

Abstract
Mitochondrial biogenesis is activated by nuclear encoded transcription co-activator peroxisome proliferator–activated receptor γ coactivator-1α (PGC-1α), which is regulated by several upstream factors including protein kinase A and Akt/protein kinase B. We have previously shown that selenoprotein H enhances the levels of nuclear regulators for mitochondrial biogenesis, increases mitochondrial mass and improves mitochondrial respiratory rate, under physiological condition. Furthermore, overexpression of selenoprotein H protects neuronal HT22 cells from ultraviolet B irradiation-induced cell damage by lowering reactive oxygen species production, and inhibiting activation of caspase-3 and -9, as well as p53. The objective of this study is to identify the cell signaling pathways by which selenoprotein H initiates mitochondrial biogenesis. We first confirmed our previous observation that selenoprotein H transfected HT22 cells increased the protein levels of nuclear-encoded mitochondrial biogenesis factors, peroxisome proliferator–activated receptor γ coactivator-1α, nuclear respiratory factor 1 and mitochondrial.transcription factor A, We then observed that total and phosphorylation of protein kinase A, Akt/protein kinase B and cyclic adenosine monophosphate response element-binding protein (CREB) were significantly increased in selenoprotein H transfected cells compared to vector transfected HT22 cells. To verify whether the observed stimulating effects on mitochondrial biogenesis pathways are caused by selenoprotein H and mediated through CREB, we knocked down selenoprotein H mRNA level using siRNA and inhibited CREB with napthol AS-E phosphate in selenoprotein H transfected cells and repeated the measurements of the aforementioned biomarkers. Our results revealed that silencing of selenoprotein H not only decreased the protein levels of PGC-1α, nuclear respiratory factor 1 and mitochondrial transcription factor A, but also decreased the total and phosphorylation levels of protein kinase A, protein kinase B, and CREB. Similarly, CREB inhibition reduced CREB activation and PGC-1α protein levels in selenoprotein H transfected cells. Moreover, selenoprotein H transfection increased the activity of mitochondrial complexes and prevented the ultraviolet B induced fall of mitochondrial membrane potential. We conclude that the effects of selenoprotein H on mitochondrial biogenesis and mitochondrial function are probably mediated through protein kinase A- CREB- PGC-1α and Akt/proetin kinase B- CREB- PGC-1α pathways.
Keywords: Akt, Mitochondria, Mitochondrial Biogenesis, Ptotein Kinase A, Protein Kinase B, PGC-1α, Selenoprotein, Transfection, Ultraviolet irradiation
1. Introduction
Selenoproteins are a diverse group of proteins that contain the trace element selenium. Selenium is co-translationally incorporated in the selenocysteine (Sec) amino acid residue of these proteins. To date about 25 selenoprotein genes have been identified in the human genome (Kryukov et al., 2003). The function of these selenoproteins are yet to be fully defined however, many of these proteins are associated with various biological functions such as oxidoreductions, redox signaling, antioxidant defense, thyroid hormone metabolism, immune responses and development (Arteel et al., 1998; Yan et al 1998; Saito et al., 1999; Takebe et al 2002; Hirashima et al., 2003; Kasaikina et al., 2012).
Selenoprotein H (SelH), which is one of the members of selenoprotein family, is a novel 14 kDa nucleolar oxidoreductase. It has a redox-responsive DNA-binding property owing to its high content of basic amino acid residues (Novoselov et al., 2007). SelH was initially identified in the Drosophila melanogaster and subsequently in the human and mouse genomes (Kryukov et al., 2003). The SelH mRNA is moderately expressed in many tissues and organs including brain during development, whereas SelH is only translated abundantly in spleen and to a lower extent in the brain in human and mouse under normal physiology (Novoselov et al., 2007). Interestingly, expression of SelH mRNA is found elevated in some tumors in humans, and various embryonic, carcinoma and bone marrow cell lines (Novoselov et al., 2007). Although the precise role of SelH is yet to be defined, available literature points out its importance in cellular proliferation during development or tumor growth.
Recent reports have demonstrated that SelH is involved in regulating expression levels of genes involved in de novo glutathione synthesis and phase II detoxification in response to redox status (Panee et al., 2007). Similarly, SelH has been shown to increase cell viability and antioxidant concentrations in Drosophila (Morozova et al., 2003), to enhance the levels of glutathione, glutathione peroxidase and total antioxidant capacities in murine hippocampal neuronal HT22 cells (Panee et al., 2007), and to protect cells from UVB-irradiation induced cell death (Ben Jilani et al., 2007). The protective effects of SelH are mediated by ameliorating formation of superoxide (Ben Jilani et al., 2007), blocking UVB-induced activations of caspase-3 and -9, as well as p53, stabilizing mitochondrial membrane potential (Mendelev et al., 2009), and enhancing mitochondrial function (Mendelev et al., 2011). However, it is not known through which cell signaling pathway(s) SelH exerts its protective effect.
We have recently reported that overexpression of SelH increases the protein levels of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and nuclear respiratory factor 1 (NRF1), two key nuclear factors that regulate mitochondrial biogenesis (Mendelev et al., 2011). It has been suggested that the transcription factor cyclic adenosine monophosphate response element (CRE)-binding protein (CREB) is a candidate sensor for energy insufficiency and PGC-1α gene possesses a binding site for CREB which thereby activates mitochondrial biogenesis by enhancing the expression of PGC-1α (Herzig et al., 200111; Fernandez-Marcos and Auwerx, 2011). In neurons, several kinases, including protein kinase A (PKA) and Akt (also known as protein kinase B) regulate the activity of CREB via phosphorylation (Vo and Goodman, 2001; Lonze and Ginty, 2002). The objectives of this study are to identify the cell signaling pathways by which SelH exerts its protective role and to further explore the effect of SelH on mitochondrial respiratory complex activities. We first confirmed our previous observation that SelH-transfected HT22 (SelH-HT22) cells increased the protein levels of nuclear-encoded mitochondrial biogenesis factors, PGC-1α, NRF1 and mitochondrial transcription factor A, (Tfam). We then knocked down SelH mRNA level using siRNA in SelH-HT22 cells to verify whether the observed stimulating effects on mitochondrial biogenesis markers are due to upregulation of SelH. Finally, we measured the activities of mitochondrial respiratory complexes in SelH- and vector-HT22 cells. Our results revealed that the stimulating effect of SelH on mitochondrial biogenesis is associated with mitochondrial functional improvement and protects cells depolarization from ultraviolet B (UVB) challenge. In addition, the present result revealed that SelH-induced mitochondrial biogenesis is mediated through phosphorylation of CREB since inhibition of CREB reduced the protein levels of PGC-1α.
2. Materials and Methods
2.1. Cell Maintenance and Treatment
Stably transfected murine hippocampal HT22 neuronal cells which carried either the MSCV expression vector alone (vector-HT22) or encoded hSelH (SelH-HT22) were used in the experiments. The transfection procedures and efficacy of transfection have been previously reported (Panee et al., 2007; Ben Jilani et al., 2007). The expression level of hSelH mRNA in SelH-transfected cells is about 30-fold higher than endogenous mSelH (Panee et al., 2007; Ben Jilani et al., 2007). Cells were propagated in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mM glutamine and 200 mM streptomycin/penicillin (Invitrogen), and maintained at 90%–95% relative humidity in 5% CO2 at 37°C. The culture medium was renewed every 3 days. For cell viability assays, cells were seeded in 6-well cell culture plates (Corning, Aton, MA, USA) and were allowed to reach 80 % optical confluence. All experiments were performed in triplicate or repeated on at least three occasions.
2.2. RNA Interference
Silencing of SelH expression in SelH-transfected HT22 cells was achieved using small interfering RNAs (siRNAs) FlexiTube GeneSolution (cat. #GS280636, Qiagen). Allstars Negative Control siRNA (Qiagen) was used as a non-silencing control (cat. #1027280, Qiagen). Transfection was carried out with Amaxa Mouse Neuron Nucleofection kit (VPG-1001) according to the manufacturer’s protocol in a Nucleofector II Device (Lonza). Briefly, 3×106 SelH overexpressing cells were spun down and added with 100 μl of nucleofection solution and 30 pmol of siRNA. The cell suspension was then transferred to a cuvette and transfection was run using Nucleofection program O-005 for high efficiency. Cells were resuspended in DMEM and plated in 10 mm plates. Protein lysates were collected at 7 and 24 hours post-transfection for further analysis.
2.3. Treatment of cell culture with Inhibitors
Various inhibitors were used to identify the pathways involved in mitochondrial biogenesis in SelH overexpressing cells. Briefly, once reached to 70–80% confluency, SelH-transfected HT22 cells were treated with PKA inhibitor 14–22 amide (PKI, 20 μM, EMD Millipore), CREB inhibitor napthol AE-S phosphate (50 and 100 μM, Research Organics, Cleveland, OH), nitric oxide synthase (NOS) inhibitor (4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, (TFA, 0.8 and 4 μM, Calbiochem), calcium calmodulin (CaMK) inhibitor KN-93 (20 μM, Calbiochem), NF-κB inhibitor 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline (QNZ, 20 and 40 nm, Calbiochem), AMPK inhibitor Compound C (20 and 40 μM, Calbiochem), and calcineurin inhibitor calcineurin autoinhibitory peptide (4 and 10 μM, Calbiochem) and measured the protein levels of mitochondrial biogenesis regulators.
2.4. Subcellular fractionation and Western Blotting
Cells were collected and lysed on ice in lysis buffer containing 20 mM Tris pH7.4, 10 mM KCl, 3 mM MgCl2, 0.5% NP40 and complete inhibitors (Mendelev et al., 2009). Lysates were centrifuged at 2,000 g for 10 min, and the resulting supernatants were collected as a cytosolic fraction. The pellets were washed twice, resuspended in lysis buffer containing 1% SDS and sonicated briefly on ice (Misonix, Ultrasonic Cell Disrupter). Lysates were then centrifuged at 20,800 g for 30 min and supernatants were collected as a nuclear fraction. Protein lysates (cytosolic and nuclear) were separated in 4–12% NuPAGE BT gels (Invitrogen), transferred to PVDF membrane (Millipore) and probed with antibodies against SelH (1:500, Santa Cruz Biotechnology, CA USA), PGC-1α (1:1000, Cell Signaling Technology, MA USA). NRF1 (1:500, Santa Cruz Biotechnology, CA USA), TFAM (1:300, Santa Cruz Biotechnology, CA USA), Akt, phosphor-Akt, PKA, phosphor-PKA, CREB, and pphospho-CREB (1:1000 each, Cell Signaling Technology, MA USA). Membranes were stripped and re-probed with β-actin or histone H3 antibody for protein loading control. Ratios of target protein band intensities to the loading control were calculated and presented.
2.5. UVB Irradiation
Cells were exposed to UVB as described previously (Mendelev et al., 2009). Briefly, cells were seeded in 24 or 96 well plates and cultured to 80% cell confluence, washed twice with cold PBS and exposed to 7 mJ/cm2 dose UVB radiations in serum-free medium at room temperature for 5 min over a UV Transilluminator (312 nm emission, Fisherbiotech FB-TI-88A). After UVB radiation, cells were returned to the culture incubator for various periods of recovery at 37°C.
2.6. Measurements of mitochondrial membrane potential
Mitochondrial membrane potential was measured spectroflorometrically using the tetramethylrhodamine methyl ester (TMRM). Briefly, vector and SelH-transfected viable cells (1×106/ml) were harvested and incubated with 30 nM TMRM at 37°C for one hour. Cells were washed in PBS and fluorescence measurement was performed at excitation and emission wavelengths of 530 and 573 nm respectively using Fluoromax-4 spectroflorometer (HORIBA Jobin Yvon Inc, Edison, NJ). Carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP 5 μM) was used to dissipate mitochondrial potential and used as a positive control.
2.7. Measurements of mitochondrial respiratory complex activities
Mitochondrial respiration at different complexes was performed polarographically to analyze activity of each complex using multiple substrate-inhibition protocol (Chen et al., 2006). Measurement was done in the presence of 0.5 M ADP using a high resolution respirometer equipped with a peltier thermostat and electromagnetic stirrer at 37°C (Oxygraph, Oroboros Instrument). Briefly, digitonin-permeabilized vector and SelH-transfected cells (1×107) were incubated in 2 ml mitochondrial respiration medium (MiR05) containing 110 mM sucrose, 0.5 mM EGTA, 3.0 mM MgCl2, 60 mM K- lactobionate, 10 mM KH2PO4, 20 mM Taurine, 20 mM HEPES, 1.0 g/l BSA, pH 7.1). The following substrates and inhibitors were used for complex I; glutamate (10 mM) malate (5 mM) rotenone (0.5 μM); for complex II+III: succinate (10 mM) and antimycin A (2.5 μM) and for complex IV: N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD, 0.5 mM), ascorbate (2 mM) and potassium cyanide (KCN, 1.0 mM). The outer mitochondrial membrane integrity following digitonin permeabilization was confirmed with cytochrome C (10 mM).
2.8. Statistical analysis
All data were presented as means±SD. Data presented in Figure 1–4 were analyzed by Mann-Whitney U-test. Data in Figure 5 was analyzed using Kruskal-Wallis test and followed by Dunn’s multiple comparison test. Data for Figure 6 were analyzed with Student-t-test and data in Figure 7 is analyzed by One Way ANOVA followed by Bonferroni’s Multiple Comparison Test. A p value <0.05 was considered as significant.
Fig. 1.
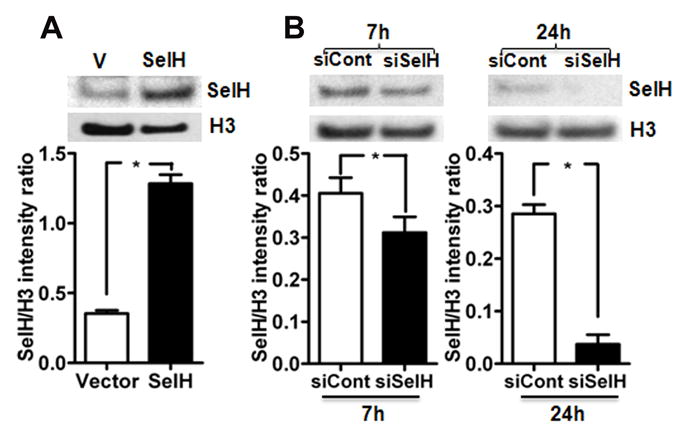
Effect of transfection and silencing of hSelH gene in HT22 cells. A) Protein level and quantitative analysis of SelH in nuclear fraction of vector and SelH-transfected cells. Transfection of SelH increased its protein level as compared to vector control. B) SelH gene silencing decreased the protein level of SelH in HT22 cells. Quantitative analysis of silencing of SelH gene on its protein level at 7 and 24 hours post- transfection. Protein level of SelH decreased after 7 and 24 hours post siRNA transfection. The decrease was more pronounced after 24 hours of post-siRNA silencing. The data are collected from three independent experiments and presented as mean±SD. *P<0.05 vs. respective control. V, vector transfected cells; SelH, SelH-transfected cells; H3, histone H3; siCont, control siRNA; siSelH, SelH siRNA; 7h, 7 hours; 24h, 24 hours.
Fig. 4.

Effect of SelH siRNA silencing on total and phospho levels of Akt, PKA and CREB. SelH silencing decreased the total protein and phospho product of Akt, PKA and CREB in SelH transfected cells. Quantitative analysis revealed that the silencing has significant effect on protein and phosphorylation of Akt, PKA and CREB in SelH transfected cells. The data are collected from three independent experiments and presented as mean±SD. *P<0.05 vs. respective control. SelH, SelH-transfected cells; H3, histone H3; siCont, control siRNA; siSelH, SelH siRNA; 7h, 7 hours; 24h, 24 hours.
Fig. 5.
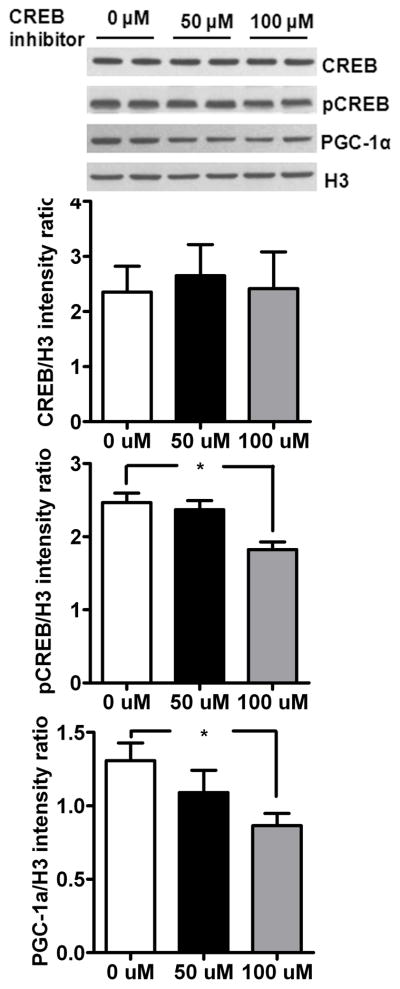
Inhibition of CREB phosphorylation blocks mitochondrial biogenesis in SelH-transfected cells. Incubation of SelH-HT22 cells with 50 and 100 μM of CREB inhibitor for 24 hours decreased phosphor-CREB and PGC-1α protein levels. The data are collected from three independent experiments and presented as mean±SD. *P<0.05 and **p<0.01 vs. respective control. SelH, SelH transfected cells; H3, histone H3.
Fig. 6.

Effect of SelH transfection on mitochondrial respiratory rate at different complexes. A) Typical oxygraph curve of digitonin permeabilized cells from vector and SelH transfected cells. Oxygen consumption hence respiratory activity (pmol/s/Mill cells) increased with the addition of specific substrates and decreased with the addition of inhibitors. Arrow indicates the addition of substrates/inhibitor to activate or inhibit specific mitochondrial complex. The activity of each complex was calculated from the difference in oxygen flow in the presence of specific substrate(s) and specific inhibitor. B–D) Activities of mitochondrial complex I (B), II+III (C) and IV (D). Note the increase in complex activities in SelH as compared to vector transfected cells. The data are collected from four independent experiments and presented as mean±SD. *P<0.05, ***P<0.001 vs. respective complex. AA, antimycin A; As, ascorbate; GM, glutamate and malate; Rot, rotenone; Sc, succinate; TMPD, N,N m,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride.
Fig. 7.

Mitochondrial membrane potential (Δψm) in vector and SelH transfected HT22 cells challenged to UVB. UVB- irradiation significantly lowered Δψ in vector transfected cells. In contrast, SelH transfection preserved mitochondrial membrane potential in UVB challenged SelH transfected cells as compared to vector cells. Data are presented as means ± SD. The data are collected from three independent experiments and presented as mean±SD. **p<0.01 vs. vector. V, vector-transfected cells and SelH, SelH-transfected cells.
3. Results
3.1. Overexpression of SelH increases and knockdown decreases SelH protein level
We have previously shown that SelH mRNA level increased by more than 30-fold in hSelH-transfected HT 22 cells (Ben Jilani et al., 2007). Presently, we studied whether transcription of SelH gene is well translated to SelH protein levels and whether silencing of SelH has the opposite effect. As expected, results revealed that overexpression of hSelH in HT22 neuronal cells significantly increased the protein level of SelH in the nuclear fraction of the SelH-transfected as compared to vector control cells (Fig. 1A). On the contrarary, silencing of SelH in SelH overexpressing cells decreased SelH protein after 7 and 24 hours post siRNA transfection (Fig. 1B). Quantative analysis of Western blots revealed that the decrease in SelH protein levels was more pronounced after 24 hours of siRNA silencing.
3.2. SelH regulates the levels of mitochondrial biogenesis regulators
Protein levels of mitochondrial biogenesis regulators PGC-1α, NRF1 and Tfam were measured in the nuclear lysates of SelH-HT22 and vector-HT22 cells. The results showed that protein levels of all three regulators of mitochondrial biogenesis, PGC-1α NRF1 and Tfam increased significantly in SelH-HT22 cells compared with vector-HT22 cells (Fig. 2A). These data are consistent with our previous published results [9]. To further verify whether the observed increase in mitochondrial biogenesis regulators is caused by SelH, we knocked down SelH mRNA level using siRNA in SelH overexpressing cells and then measured the protein levels of PGC-1α, NRF1 and Tfam levels in the nuclear fraction using Western blotting (Fig. 2B). Silencing of SelH reduced hSelH mRNA to nearly 20% of the SelH overexpressed levels as detected by RT-PCR (data not shown). Similarly, SelH siRNA silencing decreased the protein levels of PGC- 1α, NRF1 and Tfam in SelH- HT22 cells as compared to the levels obtained from SelH-HT22 cells when measured at 7 and 24 hours after knockdown (Figure 2B). SelH siRNA silencing showed no change in NRF1 and Tfam levels at 7 hours but resulted in a significant decrease in PGC-1α, NRF1 and Tfam at 24 hours post silencing as compared to the levels in SelH-HT22 cells. These results indicate the role of SelH in controlling mitochondrial biogenesis regulators.
Fig. 2.

Effect of SelH transfection and SelH silencing on the protein levels of mitochondrial biogenesis regulators. A) Protein levels of mitochondrial biogenesis regulators, PGC-1α, NRF1 and Tfam increased after SelH transfection whereas B) silencing of SelH reduced the protein levels of PGC-1α, NRF1 and Tfam as compared to control in nuclear fraction. The data are collected from three independent experiments and presented as mean±SD. *P<0.05 vs. respective control. V, vector transfected cells; SelH, SelH transfected cells; H3, histone H3; siCont, control siRNA; siSelH, SelH siRNA; 7h, 7 hours; 24h, 24 hours.
3.3. Activation of mitochondrial biogenesis signaling by SelH is associated with phosphorylation of Akt, PKA and CREB
Previous literature suggests that mitochondrial biogenesis mediated by PGC-1α and NRF1 is under the control of Akt and PKA dependant phosphorylated CREB (Than et al., 2011; Herzig et al., 2001; Vo and Goodman, 2001). Therefore, to identify SelH induced mitochondrial biogenesis pathways, we measured protein levels of total and phosphorylated (p) Akt and CREB in the nuclear fraction, and PKA in the cytosolic fraction of vector-HT22 and SelH-HT22 cells (Fig. 3). The results showed that overexpression of SelH significantly increased the protein levels of total and pPKA, pAkt, and pCREB compared to vector-HT22 cells (Fig. 3A&B). To further verify whether the observed phosphorylation changes are caused by SelH, we silenced SelH in SelH-HT22 cells using SelH siRNA and repeated the protein measurements. The results showed that downregulation of SelH not only reduced the total protein levels of PKA and Akt but also lowered the phosphorylation of PKA, Akt and CREB in these cells (Fig. 4). Interestingly, PKA inhibition with PKA inhibitor 14–22 amide (20 μM) in SelH-HT22 cells decreased the protein level of mitochondrial biogenesis regulators (Data not shown), further confirming the association of these factors in SelH-induced mitochondrial biogenesis.
Fig. 3.
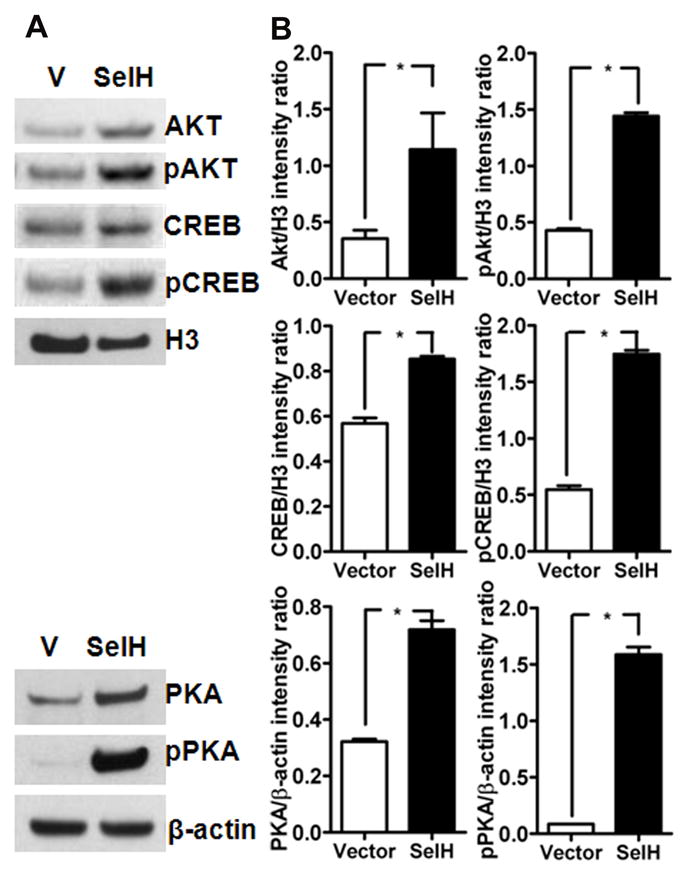
SelH regulate mitobiogenesis by Akt, PKA and CREB. A) Protein and phosphorylation levels of Akt, PKA and CREB in vector and SelH transfected cells. Both, total protein and phospho- product of Akt (nuclear), PKA (cytosolic) and CREB (nuclear) increased in SelH cells as compared to vector transfected cells. B) Quantitative western analysis of Akt, PKA and CREB revealed that SelH transfection not only increased the protein levels of Akt, PKA and CREB but also significantly elevated the phosphorylation of these proteins. The data are collected from three independent experiments and presented as mean±SD. *P<0.05 vs. respective control. V, vector transfected cells; SelH, SelH-transfected cells; H3, histone H3.
3.4. CREB inhibition prevents SelH-induced mitochondrial biogenesis by down regulating PGC-1α
To confirm whether CREB plays a role in SelH induced mitochondrial biogenesis, we inhibited CREB and measured protein levels of CREB, pCREB and PGC-1α (Fig 5). Result shows that CREB inhibition does not affect total CREB but interestingly lowers phosphorylation of CREB in SelH-transfected cells. Likewise CREB inhibition reduced the protein levels of PGC-1α after treatment. These results clearly suggest that CREB dependant PGC-1α activation regulate SelH-induced mitochondrial biogenesis.
Previous studies have also suggested that CaMK-CREB, NOS-nitric oxide (NO)-cGMP NF-κB and AMPK could activate PGC-1α (Alvarez-Guardia et al., 2010; Kelly, and Scarpulla 2004; Scarpulla 2002a,b). To determine whether these factors are also involved in mediating the SelH-induced mitochondrial biogenesis effects, we incubated the SelH-HT22 cells with nNOS inhibitor TFA (0.8 and 4 μM), CaMK inhibitor KN-93 (20 μM), NF-κB inhibitor QNZ (20 and 40 nm), AMPK inhibitor Compound C (20 and 40 μM) and calcineurin inhibitor calcineurin autoinhibitory peptide (4 and 10 μM) and measured the protein levels of mitochondrial biogenesis regulators. Result revealed no change in the protein levels of mitochondrial biogenesis regulators following inhibition with above mentioned inhibitor (data not shown) suggesting that nNOS, CaMK, NF-κB, AMPK and calcineurin- dependant mechanisms are not involved in the activation of mitochondrial biogenesis in SelH-transfected cells.
3.5. Overexpression of SelH increases mitochondrial respiratory chain complex activities
To investigate the role of SelH in regulating mitochondrial function, we used oxygen consumption assay to determine activity of respiratory complexes using complex specific substrate-inhibitors. Fig. 6A, shows the typical Oxygraph curve of permeabilized cells from vector and SelH-transfected cells. Oxygen consumption hence respiratory activity increased with the addition of specific substrates and decreased with the addition of inhibitors. As shown in Fig. 6B–D, overexpression of SelH increases the respiratory function of mitochondria. Therefore, the activity of mitochondrial complex I, II+III and IV increased by 67 (p<0.001), 105 (p<0.05) and 54 % respectively in SelH-HT22 cells as compared to vector-HT22 cells. These data indicates that overexpression of SelH has a distinct effect on mitochondria functioning in these cells.
3.6. SelH prevents stress induced collapse of mitochondrial membrane potential (Δψm)
As shown in the previous section, SelH overexpression modulates mitochondrial function. Therefore, we challenged vector and SelH-transfected cells to UVB-irradiation to ascertain whether the positive influence of SelH overexpression on mitochondrial function is also beneficial in preventing mitochondrial dysfunction under stress conditions. We measured mitochondrial membrane potential (Δψm) in vector- and SelH-transfected HT22 cells under normal and UVB challenged conditions. As shown in Fig. 7, UVB-irradiation induced a significant fall in Δψm. interestingly, overexpression of SelH ameliorated the UVB-induced Δψm depolarization compared to vector-HT22 cells (90.6±7% vs. 66.4±15%, p<0.01) suggesting that overexpression of SelH also offer beneficial outcome under stress conditions. Application of FCCP to the cell media caused same level of membrane depolarization in both vector- and selH-transfected HT22 cells, validating the method.
4. Discussion
The present study revealed that mitochondrial biogenesis regulators PGC-1α, NRF1 and Tfam increased in SelH overexpressing cells. Previous studies have suggested that PGC-1α is capable of activating NRF1, -2 and increasing mitochondrial respiratory function by activation of Tfam (Puigserver et al., 1998; Wu et al., 1999). Therefore PGC-1α plays an important role in mitochondrial respiration and biogenesis. Likewise, increase of mitochondrial mass in SelH overexpressing cells as reported previously (Mendelev et al., 2011), together with the increase in levels of mitochondrial biogenesis regulators clearly indicates the involvement of SelH in inducing mitochondrial biogenesis.
This study examines the mechanisms through which SelH regulates mitochondrial biogenesis and function. We demonstrate that transfection of human SelH gene into neuronal HT22 cells stimulates mitochondrial biogenesis signaling pathway by significantly elevating the protein level of nuclear encoded mitochondrial biogenesis regulators, PGC-1α, NRF1 and Tfam. This effect is specifically induced by SelH overexpression because knockdown of SelH using siRNA diminished the elevation of the mitochondrial biogenesis regulators in SelH-HT22 cells. Our results further revealed that the stimulating effects of SelH overexpression on mitochondrial biogenesis regulators is associated with PKA-CREB and Akt-CREB pathways, since overexpression of SelH increased the protein levels and phosphorylation of Akt, PKA and CREB. Consistently, siRNA knockdown of SelH not only prevented the phosphorylation of Akt, PKA and CREB but also reduced the protein levels of mitochondrial biogenesis regulators, PGC-1α, NRF1 and Tfam.
PGC-1α is activated by various complex signaling pathways including transcription factor CREB (Herzig et al., 2001) and present increase in the protein and phosphorylation levels of CREB seems to be well linked to the elevated levels of PGC-1α reported in the study. Wu et al. (2006) has demonstrated by screening 10,000 human full-length cDNAs for induction of the PGC-1α promoter that a number of potent activators of PGC-1α transcription were the transducer of regulated CREB binding protein (TORC). TORC induces PGC-1α transcription through CREB, thereby leading to increased mitochondrial oxidative function and fatty acid oxidation (Wu et al., 2006). It is not clear from the present study whether CREB induces PGC-1α activation directly or through various CREB partners such as PGC-1-related co-activator (PRC), CREB-Binding Protein (CBP), CREB-regulated transcription co-activators (CRTC) or TORC. The present study, however, revealed that inhibition of CREB not only reduced its phosphorylation but also blocked the SelH-induced increase in PGC-1α, suggesting the participation of CREB in mediating the effect of SelH. Interestingly, siRNA knock down of SelH diminished the changes in CREB and subsequently in PGC-1α, NRF1 and Tfam levels, indicating that CREB is the central regulator onto which various pathways are converged during mitochondrial biogenesis (Herzig et al., 2001).
Mitochondrial biogenesis and CREB activation is dependent upon its phosphorylation by various signaling including activation of PKA and Akt pathways (Vo and Goodman, 2001; Lonze and Ginty, 2002). In the present study, we observed increased protein levels and phosphorylation of PKA and Akt, which are consistent with the elevated levels of PGC-1α, NRF1 and Tfam in SelH-transfected cells as compared to vector control cells. We have also observed that PKA inhibition rescued the changes in mitochondrial biogenesis regulators (Data not shown). Published literature suggests that CREB is one of the best-characterized targets of PKA. It appears quite likely that one of the pathways that initiate SelH-induced mitochondrial biogenesis via PGC-1α is mediated through phosphorylation and activation of CREB by PKA. Moreover, present results are also further confirmed by the reports which suggest that mitochondrial biogenesis is regulated by activation of CREB and that cAMP-stimulating compound forskolin or pyrroloquinoline quinine stimulates phosphorylation of CREB and enhances PGC-1a expression, whereas dominant negative mutant of CREB or siRNA interference blocks this activation (Handschin et al., 2003; Chowanadisai et al., 2010). In addition, recent evidence indicates that PKA inhibitor H89 could reduce, whereas cAMP could rescue the reduced expression of pCREB and PGC-1α, suggesting the critical role of PKA-CREB pathway in the regulation of PGC-1α expression (Sheng et al., 2012). Thus, PKA-CREB mediated mitochondrial biogenesis is important in maintaining neuronal activity and survival in various stress conditions including neurodegenerative diseases (Sheng et al., 2012; Ryu et al., 2005; Lopez-Lluch et al., 2008).
Similarly, CREB has been shown to be a regulatory target of Akt [23–24]. Akt phosphorylates CREB at Ser-133, the pCREB band detected in the present study. Phosphorylation of CREB at Ser-133 residue stimulates recruitment of CREB-binding protein (CBP) to the promoter, and activates cellular gene expression via a cAMP response element (CRE)-dependent mechanism (Du and Montminy, 1998). Increased protein and phosphorylation of Akt observed in the present study is therefore in direct correlation of its (Akt) involvement in activation of SelH- induced mitochondrial biogenesis. Although, it is not clear from the present study whether SelH induced activation of mitochondrial biogenesis through CREB is the additive effect of PKA-CREB and Akt-CREB signaling, silencing of SelH decreased the protein and phosphorylation levels of PKA, Akt and CREB.
Mitochondrial biogenesis is activated by various signaling mechanisms involving NOS, CaMK, NF-κB, AMPK, calcineurin Akt, PKA and CREB. To determine their involvement in the present study we used the inhibitors of these molecules and results revealed that nNOS, CaMK, NF-κB, AMPK and calcineurin- dependant mechanisms are not involved in the activation of mitochondrial biogenesis in SelH-transfected cells. Further, our results clearly implicate the involvement of PKA, Akt and CREB in SelH-induced mitochondrial biogenesis.
Previous studies have shown that PGC-1α induce mitochondrial biogenesis, which increases oxygen consumption and activities of mitochondrial complexes (Wu et al., 1999; Suliman et al., 2003). Increased mitochondrial respiration maintains mitochondrial function and prevents any change in mitochondrial membrane potential and promotes survival under stress condition (Shen et al., 2008). Presently, we have observed significant increase in mitochondrial complex activities in SelH-transfected cells as compared to vector control cells. Increased activities are consistent with the sustained mitochondrial membrane potential in these cells. Likewise, the increased activities may be the result of increased mitochondrial mass (Mendelev et al., 2011) as a result of increase in the mitochondrial number. It is likely that SelH-induced mitochondrial biogenesis concomitantly improves mitochondrial function by increasing the activities of respiratory complexes. In addition, SelH overexpressed cells are resistant to UVB-induced loss of mitochondrial membrane potential, which clearly indicates that mitochondrial biogenesis enhances metabolic potential and thereby repress any change in mitochondrial functional performance (Garcia et al., 2005; Haden et al., 2007) in SelH-transfected cells.
In summary, we demonstrate that the SelH activates mitochondrial biogenesis signaling via PGC-1α, NRF1 and Tfam, Moreover, SelH-induced mitochondrial biogenesis is probably mediated through modulation of PKA-CREB and Akt-CREB pathway, because overexpression of SelH increases, while knockdown of SelH suppresses the protein levels and phosphorylation of PKA, Akt and CREB, and because inhibition of CREB blocks the PGC-1α elevation in SelH-HT22 cells. Finally, SelH increases the activities of mitochondrial respiratory complexes and preserves UVB stress-induced mitochondrial membrane potential depolarization.
Highlights.
SelH overexpression activates Akt, PKA and CREB.
Silencing of selenoprotein H (SelH) alters mitochondrial biogenesis by decreasing mitochondrial biogenesis regulators PGC-1α, NRF1 and Tfam.
SelH-induced mitochondrial biogenesis signaling is mediated through CREB and may involve Akt-CREB and PKA-CREB pathways.
SelH overexpression improves mitochondrial functional performance by increasing the activities of mitochondrial respiratory chain complex activities.
Acknowledgments
This work is supported by a grant from National Institute of Health to PAL (7R01DK075476). The BRITE is partially funded by the Golden Leaf Foundation.
Footnotes
Disclosures
No conflict of interest are declared by the authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, Laguna JC, Vazquez-Carrera M. The p65 subunit of NF-kB binds to PGC-1a, linking inflammation and metabolic disturbances in cardiac cells. Cardiovascular Research. 2010;87:449–58. doi: 10.1093/cvr/cvq080. [DOI] [PubMed] [Google Scholar]
- Arteel GE, Mostert V, Oubrahim H, Briviba K, Abel J, Sies H. Protection by selenoprotein P in human plasma against peroxynitrite-mediated oxidation and nitration. Biological Chemistry. 1998;379:1201–5. [PubMed] [Google Scholar]
- Ben Jilani KE, Panee J, He Q, Berry MJ, Li PA. Overexpression of selenoprotein H reduces Ht22 neuronal cell death after UVB irradiation by preventing superoxide formation. International Journal Biological Sciences. 2007;3:198–204. doi: 10.7150/ijbs.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hattori Y, Nakajima K, Eizawa T, Ehara T, Koyama M, Hirai T, Fukuda Y, Kinoshita M, Sugiyama A, Hayashi J, Onaya T, Kobayashi T, Tawata M. Mitochondrial complex I activity is significantly decreased in a patient with maternally inherited type 2 diabetes mellitus and hypertrophic cardiomyopathy associated with mitochondrial DNA C3310T mutation: a cybrid study. Diabetes Research and Clinical Practice. 2006;74:148–53. doi: 10.1016/j.diabres.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. Journal of Biological Chemistry. 2010;285:142–52. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. Journal of Biological Chemistry. 1998;273:32377–9. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. American Journal of Clininal Nutration. 2011;93:884S–90. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García O, Almeida A, Massieu L, Bolaños JP. Increased mitochondrial respiration maintains the mitochondrial membrane potential and promotes survival of cerebellar neurons in an endogenous model of glutamate receptor activation. Journal Neurochemistry. 2005;92:183–90. doi: 10.1111/j.1471-4159.2004.02851.x. [DOI] [PubMed] [Google Scholar]
- Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. American Journal of Respirtory and Critical Care Medicine. 2007;176:768–77. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proceedings of National Academy Sciences USA. 2003;100:7111–6. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–83. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Hill KE, Zhou J, McMahan WJ, Motley AK, Atkins JF, Gesteland RF, Burk RF. Deletion of selenoprotein P alters distribution of selenium in the mouse. Journal of Biological Chemistry. 2003;278:13640–6. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- Hirashima M, Naruse T, Maeda H, Nozaki C, Saito Y, Takahashi K. Identification of selenoprotein P fragments as a cell-death inhibitory factor. Biological and Pharmaceutocal Bulletin. 2003;26:794–8. doi: 10.1248/bpb.26.794. [DOI] [PubMed] [Google Scholar]
- Kasaikina MV, Hatfield DL, Gladyshev VN. Understanding selenoprotein function and regulation through the use of rodent models. Biochimica et Biophysica Acta. 2012;1823:1633–42. doi: 10.1016/j.bbamcr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Development. 2004;18:357–68. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–43. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–23. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- López-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Experimental Gerontology. 2008;43:813–9. doi: 10.1016/j.exger.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelev N, Mehta SL, Witherspoon S, He Q, Sexton JZ, Li PA. Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion. 2011;11:76–82. doi: 10.1016/j.mito.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelev N, Witherspoon S, Li PA. Overexpression of human selenoprotein H in neuronal cells ameliorates ultraviolet irradiation-induced damage by modulating cell signaling pathways. Experimental Neurology. 2009;220:328–34. doi: 10.1016/j.expneurol.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova N, Forry EP, Shahid E, Zavacki AM, Harney JW, Kraytsberg Y, Berry MJ. Antioxidant function of a novel selenoprotein in Drosophila melanogaster. Genes and Cells. 2003;8:963–71. doi: 10.1046/j.1365-2443.2003.00687.x. [DOI] [PubMed] [Google Scholar]
- Novoselov SV, Kryukov GV, Xu XM, Carlson BA, Hatfield DL, Gladyshev VN. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. Journal of Biological Chemistry. 2007;282:11960–8. doi: 10.1074/jbc.M701605200. [DOI] [PubMed] [Google Scholar]
- Panee J, Stoytcheva ZR, Liu W, Berry MJ. Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. Journal of Biological Chemistry. 2007;282:23759–65. doi: 10.1074/jbc.M702267200. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proceedings National Academic Sciences USA. 2005;102:13915–20. doi: 10.1073/pnas.0502878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Hayashi T, Tanaka A, Watanabe Y, Suzuki M, Saito E, Takahashi K. Selenoprotein P in human plasma as an extracellular phospholipid hydroperoxide glutathione peroxidase. Isolation and enzymatic characterization of human selenoprotein p. Journal of Biological Chemistry. 1999;274:2866–71. doi: 10.1074/jbc.274.5.2866. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochimica et Biophysica Acta. 2002a;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002b;286:81–9. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- Shen W, Liu K, Tian C, Yang L, Li X, Ren J, Packer L, Cotman CW, Liu J. R-alpha-lipoic acid and acetyl-L-carnitine complementarily promote mitochondrial biogenesis in murine 3T3-L1 adipocytes. Diabetologia. 2008;51:165–74. doi: 10.1007/s00125-007-0852-4. [DOI] [PubMed] [Google Scholar]
- Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. Journal of Neurochemistry. 2012;120:419–29. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. Journal of Biological Chemistry. 2003;278:41510–8. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- Takebe G, Yarimizu J, Saito Y, Hayashi T, Nakamura H, Yodoi J, Nagasawa S, Takahashi K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. Journal of Biological Chemistry. 2002;277:41254–8. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- Than TA, Lou H, Ji C, Win S, Kaplowitz N. Role of cAMP-responsive element-binding protein (CREB)-regulated transcription coactivator 3 (CRTC3) in the initiation of mitochondrial biogenesis and stress response in liver cells. Journal of Biological Chemistry. 2011;286:22047–54. doi: 10.1074/jbc.M111.240481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. Journal of Biological Chemistry. 2001;276:13505–8. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proceedings National Academic Sciences USA. 2006;103:14379–84. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yan J, Barrett JN. Purification from bovine serum of a survival-promoting factor for cultured central neurons and its identification as selenoprotein-P. Journal of Neuroscience. 1998;18:8682–91. doi: 10.1523/JNEUROSCI.18-21-08682.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]