered in addition to localization precision.
Additionally, achievable or physiologically justifiable probe densities can correspond to nearest neighbor distances of several tens of nanometers and significantly limit interpretation of complex structures. Similarly, if the targeted protein population is sparsely and stochastically labeled and detected, the inherent randomness of labeling can obscure the underlying structure further and can lead to faulty interpretation of the images obtained. The impact of this effect depends on the complexity of the structure and the frequency of occurrence of the features of interest. To decide whether or not, for example, two thin, approximately parallel microtubules touch is relatively independent of the fact that their staining might appear discontinuous along the fibers. Determining, in contrast, how much a fibrous network is fractured depends strongly on this detail. Similarly, identifying the complex (3D) folding of an organelle membrane requires not only sufficient optical resolution but also an imaged probe distribution dense enough to represent every fold or curvature. This required probe density can be seen in analogy to the Nyquist criterion, which demands a minimum sampling frequency to resolve a structure and the SNR necessary to observe a feature of interest (49). Indeed, in practice, the molecular density is often the limiting factor in obtainable resolution.
3.2. Temporal Resolution
Fluorescence image acquisition time limits the rates of dynamic processes that can be visualized. When widefield illumination and detection are used in imaging, imaging speed is often determined by the camera frame rate. In a laser-scanning system, frame rates are determined by the total number of pixels in the image and the scan rate. Therefore, in principle, faster cameras and scanning systems lead to faster acquisition times. However, in practice, the maximum frame rate in either case is ultimately limited by the SNR, which depends on photophysical probe properties such as molecular fluorescence emission rates. In diffraction-unlimited microscopy, other factors must also be considered.
As resolution increases in STED microscopy, Nyquist sampling of the specimen requires decreasing the pixel size by a corresponding factor. For a given scan speed and excitation intensity, smaller pixels result in a reduced pixel dwell time and a corresponding reduction in collected signal. Fast scanning therefore requires increased excitation intensities to maintain a given SNR. Eventually, saturation of the fluorophore emission will limit the detected signal as scan speeds increase. However, by imaging a small field of view with fast scanning, STED microscopy has been demonstrated at video rate on living neurons (50) and at up to 200 frames per second on colloidal crystals (51). Additionally, STED microscopy can be combined with fluorescence correlation spectroscopy to access dynamics on the microsecond scale in observation volumes smaller than the diffraction limit (52).
In LM, two aspects of temporal resolution must be considered. First, for reliable localization of molecules during analysis, individual frames must be recorded quickly enough so that single fluorophore images are not significantly distorted by directed motion or diffusion (53). When this criterion holds, molecular trajectories can also be assembled using coordinates of molecules imaged for several sequential frames, providing information about molecular dynamics on timescales of (typically) a few milliseconds (54). Second, the number of frames required to generate an image should be small enough that the structures of interest do not significantly change during the recording time (49).
Using simple localization routines requires that a maximum of one fluorophore be imaged per diffraction-limited area per frame. As a result, temporal resolution depends on the density required for a desired spatial resolution. Therefore, LM has an inherent trade-off between spatial and temporal resolution (49). Despite this trade-off, live-cell imaging at rates as fast as 2 Hz (each image being assembled from 250 frames) has been reported using LM with spatial resolution on the order of a few tens of nanometers (55).
3.3. Spectral Resolution
Observing protein interactions demands the ability to image multiple species. In conventional fluorescence microscopy, multiple targets are usually distinguished through the separation of their distinct emission spectra with appropriate filters. In cases of strong crosstalk between channels, computational linear unmixing or blind source separation techniques (56) can be implemented to separate the signals. Extending subdiffraction microscopy to multicolor imaging has, however, presented challenges owing to the requirement of usually more than one illumination wavelength per fluorophore and the limited availability of suitable probes.
In LM methods, numerous approaches have succeeded in imaging multiple species with diffraction-unlimited resolution. To separate two photoswitchable fluorescent proteins with some spectral overlap, sequential imaging can be used (57). However, this approach requires long acquisition times not compatible with live-cell imaging. The use of photoactivatable organic dye pairs allows distinguishing between labels by their distinct activation spectra (58) and, combined with fusion protein labeling technologies (59), allows multicolor imaging of live cells (55).
Alternatively, the single-molecule nature of LM can be exploited to distinguish among multiple species simultaneously, despite significant spectral overlap. Identified single emitters can be assigned to a probe species through an analysis of their intensity ratios detected in two spectral bands (35), an approach not applicable in ensemble imaging methods. This approach has recently been demonstrated for four-color imaging in fixed cells using organic dyes (60) and three-color imaging in living cells using genetically encodable probes (36).
STED imaging of two spectrally well-separated fluorophores with four separate laser lines has also required sequential imaging of each species owing to the overlap between the blue-shifted STED beam and the excitation spectrum of the red-shifted fluorophore (61, 62). This scheme requires imaging the red-shifted channel before the blue-shifted one, and only a single acquisition of this channel is possible because the second STED beam usually bleaches the red-shifted probe. More recently, researchers have been able to avoid this problem by deriving excitation-STED beam pairs with both STED beams red-shifted with respect to both probe emission spectra (63). Furthermore, a single STED beam can be used to image multiple species when one of the fluorophores has a long Stokes shift, and fluorophores can be distinguished by their respective excitation laser wavelengths (45). This method has also been applied in the first demonstration of dual-color STED microscopy in living cells (64). In another approach, two fluorophores with similar absorption and emission spectra were distinguished by their respective fluorescence lifetimes for simultaneous dual-channel STED microscopy (65). In this report, Buckers et al. (65) imaged a third, spectrally distinct fluorophore quasi-simultaneously by interleaving the pulses of the respective excitation-STED beam pairs.
4. DATA ANALYSIS METHODS
4.1. Data Analysis in Stimulated Emission Depletion Microscopy
As described above, the organization of STED microscopy data is similar to that of the data obtained by other laser-scanning microscopes. 2D or 3D probe density maps are readily available as the output of the microscope. So far, STED microscopy analysis methods mostly resemble those already established in confocal microscopy.
Given the improved spatial resolution, careful spatial correlation analysis between different color channels plays a central role in the analysis of multicolor STED microscopy (65). To avoid artifacts, it is crucial that spatial shifts between the different channels are characterized with a precision smaller than the desired resolution of the instrument. STED microscopy is fortunately insensitive to artificial signal shifts owing to fluorophore dipole orientation (66).
When imaging well-separated subresolution-sized objects (such as small vesicles), STED microscopy data can be used to localize the centers of these objects with a precision much better than the STED resolution. Applying algorithms similar to those used in LM, researchers have achieved localization precisions in the angstrom range for bright objects such as diamond color centers (67). Additionally, STED microscopy, with its particle localization and tracking abilities and its improved resolution, allows tracking of particles distributed more densely than those that can be tracked by diffraction-limited imaging methods (Figure 3a).
Figure 3.
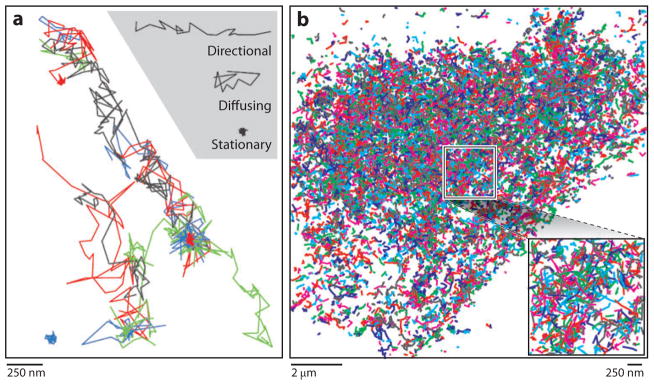
Trajectories in diffraction-unlimited microscopy. (a) Trajectories of synaptic vesicles imaged at video rate by stimulated emission depletion microscopy. Adapted from Reference 50; reprinted with permission from AAAS. (b) Trajectories of Dendra2-HA molecules expressed in a living fibroblast cell imaged at 37°C using localization microscopy. Inset shows zoom-in of boxed region.
Furthermore, owing to depletion efficiencies that do not fully reach 100% and through excitation by the STED beam, highly resolved structures can be surrounded by a usually low but often visible haze. Therefore, application of spatial filtering and deconvolution techniques is often employed to improve STED images for presentation. Often, STED data is linearly deconvolved (61, 68) or subject to maximum likelihood deconvolution (62).
4.2. Data Analysis in Localization Microscopy
Postacquisition data analysis is inherent to LM. Above all else, the images of single molecules must be analyzed to determine molecular positions before a super-resolution image can be generated. The following subsections review current algorithms for identifying and localizing molecules and the subsequent analysis that can be performed using the single-molecule information provided by LM.
4.2.1. Localization algorithms
As in the analysis routines that have been used for decades in single-particle tracking experiments, in LM techniques, raw images must be analyzed to identify single molecules and determine their positions. Typically, after background subtraction, intensity peaks above a threshold are identified in each frame and then least-squares fitted with a 2D Gaussian (25–27). Intensity peaks are determined to originate from single molecules on the basis of intensity, size, shape, and quality of fit. Although not always encoded in analysis routines directly, single molecules are also identified by the observation of single-step photobleaching.
Whereas a uniform background signal can be subtracted as a baseline from a raw image or included as an offset in the fitting function, cellular imaging typically results in autofluorescence, which generates spatially and temporally varying background. To this end, more sophisticated background subtraction methods have been demonstrated, including calculating differential images (25); using an intensity-weighted, summed widefield image (53); or using a rolling-ball algorithm (69).
Sampling structures on nanometer length scales demands localizing molecules at as high a spatial density as possible. Therefore, identifying single molecules reliably is perhaps the most critical aspect of LM data analysis, and a balance must be found between false-negative and false-positive identifications. Owing to the stochastic nature of photobleaching, the former occurs when the number of detected photons from a single molecule is less than a user-determined intensity threshold. Background noise and shot noise also complicate single-molecule identification and degrade resolution. However, recent studies have shown that a combination of denoising and deconvolution (70) or smoothing raw images with a Gaussian of standard deviation approximately twice that of the single-molecule PSF (71) (for the purposes of identification only) can minimize identification errors.
After identification, molecules are localized, often with subpixel precision. Because centroid localizations in the presence of background or other sources of noise have poor localization accuracy compared with Gaussian fits (72), least-squares Gaussian fits have been most commonly used when analyzing localization-based imaging data. However, the true image of a dipole emitter with fixed orientation or restricted rotation is significantly non-Gaussian. Approximation of the single-molecule PSF by a Gaussian can result in significant position inaccuracies, which increase with the NA of the objective lens (<2.5 nm for an NA of 1.2; <10 nm for an NA of 1.4) (73). More important, when coupled with defocus, fixed molecular orientation can introduce position inaccuracies as large as 125 nm (66). Furthermore, a maximum likelihood estimator is more precise than a nonlinear least-squares algorithm (74), and optimal localization can be achieved only when using maximum likelihood estimation with the proper PSF (75). In any case, owing to their robustness (76) and easy implementation, Gaussian approximations to the PSF remain widely used for 2D localization. For 3D localization, more complex theoretical model functions (77), optionally including experimentally derived parameters (40, 78), or experimentally measured PSFs (39, 79) have been used.
Localization values are usually corrected for sample drift over the course of measurement through the implementation of drift-correction algorithms. Next to active drift compensation, either fiduciary markers in the field of view are monitored and their positions subtracted from the probe localizations (27, 80), or, for static structures, subpopulations of probe molecules recorded in different time windows are correlated with one another (40, 81).
Because the density of molecules and not the localization precision is typically the limiting factor in LM resolution, localizing large numbers of molecules is crucial to resolution improvement in LM. On the data processing side, accepting molecules with fewer detected photons results in poorer localization precision but allows higher density of localized molecules, both of which affect the localization-based resolution as explained above. Another problem can occur when localization routines fail to distinguish between images of single and multiple fluorophores that reside within a diffraction-limited area. If this situation is to be avoided, the density of visible molecules per frame generally has to be <~1 μm−2. Recent developments in multiple-fluorophore-fitting algorithms allow imaging densities of up to 10 μm−2 (82, 83) to be safely used while still achieving theoretical limits in localization precision (83). A similar approach has shown that Bayesian information criteria can be used for reliable multiple-fluorophore fitting of images with low SNRs (84). Because the recording time is roughly inversely proportional to the maximum molecular density per frame, such improvements in high-density fitting allow faster live-cell (time-lapse) imaging.
Initially, post data acquisition analysis times for LM exceeded acquisition times by orders of magnitude. This limitation has now largely been overcome for 2D LM and certain other situations. For example, there have been several reports of real-time image processing for LM data either using simplified localization algorithms (85, 86) or implementing graphics processing units (87, 88). Obviously, a fast analysis routine that does not compromise localization precision is desirable (88).
4.2.2. Trajectories
In addition to structure, analysis of measured molecular coordinates as a function of time can reveal dynamic details of biological systems. For example, if the same molecule is imaged and localized for several consecutive frames, a single-molecule trajectory can be constructed (54). Acquisition of multiple trajectories (Figure 3b) can be used to generate a super-resolution map of molecular mobility, determine molecular diffusion properties, and correlate motions of multiple species. Using typical acquisition frame rates, molecules can be observed on millisecond timescales for up to tens or hundreds of frames. For such analysis to be performed, three key criteria must be considered:
The limited photon budget of a single molecule creates a trade-off for trajectory imaging. Molecules may be imaged for larger numbers of frames, but at the price of fewer photons per frame and thus greater uncertainty in their positions and intensities within each frame.
Imaging must be performed at low enough density to avoid confusion of one molecule with another.
If a molecule moves too much within a single frame, its image accumulated over the recording time may become so distorted or blurred that it cannot be localized properly, resulting in localizations that do not represent the potentially complex trajectory of the particle (89).
Thus, to avoid significant additional localization errors for diffusing molecules, the characteristic area covered per molecule per frame should follow
| (5) |
where r0 is the radius of the PSF, D is the diffusion coefficient, and τ is the time per frame. For molecules moving actively or flowing with a velocity υ, the criteria becomes
| (6) |
Given that these criteria have been met, high-density maps of molecular trajectories can be constructed (54). Such maps can reveal dynamic details and multiple molecular populations that are often inaccessible to ensemble measurements and can form a useful complement to the structural information provided by LM images. If the molecular density is sufficiently high, maps of molecular mobility can be calculated by binning data for mean-squared displacement for many molecules within a grid of pixels. Alternatively, quantification of single-molecule motions by particle image correlation spectroscopy (90) can be used to determine the diffusion properties of one or more molecular species without knowledge of the identities of molecules to generate individual trajectories. Thus, raw images can be acquired at much higher densities and can still allow for diffusion analysis.
4.2.3. Analysis of spatial correlations
Once image data have been obtained, quantification is crucial for interpretation of the results. Numerous statistical methods for measurement of spatial correlations exist. We focus here on several methods that can be used to measure clustering or coclustering.
Colocalization is a popular method for determining spatial correlations on short length scales. Using two distinct fluorescence signals (FA and FB) as a reporter of local concentration of two molecular species of interest (A and B), the two signals can be correlated using, for example, Pearson’s correlation coefficient (91):
| (7) |
where δFi = Fi − 〈Fi 〉 and 〈Fi〉 denotes the spatial average of Fi. Such analysis is typically carried out on each pixel within a region of interest of an image. For LM data, the number of molecules of each type can be binned in a grid of pixels, and those numbers of molecules per unit area can be used for FA and FB. The value of C ranges from +1 (correlated) to −1 (anticorrelated), with zero ± uncertainty indicating uncorrelated. All results should be interpreted on the length scale of the chosen pixel size or the resolution (whichever is larger). Alternatively, other metrics such as the overlap coefficient can be used to quantify colocalization (63). A significant correlation does not necessarily imply interaction among molecules; additional tests to confirm interactions should be performed if this kind of information is needed. Performing such a colocalization analysis does not require correction for nonuniformities in sample shape as long as the two channels are sampled with uniform probability. It does, however, require correction for species misidentification. For longer length scale correlations, image correlation spectroscopy (92) is a useful option.
Ripley’s K-test quantifies clustering or coclustering as a function of length scale. Starting from molecular coordinates, the K-test compares N(r), the observed number of molecules within radius r, with the number expected from a random distribution (93, 94):
| (8) |
where ρ is the average number of molecules per unit area, and r is the radius. Instead of using πρr2 as the denominator, πρ is used, so that when N(r) = πρr2 (as expected for a random distribution), the square root expression reduces to r, and L(r) − r = 0. Thus, the metric L(r) − r gives values >0 for clustered distributions, <0 for anticlustered distributions, and 0 ± uncertainty when the distribution is random. Determination of the uncertainty requires simulation of confidence intervals and correction for the shape of the analysis area. The two-color K-test also requires correction for species misidentification rates. Because the K-test measures the numbers of molecules within a radius r, its results report on any clustering within that chosen length scale, up to a length scale where the clusters themselves are randomly distributed.
The pair correlation measures the ratio of the number of molecules at a given distance r ± (Δr/2) from another molecule (on average) to the number expected from a random distribution:
| (9) |
Thus, g(r) > 1 for correlated distributions, g(r) < 1 for anticorrelated distributions, and g(r) = 1 ± uncertainty for random distributions. This analysis typically uses molecular coordinates but can, in principle, be used on images directly. Pair correlation analysis can be applied to test clustering of one or more species but does require corrections for edges of the analysis area and for species misidentification rates. The complexity of biological systems often precludes direct interpretation of the functional dependence of g(r) beyond the amplitude and characteristic decay length. Also, intermittency of probe molecules can cause significant artifacts in cluster analysis based on localization data (95, 96). Nonetheless, these parameters do give useful information about clustering in the system: The amplitude gives cluster density relative to a uniform distribution with the same average density, and the decay length is a measure of cluster size. Recent work characterizing biological membrane heterogeneity demonstrates the utility of such analysis methods (97).
The nearest neighbor distance distribution can be obtained from molecular coordinates and used as a means to test for clustering (98). For example, in a model in which higher average density of molecules results from adding to existing clusters, nearest neighbor distances decrease as a function of average density. In models in which additional molecules cause more clusters but cluster properties are unchanged, nearest neighbor distances remain invariant as a function of average density.
Poisson statistics can be used to test whether particles are distributed randomly. Coordinates of molecules are binned into pixels, and the histogram of the numbers of particles in each bin is fitted with a Poisson distribution, which should be successful (i.e., should have a χ2 value of ~ 1) if the molecules are randomly distributed. A failed fit suggests nonrandomness. The pixel size can then be varied to test for nonrandomness as a function of length scale (99). This type of procedure gives a simple yes/no test for clustering but does not give any additional information on the type of clustering.
Single linkage cluster analysis (100) defines clusters as containing all molecules with a maximum distance dmax to at least one other member of the cluster. Clusters are identified as sets of molecules (i.e., lists of coordinates) that can then be analyzed by any desired method (Figure 4). Typical properties include area, perimeter, density (particles per unit area), and shape (i.e., ratio of length to width), but many other options exist. This method has the advantage that one obtains a distribution of values, rather than a single averaged value, so multiple populations can be detected and distinguished.
Figure 4.
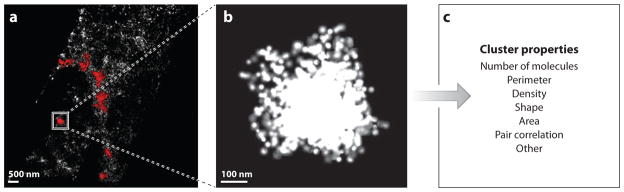
Single linkage cluster analysis (SLCA) of FPALM data set to identify and characterize membrane protein clusters. (a) FPALM image of Dendra2-tagged linker of activated T cells in HAb2 fixed fibroblast cell. Examples of clusters identified by SLCA are shown in red (maximum nearest neighbor distance 30 nm). (b) Magnified view of one such cluster contained in the box in panel a. (c) List of properties that can be analyzed once the molecules constituting each cluster have been identified.
4.3. Visualization and Rendering
The foremost objective of any imaging system is to create a meaningful visual representation of the distribution of labeled objects within the sample. In a STED microscope, image pixels are sequentially generated with intensity values proportional (or equal) to the number of photons detected from a predetermined position in the sample.
Because the data obtained in LM are composed of the coordinates of localized molecules, methods for visualizing this data are not as straightforward. In initial reports, images were generated as scatter plots or 2D Gaussian rendering of molecular positions or by binning the molecular positions into pixels (i.e., a 2D histogram) whose intensities correspond to the number of molecules localized in that region. However, a thorough investigation has shown that the multiresolution nature of localization-based images is not ideally suited for either of these rendering methods (101). Namely, scatter plots suffer from nonlinearity owing to saturation from symbol overlap, and Gaussian spots result in a loss in resolution by a factor of (101). Baddeley et al. (101) propose rendering methods based on adaptive bin size histograms and triangulation to generate images from single-molecule coordinates that better represent the resolution and noise of the data while reducing artifacts (Figure 5). Similarly, an adaptively jittered 2D histogram rendering method has been proposed; it has the advantages over the conventional 2D histogram that molecule positions are not forced onto a regular grid and that the resulting image reflects the localization precision (71).
Figure 5.

Visualization of data in diffraction-unlimited microscopy. (a) STED image of SNAP-25 on the plasma membrane of fixed neuronal cell. The area inside the blue circle has been linearly deconvolved. (b–g) A comparison of simulated LM data representing two parallel lines rendered using (b) a scatter plot of molecular coordinates, (c) Gaussian spots with standard deviation equal to the simulated localization precision (10 nm), (d ) a quad-tree-based adaptive histogram, (e) the Delaunay triangulation method, (f) randomly subsampled triangulation, and ( g) adaptively jittered and averaged triangulation. Line profiles were generated by summing data along the vertical axis. Dashed lines represent the theoretical distribution of positions. Abbreviations: LM, localization microscopy; SNAP-25, synaptosomal-associated protein 25; STED, stimulated emission depletion. Panel a from Reference 68; panels b–g from Reference 101.
5. EXAMPLES OF BIOLOGICAL APPLICATIONS
Applying LM and STED microscopy to biological questions and analyzing their data appropriately have yielded important results and demonstrate the importance of data processing to taking full advantage of these methods. The publications discussed below provide interesting examples but are not exclusive.
Willig et al. (102) examined the spatial distribution of the synaptic vesicle protein synaptotagmin I by STED microscopy. Immunostained protein distributions imaged at 45–66-nm resolution revealed clusters that were fitted by Lorentzian functions to determine their size distribution. The resulting histograms peaked between 70 and 85 nm with no significant differences among vesicle pools and between stimulation and staining protocols. The similarity between these values and the STED resolution indicated that the underlying structures are well below 70 nm, which was consistent with the 35–40-nm vesicle diameter. Comparing the brightness histograms of clusters with those of single antibodies confirmed that synaptotagmin clusters represent several antibodies. The authors concluded from their STED data that synaptotagmin stays clustered during exocytosis on the presynaptic membrane and does not diffuse, independent of the amount of stimulation.
In another STED application, Neumann et al. (63) analyzed the spatial distribution of three isoforms of the human mitochondrial voltage-dependent anion channel (hVDAC) with respect to the cytosolic protein hexokinase-I. Dual-color STED images were recorded using an interleaved pulse scheme whereby the lasers were switched between two excitation/depletion beam pairs at 40-ns time intervals. Two detection channels were recorded for each of the two illumination scenarios, and a linear unmixing algorithm was applied to correct for channel crosstalk. An iterative seeded-region-growing algorithm consisting of a series of erosion, smoothing, thresholding, and dilation operations allowed extraction of image regions that contained mitochondria. For colocalization analysis, Pearson’s correlation coefficient and the overlap coefficient revealed that a larger fraction of hexokinase-I is colocalized with hVDAC3 than with the other two isoforms, hVDAC1 and hVDAC2.
Greenfield et al. (103) mapped the cellular localization of the Tar receptor, CheY, and CheW (three proteins central to bacterial chemotaxis) in Escherichia coli by labeling the proteins with photoswitchable proteins tdEos or mEos and by imaging them by LM. To identify the molecules, image frames were filtered and areas identified where the signal was four standard deviations above the background level. To prevent multiple counts of the same blinking molecule and to achieve a better SNR, observed spots that reappeared within five frames were combined. To further improve image quality, only molecules emitting at least 100 photons and featuring a <40-nm localization error were counted. Clusters were objectively identified and characterized in a next step, through the use of a variant of single-linkage cluster algorithms that groups proteins closer than 30 nm. Groups of at least 10 proteins were defined as clusters. By comparing the observed cluster sizes and position distributions extracted from ~1.1 million localized individual molecules with a stochastic self-assembly model, the authors demonstrated that stochastic self-assembly without direct cytoskeletal involvement or active transport can explain the observed protein distributions (see Figure 6h).
Figure 6.

Examples of data visualization and processing in localization microscopy (LM). (a,b) Stacked visualization of several tubular membrane invaginations labeled for clathrin ( green), FBP17 (red ), and dynamin (blue). Multiple invaginations were aligned according to their clathrin and dynamin signals. (c) Plot of the localized probe positions along the tubules for 207 clathrin and FBP17 data sets. (d ) Projections and sections of a 3D data set of a single clathrin-coated pit in two orientations. Panels a–d adapted with permission from Reference 104. (e) Normalized K-test of PA-GFP-hemagglutinin in live and fixed fibroblasts showing clustering on length scales from ~20 nm to ~2.5 μm as indicated by the results’ being significantly above the dotted green line denoting a random distribution. Panel e reproduced with permission from Reference 53. ( f,g) LM image of integrin αv–tdEos. Single molecules are color-coded by their axial positions. (f) Top view. ( g) Side view of the focal adhesion denoted by the white box in panel f. The histogram shows the axial distribution (units: nm) of the molecules. Panels f and g adapted with permission from Reference 105. (h) Histogram of cluster sizes of Eos-Tar illustrating that smaller clusters occur much more frequently than large ones. The insets show sample cluster images and the cumulative distribution function. The red lines represent the fit to a self-assembly model. Panel h reproduced with permission from Reference 103.
Using 3D LM to image the budding of single vesicles and their fission from isolated plasma membrane sheets has provided valuable information on the coupling between clathrin-dependent endocytosis and F-BAR-dependent tubulation. Wu et al. (104) blocked fission by GTP analog GTPγS and imaged membrane sheets immunolabeled for clathrin or dynamin, colabeled for the F-BAR protein FBP17. Combining and carefully aligning the 3D localization data of 59–207 observed tubular membrane invaginations into a single image enabled the visualization of a representative membrane invagination with high molecular densities. This visualization clearly confirmed the restriction of clathrin, dynamin, and FBP17 to the coated pit, a constriction site at the neck region, and the tubule, respectively (see Figure 6a–c). From a section through the 3D LM representation of a single clathrin-coated pit, the characteristic spherical half-shell shape of the clathrin-coated pit of approximately ~100-nm diameter could be resolved (Figure 6d ).
In the first application of LM in living cells, Hess et al. (53) imaged the influenza membrane protein hemagglutinin (HA) to discriminate between membrane raft theories. On the basis of the clustering patterns and dynamics of HA observed by LM in live cells, the observations eliminated the hypothesis that line tension (and thereby lipid fluid-fluid phase coexistence) plays a dominant role in domain shape and also eliminated solid-phase membrane domain models for HA. Analysis of spatial correlations through K-test (Figure 6e) and diffusion distance histograms provided further evidence that HA clusters occur by modulation of diffusion within certain membrane regions.
Kanchanawong et al. (105) utilized the extremely high axial resolution of ~10 nm of the LM variant iPALM to decipher the spatial organization of integrin-based focal adhesions that link cells to the extracellular matrix. The authors labeled proteins known to locate in the adhesion complexes with the photoswitchable proteins tdEos or mEos2. Molecules were imaged and localized in 3D, and focal adhesions were identified manually for further characterization (Figure 6f ). To determine the absolute axial position of the focal adhesion molecules, the authors compared the molecules’ positions with those of autofluorescent molecules on the coverslip surface, which were present in the neighborhood of the focal adhesions. Center z-positions for the distributions of each molecular species, imaged individually, were extracted from z-position histograms either by fitting the histogram data with a Gaussian function or by calculating the first moment of the distribution (Figure 6g). The ~10-nm axial localization precision allowed the authors to map these protein components precisely and to generate for the first time a map of focal adhesions.
6. DISCUSSION AND OUTLOOK
The primary objective of any imaging technique is to learn something about an observed specimen. Every new microscopy method is measured against this expectation. For diffraction-unlimited microscopy, this expectation is especially high given that the information content of an image is closely related to its resolution. Ultimately, the success of optical nanoscopy will be determined by the insights that can be gleaned from analyzing the higher-resolution data, particularly in living specimens. To this extent, a need for well-established and standardized analysis methods is critical, especially in the case of LM, in which additional single-molecule information is inherently obtained. Although numerous applications have already taken advantage of this additional information, future applications will likely push these techniques even further.
Furthermore, the context of visualized structures must be considered. Atomic force microscopy (26) and electron microscopy (80, 106) have been used to complement and validate subdiffraction optical microscopy. Overlaying LM or STED images with electron microscopy micrographs not only confirms the correct localization of a staining but also provides anatomical context that may be required for data interpretation.
As diffraction-unlimited techniques continue to achieve increasingly better resolution, it is worth considering the expectations for visualizing samples at the molecular level: All biological samples are composed of finite numbers of individual molecules. Thus, structures underlying microscope images are fundamentally discrete, not continuous. At smaller and smaller length scales, the number of molecules sampled within the resolution limit becomes smaller and smaller, and the expectation that images of such samples should be continuous is increasingly problematic. Rather, we must try to shed our macroscopic intuition and develop a new nanoscopic intuition: The best we can do is to find the positions of all these molecules with subnanometer precision; this information is the visible portion of the sample. In this case, the resolution of the image as it is defined conventionally is difficult to apply. Instead, the best metric quantifying our ability to answer a particular question of interest has to include the SNR and will depend greatly on the question and the experimental setting in which it is asked.
Considering SNR, two aspects in particular, the number of photons detected from the probe molecule and the background noise observed relative to that signal, play central roles. Regarding the former, detecting fewer photons in total does not necessarily correlate with less information content. Hypothetically imaging a cluster of 10 vesicles of 40-nm diameter in a diffraction-limited volume illustrates this point: A confocal microscope image of the sample yields a much higher signal per pixel than does a STED image (assuming the same excitation intensity, pixel size, and dwell time per pixel) because the signal of all vesicles is combined in the former case, whereas the signal of only one or two vesicles adds up in the latter case. Nonetheless, the STED image that resolves all vesicles contains, without question, much more information than the confocal image, where all vesicles are blurred together. For the same reason, background can appear more prominent when higher resolution is used. Whereas individual antibodies produce nearly negligible background in a confocal image when compared with the accumulated signal of a cluster of 10 vesicles, the same number of background molecules can become quite noticeable in a STED image, where each vesicle appears at a tenth of the confocal signal intensity. This phenomenon, which can also be observed in LM, can make it difficult to distinguish specific from nonspecific labeling.
Generally, diffraction-unlimited microscopy benefits from bright fluorescent labeling even more than conventional microscopy does because the objects of interest tend to be smaller and therefore contain fewer probe molecules. This is also true for the density of detected labels. Especially in LM, data quality depends not only on particle detection and localization algorithms and the imaging protocol, but also on the number of labels and the brightness of individual probe molecules. Therefore, development of new probes—including brighter, smaller, more photostable molecules—as well as new labeling techniques plays an essential role in future improvements of diffraction-unlimited techniques.
As the use of diffraction-unlimited microscopy continues to grow, and as the corresponding instruments become ubiquitous, the experimental design and its relationship to data analysis methods will determine the ultimate success in answering a particular biological question. The maximum resolution, if it can still be defined, will serve only as a guideline for feasibility. Rather, the old scientific methods still reign: The crucial requirement is the selection of the hypothesis and the ability to conceive an experiment that addresses the hypothesis within the constraints of the instrument and analysis methods. As an ever wider range of capabilities becomes available, the question remains: Which important biological questions can be addressed today with existing nanoscopy technology? Many questions beckon but have not been addressed; this is a time of opportunity for biological understanding. As the gap between existing capabilities and their fullest use narrows, it is inspiring to imagine what new discoveries will be made.
Acknowledgments
We thank Edward Allgeyer and Fang Huang for helpful comments on the manuscript. T.J.G. is supported by the National Institute of General Medical Sciences (F32GM096859). S.T.H. is supported by grants K25AI65459 from the National Institute of Allergy and Infectious Diseases, R15GM094713 from the National Institute of General Medical Sciences, CHE0722759 from the National Science Foundation, and MTAF 1106 and MTAF 2061 from the Maine Technology Institute, as well as by University of Maine startup funds from the Maine Economic Improvement Fund. J.B. is supported by the National Institute of General Medical Sciences (RC1 GM091791).
Glossary
- Resolution
the minimum distance at which objects can be distinguished when imaged with a microscope; often characterized by the full width at half maximum of the point-spread function
- Stimulated emission
photophysical process that drives an excited fluorophore to its ground state through stimulation by light
- Stimulated emission depletion (STED)
process by which spontaneous fluorescence can be switched off in a targeted manner
- Localization microscopy (LM)
family of diffraction-unlimited imaging techniques that rely on single-molecule localization and image reconstruction
- Point-spread function (PSF)
spatial distribution of light emitted by a point source when observed in a microscope
- FWHM
full width at half maximum
- Localization precision
statistical error in determining a position
- Diffraction limit
fundamental physical phenomenon that restricts how tightly light can be focused, thereby limiting the resolution of microscopes
- SNR
signal-to-noise ratio
- Localization accuracy
systematic error in determining a position
Footnotes
DISCLOSURE STATEMENT
The authors hold patents in the field and have financial interest in Vutara, Inc.
Contributor Information
Travis J. Gould, Email: travis.gould@yale.edu.
Samuel T. Hess, Email: sam.hess@umit.maine.edu.
Joerg Bewersdorf, Email: joerg.bewersdorf@yale.edu.
LITERATURE CITED
- 1.Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–58. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 2.Toomre D, Bewersdorf J. A new wave of cellular imaging. Annu Rev Cell Dev Biol. 2010;26:285–314. doi: 10.1146/annurev-cellbio-100109-104048. [DOI] [PubMed] [Google Scholar]
- 3.Abbe E. Beiträge zur Theorie des Mikroskops und der Mikroskopischen Wahrnehmung. Arch Mikrosk Anat. 1873;9:413–68. [Google Scholar]
- 4.Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–82. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 5.Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- 6.Hell SW, Jakobs S, Kastrup L. Imaging and writing at the nanoscale with focused visible light through saturable optical transitions. Appl Phys A. 2003;77:859–60. [Google Scholar]
- 7.Bretschneider S, Eggeling C, Hell SW. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys Rev Lett. 2007;98:218103. doi: 10.1103/PhysRevLett.98.218103. [DOI] [PubMed] [Google Scholar]
- 8.Hell SW, Kroug M. Ground-state-depletion fluorescence microscopy: a concept for breaking the diffraction resolution limit. Appl Phys B. 1995;60:495–97. [Google Scholar]
- 9.Dedecker P, Hotta J, Flors C, Sliwa M, Uji-i H, et al. Subdiffraction imaging through the selective donut-mode depletion of thermally stable photoswitchable fluorophores: numerical analysis and application to the fluorescent protein Dronpa. J Am Chem Soc. 2007;129:16132–41. doi: 10.1021/ja076128z. [DOI] [PubMed] [Google Scholar]
- 10.Grotjohann T, Testa I, Leutenegger M, Bock H, Urban NT, et al. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature. 2011;478:204–8. doi: 10.1038/nature10497. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA. 2005;102:17565–69. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harke B, Keller J, Ullal CK, Westphal V, Schonle A, Hell SW. Resolution scaling in STED microscopy. Opt Express. 2008;16:4154–62. doi: 10.1364/oe.16.004154. [DOI] [PubMed] [Google Scholar]
- 13.Westphal V, Hell SW. Nanoscale resolution in the focal plane of an optical microscope. Phys Rev Lett. 2005;94:143903. doi: 10.1103/PhysRevLett.94.143903. [DOI] [PubMed] [Google Scholar]
- 14.Barak LS, Webb WW. Diffusion of low density lipoprotein-receptor complex on human fibroblasts. J Cell Biol. 1982;95:846–52. doi: 10.1083/jcb.95.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt T, Schutz GJ, Baumgartner W, Gruber HJ, Schindler H. Imaging of single molecule diffusion. Proc Natl Acad Sci USA. 1996;93:2926–29. doi: 10.1073/pnas.93.7.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ram S, Ward ES, Ober RJ. Beyond Rayleigh’s criterion: a resolution measure with application to single-molecule microscopy. Proc Natl Acad Sci USA. 2006;103:4457–62. doi: 10.1073/pnas.0508047103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–83. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-overhand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–65. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 19.Bornfleth H, Sätzler K, Eils R, Cremer C. High-precision distance measurements and volume- conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy. J Microsc. 1998;189:118–36. [Google Scholar]
- 20.Lacoste TD, Michalet X, Pinaud F, Chemla DS, Alivisatos AP, Weiss S. Ultrahigh-resolution multicolor colocalization of single fluorescent probes. Proc Natl Acad Sci USA. 2000;97:9461–66. doi: 10.1073/pnas.170286097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon MP, Ha T, Selvin PR. Single-molecule high-resolution imaging with photobleaching. Proc Natl Acad Sci USA. 2004;101:6462–65. doi: 10.1073/pnas.0401638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu X, Wu D, Mets L, Scherer NF. Nanometer-localized multiple single-molecule fluorescence microscopy. Proc Natl Acad Sci USA. 2004;101:11298–303. doi: 10.1073/pnas.0402155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagerholm BC, Averett L, Weinreb GE, Jacobson K, Thompson NL. Analysis method for measuring submicroscopic distances with blinking quantum dots. Biophys J. 2006;91:3050–60. doi: 10.1529/biophysj.105.079178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lidke KA, Rieger B, Jovin TM, Heintzmann R. Superresolution by localization of quantum dots using blinking statistics. Opt Express. 2005;13:7052–62. doi: 10.1364/opex.13.007052. [DOI] [PubMed] [Google Scholar]
- 25.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–45. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 26.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–72. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–96. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baddeley D, Jayasinghe ID, Cremer C, Cannell MB, Soeller C. Light-induced dark states of organic fluorochromes enable 30 nm resolution imaging in standard media. Biophys J. 2009;96:L22–24. doi: 10.1016/j.bpj.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biteen JS, Thompson MA, Tselentis NK, Bowman GR, Shapiro L, Moerner WE. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat Methods. 2008;5:947–49. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Folling J, Bossi M, Bock H, Medda R, Wurm CA, et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–45. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- 31.Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47:6172–76. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 32.Vogelsang J, Cordes T, Forthmann C, Steinhauer C, Tinnefeld P. Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc Natl Acad Sci USA. 2009;106:8107–12. doi: 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci USA. 2006;103:18911–16. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gould TJ, Gunewardene MS, Gudheti MV, Verkhusha VV, Yin S, et al. Nanoscale imaging of molecular positions and anisotropies. Nat Methods. 2008;5:1027–30. doi: 10.1038/nmeth.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bossi M, Folling J, Belov VN, Boyarskiy VP, Medda R, et al. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Lett. 2008;8:2463–68. doi: 10.1021/nl801471d. [DOI] [PubMed] [Google Scholar]
- 36.Gunewardene M, Subach FV, Gould TJ, Penoncello GP, Gudheti MV, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–28. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gould TJ, Myers JR, Bewersdorf J. Total internal reflection STED microscopy. Opt Express. 2011;19:13351–57. doi: 10.1364/OE.19.013351. [DOI] [PubMed] [Google Scholar]
- 38.Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97:8206–10. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, et al. Three-dimensional sub–100 nm resolution fluorescence microscopy of thick samples. Nat Methods. 2008;5:527–29. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- 40.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–13. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavani SR, Thompson MA, Biteen JS, Lord SJ, Liu N, et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc Natl Acad Sci USA. 2009;106:2995–99. doi: 10.1073/pnas.0900245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baddeley D, Cannell M, Soeller C. Three-dimensional sub–100 nm super-resolution imaging of biological samples using a phase ramp in the objective pupil. Nano Res. 2011;4:589–98. [Google Scholar]
- 43.Tang J, Akerboom J, Vaziri A, Looger LL, Shank CV. Near-isotropic 3D optical nanoscopy with photon-limited chromophores. Proc Natl Acad Sci USA. 2010;107:10068–73. doi: 10.1073/pnas.1004899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aquino D, Schonle A, Geisler C, Middendorff CV, Wurm CA, et al. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat Methods. 2011;8:353–59. doi: 10.1038/nmeth.1583. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt R, Wurm CA, Jakobs S, Engelhardt J, Egner A, Hell SW. Spherical nanosized focal spot unravels the interior of cells. Nat Methods. 2008;5:539–44. doi: 10.1038/nmeth.1214. [DOI] [PubMed] [Google Scholar]
- 46.Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–30. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gugel H, Bewersdorf J, Jakobs S, Engelhardt J, Storz R, Hell SW. Cooperative 4Pi excitation and detection yields sevenfold sharper optical sections in live-cell microscopy. Biophys J. 2004;87:4146–52. doi: 10.1529/biophysj.104.045815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hell S, Steltzer EHK. Properties of a 4Pi confocal fluorescence microscope. J Opt Soc Am A. 1992;9:2159–67. [Google Scholar]
- 49.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–23. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westphal V, Rizzoli SO, Lauterbach MA, Kamin D, Jahn R, Hell SW. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–49. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- 51.Lauterbach MA, Ullal CK, Westphal V, Hell SW. Dynamic imaging of colloidal-crystal nanostructures at 200 frames per second. Langmuir. 2010;26:14400–4. doi: 10.1021/la102474p. [DOI] [PubMed] [Google Scholar]
- 52.Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–62. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- 53.Hess ST, Gould TJ, Gudheti MV, Maas SA, Mills KD, Zimmerberg J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc Natl Acad Sci USA. 2007;104:17370–75. doi: 10.1073/pnas.0708066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–57. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 55.Jones SA, Shim SH, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neher RA, Mitkovski M, Kirchhoff F, Neher E, Theis FJ, Zeug A. Blind source separation techniques for the decomposition of multiply labeled fluorescence images. Biophys J. 2009;96:3791–800. doi: 10.1016/j.bpj.2008.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci USA. 2007;104:20308–13. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–53. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein T, Loschberger A, Proppert S, Wolter S, van de Linde S, Sauer M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat Methods. 2011;8:7–9. doi: 10.1038/nmeth0111-7b. [DOI] [PubMed] [Google Scholar]
- 60.Testa I, Wurm CA, Medda R, Rothermel E, von Middendorf C, et al. Multicolor fluorescence nanoscopy in fixed and living cells by exciting conventional fluorophores with a single wavelength. Biophys J. 2010;99:2686–94. doi: 10.1016/j.bpj.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Donnert G, Keller J, Wurm CA, Rizzoli SO, Westphal V, et al. Two-color far-field fluorescence nanoscopy. Biophys J. 2007;92:L67–69. doi: 10.1529/biophysj.107.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer L, Wildanger D, Medda R, Punge A, Rizzoli SO, et al. Dual-color STED microscopy at 30-nm focal-plane resolution. Small. 2008;4:1095–100. doi: 10.1002/smll.200800055. [DOI] [PubMed] [Google Scholar]
- 63.Neumann D, Buckers J, Kastrup L, Hell SW, Jakobs S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010;3:4. doi: 10.1186/1757-5036-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pellett PA, Sun X, Gould TJ, Rothman JE, Xu MQ, et al. Two-color STED microscopy in living cells. Biomed Opt Express. 2011;2:2364–71. doi: 10.1364/BOE.2.002364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buckers J, Wildanger D, Vicidomini G, Kastrup L, Hell SW. Simultaneous multi-lifetime multi-color STED imaging for colocalization analyses. Opt Express. 2011;19:3130–43. doi: 10.1364/OE.19.003130. [DOI] [PubMed] [Google Scholar]
- 66.Engelhardt J, Keller J, Hoyer P, Reuss M, Staudt T, Hell SW. Molecular orientation affects localization accuracy in superresolution far-field fluorescence microscopy. Nano Lett. 2011;11:209–13. doi: 10.1021/nl103472b. [DOI] [PubMed] [Google Scholar]
- 67.Rittweger E, Han KY, Irvine SE, Eggeling C, Hell SW. STED microscopy reveals crystal colour centres with nanometric resolution. Nat Photonics. 2009;3:144–47. [Google Scholar]
- 68.Willig KI, Keller J, Bossi M, Hell SW. STED microscopy resolves nanoparticle assemblies. New J Phys. 2006;8:106. [Google Scholar]
- 69.Gould TJ, Verkhusha VV, Hess ST. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 2009;4:291–308. doi: 10.1038/nprot.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsuda A, Shao L, Boulanger J, Kervrann C, Carlton PM, et al. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP-histones. PLoS ONE. 2010;5:e1 2768. doi: 10.1371/journal.pone.0012768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krizek P, Raska I, Hagen GM. Minimizing detection errors in single molecule localization microscopy. Opt Express. 2011;19:3226–35. doi: 10.1364/OE.19.003226. [DOI] [PubMed] [Google Scholar]
- 72.Cheezum MK, Walker WF, Guilford WH. Quantitative comparison of algorithms for tracking single fluorescent particles. Biophys J. 2001;81:2378–88. doi: 10.1016/S0006-3495(01)75884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enderlein J, Toprak E, Selvin PR. Polarization effect on position accuracy of fluorophore localization. Opt Express. 2006;14:8111–20. doi: 10.1364/oe.14.008111. [DOI] [PubMed] [Google Scholar]
- 74.Abraham AV, Ram S, Chao J, Ward ES, Ober RJ. Quantitative study of single molecule location estimation techniques. Opt Express. 2009;17:23352–73. doi: 10.1364/OE.17.023352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 2010;7:377–81. doi: 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang B, Zerubia J, Olivo-Marin JC. Gaussian approximations of fluorescence microscope point-spread function models. Appl Opt. 2007;46:1819–29. doi: 10.1364/ao.46.001819. [DOI] [PubMed] [Google Scholar]
- 77.Ram S, Prabhat P, Chao J, Ward ES, Ober RJ. High accuracy 3D quantum dot tracking with multifocal plane microscopy for the study of fast intracellular dynamics in live cells. Biophys J. 2008;95:6025–43. doi: 10.1529/biophysj.108.140392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kao HP, Verkman AS. Tracking of single fluorescent particles in three dimensions: use of cylindrical optics to encode particle position. Biophys J. 1994;67:1291–300. doi: 10.1016/S0006-3495(94)80601-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mlodzianoski MJ, Juette MF, Beane GL, Bewersdorf J. Experimental characterization of 3D localization techniques for particle-tracking and super-resolution microscopy. Opt Express. 2009;17:8264–77. doi: 10.1364/oe.17.008264. [DOI] [PubMed] [Google Scholar]
- 80.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–45. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 81.Mlodzianoski MJ, Schreiner JM, Callahan SP, Smolkova K, Dlaskova A, et al. Sample drift correction in 3D fluorescence photoactivation localization microscopy. Opt Express. 2011;19:15009–19. doi: 10.1364/OE.19.015009. [DOI] [PubMed] [Google Scholar]
- 82.Holden SJ, Uphoff S, Kapanidis AN. DAOSTORM: an algorithm for high-density super-resolution microscopy. Nat Methods. 2011;8:279–80. doi: 10.1038/nmeth0411-279. [DOI] [PubMed] [Google Scholar]
- 83.Huang F, Schwartz SL, Byars JM, Lidke KA. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed Opt Express. 2011;2:1377–93. doi: 10.1364/BOE.2.001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quan T, Zhu H, Liu X, Liu Y, Ding J, et al. High-density localization of active molecules using Structured Sparse Model and Bayesian Information Criterion. Opt Express. 2011;19:16963–74. doi: 10.1364/OE.19.016963. [DOI] [PubMed] [Google Scholar]
- 85.Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–40. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- 86.Wolter S, Schuttpelz M, Tscherepanow M, van de Linde S, Heilemann M, Sauer M. Real-time computation of subdiffraction-resolution fluorescence images. J Microsc. 2010;237:12–22. doi: 10.1111/j.1365-2818.2009.03287.x. [DOI] [PubMed] [Google Scholar]
- 87.Quan T, Li P, Long F, Zeng S, Luo Q, et al. Ultra-fast, high-precision image analysis for localization-based super resolution microscopy. Opt Express. 2010;18:11867–76. doi: 10.1364/OE.18.011867. [DOI] [PubMed] [Google Scholar]
- 88.Smith CS, Joseph N, Rieger B, Lidke KA. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods. 2010;7:373–75. doi: 10.1038/nmeth.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wieser S, Schutz GJ. Tracking single molecules in the live cell plasma membrane—do’s and don’t’s. Methods. 2008;46:131–40. doi: 10.1016/j.ymeth.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 90.Semrau S, Schmidt T. Particle image correlation spectroscopy (PICS): retrieving nanometer-scale correlations from high-density single-molecule position data. Biophys J. 2007;92:613–21. doi: 10.1529/biophysj.106.092577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pyenta PS, Holowka D, Baird B. Cross-correlation analysis of inner-leaflet-anchored green fluorescent protein co-redistributed with IgE receptors and outer leaflet lipid raft components. Biophys J. 2001;80:2120–32. doi: 10.1016/S0006-3495(01)76185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petersen NO, Hoddelius PL, Wiseman PW, Seger O, Magnusson KE. Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys J. 1993;65:1135–46. doi: 10.1016/S0006-3495(93)81173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diggle PJ. On parameter estimation and goodness-of-fit testing for spatial point patterns. Biometrics. 1979;38:87–101. [Google Scholar]
- 94.Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–70. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PLoS ONE. 2011;6:e2 2678. doi: 10.1371/journal.pone.0022678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods. 2011;8:527–28. doi: 10.1038/nmeth.1627. [DOI] [PubMed] [Google Scholar]
- 97.Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–75. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–16. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 99.Hess ST, Kumar M, Verma A, Farrington J, Kenworthy A, Zimmerberg J. Quantitative electron microscopy and fluorescence spectroscopy of the membrane distribution of influenza hemagglutinin. J Cell Biol. 2005;169:965–76. doi: 10.1083/jcb.200412058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gower JC, Ross GJS. Minimum spanning trees and single linkage cluster analysis. J R Stat Soc Ser C (Appl Stat) 1969;18:54–64. [Google Scholar]
- 101.Baddeley D, Cannell MB, Soeller C. Visualization of localization microscopy data. Microsc Microanal. 2010;16:64–72. doi: 10.1017/S143192760999122X. [DOI] [PubMed] [Google Scholar]
- 102.Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–39. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- 103.Greenfield D, McEvoy AL, Shroff H, Crooks GE, Wingreen NS, et al. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009;7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu M, Huang B, Graham M, Raimondi A, Heuser JE, et al. Coupling between clathrin-dependent endocytic budding and F-BAR-dependent tubulation in a cell-free system. Nat Cell Biol. 2010;12:902–8. doi: 10.1038/ncb2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–84. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Watanabe S, Punge A, Hollopeter G, Willig KI, Hobson RJ, et al. Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods. 2011;8:80–84. doi: 10.1038/nmeth.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]