

Abstract
Atrial natriuretic peptide (ANP) inhibits agonist-induced pulmonary edema formation, but the signaling pathway responsible is not well defined. To investigate the role of the particulate guanylate cyclase-linked receptor, natriuretic peptide receptor-A (NPR-A), we measured acute lung injury responses in intact mice and pulmonary microvascular endothelial cells (PMVEC) with normal and disrupted expression of NPR-A. NPR-A wild-type (NPR-A+/+), heterozygous (NPR-A+/−), and knockout (NPR-A−/−) mice were anesthetized and treated with thrombin receptor agonist peptide (TRAP) or lipopolysaccharide (LPS). Lung injury was assessed by lung wet-to-dry (W/D) weight and by protein and cell concentration of bronchoalveolar lavage (BAL) fluid. No difference in pulmonary edema formation was seen between NPR-A genotypes under baseline conditions. TRAP and LPS increased lung W/D weight and BAL fluid cell counts more in NPR-A−/− mice than in NPR-A+/− or NPR-A+/+ mice, but no genotype-related differences were seen in TRAP-induced increases in bloodless lung W/D weight or LPS-induced increases in BAL protein concentration. Pretreatment with ANP infusion completely blocked TRAP-induced increases in lung W/D weight and blunted LPS-induced increases in BAL cell counts and protein concentration in both NPR-A−/− and NPR-A+/+ mice. Thrombin decreased transmembrane electrical resistance in monolayers of PMVECs in vitro, and this effect was attenuated by ANP in PMVECs isolated from both genotypes. Administration of the NPR-C-specific ligand, cANF, also blocked TRAP-induced increases in lung W/D weight and LPS-induced increases in BAL cell count and protein concentration in NPR-A+/+ and NPR-A−/− mice. We conclude that ANP is capable of attenuating agonist-induced lung edema in the absence of NPR-A. The protective effect of ANP on agonist-induced lung injury and pulmonary barrier function may be mediated by NPR-C.
Keywords: endothelium, natriuretic peptides, pulmonary edema, receptors, vascular permeability
atrial (anp) and brain natriuretic peptide (BNP) are part of a family of proteins that have myriad effects on cardiovascular homeostasis, including natriuresis, antagonism of the renin-angiotensin system, and inhibition of vascular smooth muscle tone (44). These effects serve to limit intravascular fluid accumulation and protect against elevation in ventricular filling volume and systemic arterial pressure (8). ANP has also been shown to cause an acute decrease in plasma volume that is maintained even in nephrectomized animals and is associated with an increase in transluminal movement of fluid and albumin into the interstitial space (1, 12, 48).
Most of the biological effects of the natriuretic peptides are mediated by binding to one of three natriuretic peptide receptors (NPR) (41). NPR-A and NPR-B are membrane-bound guanylate cyclase-linked receptors that mediate most of the known biological effects of the natriuretic peptides by increasing intracellular cGMP synthesis in response to ligand binding. The binding affinities of ANP and BNP for NPR-A are nearly three orders of magnitude greater than they are for NPR-B. The reverse is true for C-type natriuretic peptide (28). A third receptor, NPR-C, has similar binding affinity for all three peptides, but does not possess a guanylate cyclase domain, and was initially thought to act primarily as a clearance receptor (32). However, subsequent studies have shown that NPR-C is capable of inhibiting adenylate cyclase via a G-inhibitory protein coupled receptor (Gi) and activating phospholipase C, suggesting that NPR-C is capable of mediating unique effects of the natriuretic peptides by non-guanylate cyclase-linked pathways (2, 36).
NPR-A knockout mice have elevated circulating plasma volumes, cardiac hypertrophy, and systemic hypertension (37). Targeted deletion of NPR-A to vascular endothelial cells resulted in normal vasodilatory responses to ANP, but a similar degree of systemic hypertension and cardiac hypertrophy as seen in nonselective models of NPR-A knockout mice (42). These findings suggest that ANP modulates vascular pressure and volume via its effects on vascular permeability, as opposed to its diuretic or vasodilatory effects.
In contrast to the systemic circulation, ANP does not appear to increase permeability in the lung and paradoxically inhibits increases in both systemic and pulmonary vascular permeability caused by a variety of agonists. For example, ANP blunts the permeability of systemic and pulmonary vascular endothelial cell (PVEC) monolayers to thrombin, vascular endothelial growth factor, H2O2, hypoxia, tumor necrosis factor-α, and lipopolysaccharide (LPS) (3a, 7, 22, 39, 43, 49). Infusion of ANP reduces agonist-induced lung edema in isolated lungs (19, 20) and oxidant-induced lung injury in intact animals (31). ANP has also been shown to improve oxygenation and lung injury score in humans with high-altitude pulmonary edema or acute respiratory distress syndrome (ARDS) (34, 46). Studies showing that agonist-induced pulmonary edema formation is worse in response to ANP-specific antibodies and in ANP knockout mice suggest a physiological role for ANP in protecting against acute lung injury (5, 21, 45). Although the effect of ANP on pulmonary edema in vivo may be due in part to a reduction in plasma volume or vascular tone, its ability to mitigate agonist-induced increases in the permeability of PVEC monolayers and isolated perfused lungs suggests that ANP has a direct effect on enhancement of pulmonary endothelial barrier function. This hypothesis is supported by the observation that ANP has a greater inhibitory effect on oleic acid-induced lung edema in dogs than diuretics, despite a similar degree of diuresis and reduction in pulmonary arterial pressure (33).
The signal transduction pathways that mediate the protective effect of ANP on acute lung injury are not well understood. Considering that most biological effects of ANP are mediated by cGMP (40, 41) and that the inhibitory effect of ANP on permeability can be mimicked by cGMP administration (10, 35, 43, 46), we hypothesized that ANP mitigates pulmonary edema formation via NPR-A. To test this hypothesis, we examined the effect of agonist-induced lung injury in mice with normal and disrupted NPR-A expression and the ability of ANP to protect against acute lung injury in these mice and against barrier dysfunction in PVECs isolated from these animals.
METHODS
Animals.
All animal housing and experiments were conducted in accordance with protocols approved by the Providence Veterans Affairs Medical Center Institutional Animal Care and Use Committee. Heterozygous breeding pairs of mice with gene-targeted disruption of Npr1 were obtained from Jackson Laboratories (Bar Harbor, ME).
Thrombin receptor agonist peptide-induced lung injury.
Mice were anesthetized, and the external jugular vein was catheterized. Mice were given 100 μl of saline vehicle or 2–10 μg/kg ANP in 100 μl of saline intravenously (iv), followed 30 min later by 100 μl of thrombin receptor agonist peptide (TRAP) (4 mg/kg iv) or saline vehicle. Mice were killed 30 min later, and blood, heart, lungs, and kidneys were collected for analysis.
LPS-induced lung injury.
Mice were anesthetized and given LPS (2.5 mg/kg) or an equal volume of saline vehicle by intratracheal installation. Four hours later, animals either underwent bronchoalveolar lavage or were euthanized for blood and organ collection.
Measurement of lung injury.
For measurement of standard lung wet-to-dry (W/D) weight ratio, lungs were removed, blotted free of excess fluid, and weighed immediately (wet weight) and then again after drying at 90°C for 48 h. Bloodless W/D weight ratios were obtained by measuring hemoglobin concentration of blood and lung homogenate supernatant and then calculating the blood volume (QB) and water volume (QW) of the lung as previously described (13). Briefly, we measured the water fraction in the lung homogenate (WFH), the supernatant of the homogenate (WFS), and the blood (WFB) by (Wwet − Wdry)/Wwet, where Wwet and Wdry are wet and dry weight, respectively. We then measured QB of the lung by:
where 1.039 is the density of blood, QH is the weight of the lung homogenate, HbS is the hemoglobin concentration of supernatant of lung homogenate, and HbB is the hemoglobin concentration of blood. QW of the lung was obtained by:
where Ww is the weight of the distilled water added when the lung is homogenized. QW was then divided by the calculated lung dry weight (lung wet weight − QB − QW) to obtain bloodless W/D weight ratios. Bronchoalveolar lavage (BAL) fluid was collected from anesthetized, tracheotomized mice using 600 μl of 1× PBS. Cell counts were obtained using a standard hemocytometer at ×10 magnification. Protein concentration of the BAL fluid was measured using a modified Bradford assay at an absorbance at 750 nm.
Infusion of natriuretic peptides.
ANP (360 ng·kg−1·min−1), NPR-C-specific ligand cANF (500 ng·kg−1·min−1), or saline vehicle was delivered via an external jugular vein catheter at an average rate of 2 μl/min, starting 30 min before TRAP or LPS administration and continuing an additional 30 min after. Animals treated with TRAP were killed for blood and organ collection immediately after completion of ANP infusion. Animals treated with LPS were killed for blood and organ collection 4 h after completion of ANP infusion.
Measurement of plasma ANP levels.
Plasma samples were extracted on Sep Pak C18 cartridges and reconstituted in assay buffer, and ANP concentrations were measured by enzyme immunoassay following the manufacturer's recommendation (Peninsula Laboratories, San Carlos, CA).
Isolation of pulmonary microvascular endothelial cells.
Pulmonary microvascular endothelial cells (PMVEC) were isolated from NPR-A−/−and NPR-A+/+ mice using a protocol established by Atkinson and colleagues (Brigham and Women's Hospital) (http://vrd.bwh.harvard.edu/core_facilities/mlec.html). Endothelial cells were characterized by staining for VE-cadherin; uptake of fluorescently conjugated acetylated low-density lipoprotein; and ability to form tubular structures when grown in Matrigel. PMVEC were used during passages 2–6.
Measurement of NPR expression.
Total RNA was extracted from lung, kidney, and isolated PMVECs. Transcripts for NPR-A, NPR-B, and NPR-C were amplified using primer sequences designed with the MIT Primer3 program.
Measurement of cGMP.
Equivalent numbers of PMVEC were serum starved for 2 h and then incubated with 10−8, 10−7, or 10−6 mol/l ANP or vehicle for 20 min, before being lysed in 0.1 mol/l HCl. Cell lysate cGMP and cAMP levels were measured by enzyme immunoassay following the manufacturer's recommendations (Cayman Chemical, Ann Arbor, MI).
Measurement of endothelial monolayer permeability.
Endothelial monolayer permeability was assayed by measurement of transendothelial electrical resistance (TER) using electrical cell impedance sensing (Applied Biophysics, Troy, NY), as previously described (25). The TER was measured in Ohms every 1.12 min, and a baseline TER was established for 1 h before addition of ANP (1 μmol/l) or vehicle. Thirty minutes later, cells were treated with ANP plus vehicle, thrombin (2 U/ml) plus vehicle, or thrombin plus ANP. Thrombin-induced fall in TER was defined as the difference between the last TER measured before addition of thrombin and the lowest TER measured after addition of thrombin and occurred between 3 and 12 min after addition of thrombin. Thrombin-induced fall in TER was normalized by dividing the resistance in Ohms at any given point by the resistance in Ohms at the last time point measured immediately before the addition of thrombin. The effect of ANP was expressed as a percentage of thrombin-induced fall in TER in the absence of ANP or the difference in Ohms.
Statistical analysis.
Differences in mean values of lung W/D weight and BAL fluid protein and cell concentration between experimental groups were compared by two-way ANOVA. Where significant differences were found, multiple pairwise comparisons were done using the Tukey test. For changes in lung W/D weight in mice given ANP or cANF, mean and 95% confidence intervals for W/D ratios were calculated and compared between groups using a generalized linear mixed model. Lung W/D weight was treated as a random effect within animal, with genotype, ANP, or cANF (yes/no), and TRAP or LPS (yes/no) treated as factorial fixed effects, including main and all two- and three-way interactions, and covarying for the animal's live weight to remove variability in lung size attributable to overall size. Individual group estimates and comparisons were carried out using orthogonal linear estimates based on the model parameters. Alpha was maintained at 0.05 across all comparisons using the Holm test to adjust the individual P values resulting from comparisons. Differences in plasma ANP levels and TER were assessed using t-test. Differences in BAL fluid protein and cell counts between LPS-treated mice given saline and LPS-treated mice given ANP or cANF were assessed using t-test. Differences were considered statistically significant at P ≤ 0.05. Values shown are means ± SD.
RESULTS
Characterization of disrupted NPR-A signaling.
Expression of Npr1, the gene that encodes for NPR-A, was demonstrated in the lung, kidney, and PMVEC from NPR-A+/+, but not NPR-A−/−, mice (Fig. 1A). Transcript levels of NPR-B and NPR-C did not differ appreciably between NPR-A+/+ and NPR-A−/− mice (Fig. 1A). Previously reported phenotypic differences in cardiac mass (37) were also observed between NPR-A+/+ and NPR-A−/− mice (Fig. 1B). Functionality of NPR-A in knockout mice was assessed by examining cGMP response to ANP. ANP produced a dose-dependent increase in intracellular cGMP concentration in PMVECs isolated from NPR-A+/+ but not NPR-A−/− mice (Fig. 2). ANP had no appreciable effect on intracellular cAMP in PMVEC isolated from mice of either genotype (Fig. 2B).
Fig. 1.

Characterization of natriuretic peptide receptor (NPR) transcript expression and heart weight-to-body weight phenotype in NPR-A−/− mice. A: expression of transcripts for NPR-A, NPR-B, and NPR-C in lung, and pulmonary microvascular endothelial cells (PMVEC) isolated from NPR-A wild-type (NPR-A+/+) and knockout mice (NPR-A−/−). B: heart weight-to-body weight ratio in NPR-A wild-type, heterozygote (NPR-A+/−), and knockout mice. Values are means ± SD; n = 12 per group. *P < 0.05 vs. NPR-A+/+.
Fig. 2.

cGMP and cAMP production in NPR-A+/+ and NPR-A−/− mouse PVMEC in response to atrial natriuretic peptide (ANP). Shown are intracellular levels of cGMP (A) or cAMP (B) in cell lysates from PMVECs isolated from two different NPR-A+/+ mice [referred to as wild-type (WT) 6 and WT8] and two different NPR-A−/− mice [referred to as knockout (KO) 4 and KO6] in response to increasing concentrations of ANP.
Effect of NPR-A expression on agonist-induced lung injury.
No genotypic differences in lung W/D weight ratio were seen in mice treated with saline alone (Fig. 3, A and B). Administration of TRAP significantly increased lung W/D weight ratio in NPR-A−/− mice. There was a trend toward an increase in lung W/D weight in NPR-A+/+ or NPR-A+/− mice given TRAP, but the difference was not statistically significant (Fig. 3A).
Fig. 3.
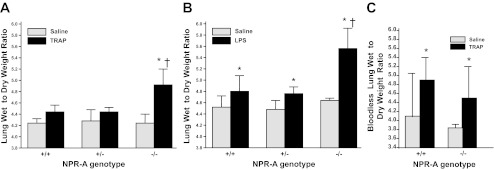
Lung wet-to-dry weight ratios in NPR-A wild-type and gene-targeted mice with agonist-induced lung injury. A: lung wet weight-to-dry weight ratios in NPR-A+/+, NPR-A+/−, and NPR-A−/− mice treated with the thrombin receptor agonist peptide (TRAP; 4 mg/kg) or equal volume of saline. Values are means ± SD; n = 5–6 per group. *P < 0.001 vs. NPR-A−/− mice given saline vehicle. †P < 0.01 vs. NPR-A+/+ and NPR-A+/− mice given TRAP. B: lung wet-to-dry weight ratios in NPR-A+/+, NPR-A+/−, and NPR-A−/− mice treated with lipopolysaccharide (LPS; 2.5 mg/kg) or equal volume of saline. Values are means ± SD; n = 6 per group. *P < 0.001 vs. mice of same genotype given saline vehicle. †P < 0.01 vs. NPR-A+/+ and NPR-A+/− mice given LPS. C: bloodless lung wet weight-to-dry weight ratios in NPR-A+/+ and NPR-A−/− mice treated with TRAP (4 mg/kg) or equal volume of saline. Values are means ± SD; n = 4–6 per group. *P < 0.05 vs. saline.
To determine whether the effect of NPR-A genotype on lung W/D weight ratio was unique to thrombin-induced injury, we repeated the above experiments using an LPS model of acute lung injury. LPS increased lung W/D weight in all NPR-A genotypes, compared with saline vehicle (Fig. 3B). As observed with TRAP administration, W/D weight ratio was greater in NPR-A−/− mice treated with LPS, than in NPR-A+/+ or NPR-A+/− mice (Fig. 3B). There was a statistically significant interaction between NPR-A genotype and TRAP and NPR-A genotype and LPS on lung W/D weight ratio (P = 0.018 and 0.001, respectively). To determine whether genotype-related differences in lung W/D weight response to TRAP or LPS were due to differences in lung blood volume, additional experiments were conducted using bloodless W/D weight ratios. Administration of TRAP increased bloodless W/D ratios in both genotypes, but the TRAP-induced increase in bloodless lung W/D ratios was no greater in NPR-A−/− mice than in NPR+/+ mice (Fig. 3C). There was no significant interaction between NPR-A genotype and TRAP on bloodless lung W/D weight ratio.
Effect of NPR-A genotype on alveolar protein and cell concentration.
To determine whether increases in lung W/D weight ratio were associated with increases in permeability to intravascular proteins and cells, we next examined the effect of NPR-A expression on LPS-induced changes in total protein and cell concentration in BAL fluid. LPS increased protein concentration of BAL fluid in all NPR-A genotypes; however, the interaction between NPR-A genotype and protein concentration was not statistically significant (Fig. 4A). LPS increased BAL fluid cell count in all NPR-A genotypes. Cell count of BAL fluid was greater in NPR-A−/− than in NPR-A+/+ or NPR-A+/− mice (Fig. 4B). There was a significant interaction between genotype and LPS on cell count of BAL fluid (P = 0.009).
Fig. 4.

LPS increases protein and cell concentration in bronchoalveolar lavage fluid (BALF) of NPR-A wild-type and gene-targeted mice. A: protein concentration in BALF in NPR-A+/+, NPR-A+/−, and NPR-A−/− mice treated with LPS (2.5 mg/kg) or equal volume of saline. B: cell count in same BALF as shown in A. Values are means ± SD; n = 8–9 per group. *P < 0.005 vs. mice of same genotype given vehicle. †P < 0.001 vs. NPR-A+/+ and NPR-A+/− mice given LPS.
Effect of ANP administration on agonist-induced lung injury.
To determine whether the protective effects of ANP on lung injury are mediated through NPR-A, we next examined the effect of ANP administration on TRAP- and LPS-induced increases in lung W/D weight in NPR-A+/+ and NPR-A−/− mice. No genotypic differences in plasma ANP levels were seen at baseline. Bolus injection of ANP at doses of 2, 5, or 10 μg/kg given 30 min before TRAP did not raise plasma ANP levels at the end of the experiment or protect against TRAP-induced increases in lung W/D weight ratio (data not shown). To maintain elevated circulating ANP levels throughout the study period, mice were given a continuous infusion of ANP (360 ng·kg−1·min−1) or equal volume of saline 30 min before TRAP or LPS and continuing for 30 min afterwards. Intravenous infusion of ANP resulted in an increase in plasma ANP levels, compared with saline infusion, in both the NPR-A+/+ mice (202 ± 42 vs. 303 ± 169 pg/ml) and NPR-A−/− mice (199 ± 46 vs. 392 ± 168 pg/ml). Administration of TRAP + ANP did not further increase plasma ANP levels. Compared with saline, infusion of ANP for 60 min had no effect on lung W/D weight in either NPR-A genotype (Fig. 5). However, ANP infusion completely blocked TRAP-induced increases in lung W/D weight in both NPR-A+/+ and NPR-A−/− mice (Fig. 5). ANP infusion also blocked LPS-induced increases in lung W/D weight in NPR-A+/+ mice. There was a trend toward an ANP inhibition of LPS-induced increase in lung W/D weight in NPR-A−/− mice, but the difference was not statistically significant (Fig. 5). Infusion of ANP also decreased BAL fluid cell count and protein concentration in LPS-injured mice, and this effect was similar in NPR-A+/+ and NPR-A−/− mice (Fig. 6, A and B, respectively). To determine whether the protective effect of ANP on LPS-induced lung edema in NPR-A knockout mice was mediated by NPR-C, we repeated experiments using the NPR-C-specific ligand cANF. In both NPR-A+/+ and NPR-A−/− mice, TRAP administration caused a significant increase in lung W/D weight when animals were pretreated with saline, but not when they were treated with intravenous infusion of cANF (Fig. 7). Infusion of cANF also decreased BAL fluid cell counts and protein concentration following LPS-induced lung injury in both NPR-A+/+ and NPR-A−/− mice (Fig. 8, A and B, respectively).
Fig. 5.

ANP attenuates agonist-induced pulmonary edema in NPR-A wild-type and knockout mice. Effect of ANP infusion on lung wet-to-dry weight ratios in NPR-A+/+ or NPR-A−/− mice treated with saline, TRAP, or LPS in the presence or absence of ANP is shown. Values are means ± SD; n = 5 per group, except for saline + ANP (n = 3). *P < 0.05 vs. TRAP + ANP. †P < 0.05 vs. LPS + ANP.
Fig. 6.

ANP attenuates LPS-induced increases in protein concentration and cell count of BALF in NPR-A wild-type and knockout mice. Protein (A) and cell (B) concentration in BALF in NPR-A+/+ and NPR-A−/− mice treated with infusion of ANP or saline vehicle before treatment with LPS (2.5 mg/kg intratracheally) are shown. Values are means ± SD; n = 5–8 per group. *P < 0.05 vs. saline + LPS-treated mice.
Fig. 7.

The NPR-C-specific ligand cANF attenuates TRAP-induced increases in lung wet weight-to-dry weight ratio. Effect of TRAP or saline vehicle on lung wet-to-dry weight ratios in NPR-A+/+ or NPR-A−/− mice pretreated with an infusion of saline or cANF is shown. Values are means ± SD; n = 3–6 per group. *P < 0.05 vs. saline + saline-treated mice.
Fig. 8.

cANF attenuates LPS-induced increases in protein concentration and cell count of BALF in NPR-A wild-type and knockout mice. Protein (A) and cell (B) concentration in BALF in NPR-A+/+ and NPR-A−/− mice treated with infusion of cANF or saline vehicle before treatment with LPS (2.5 mg/kg intratracheally) is shown. Values are means ± SD; n = 3–4 per group. *P < 0.05 vs. saline + LPS-treated mice.
Effect of ANP on thrombin-induced permeability of PMVEC.
To examine the role of intact NPR-A signaling on endothelial barrier function, we isolated PMVECs from NPR-A+/+ and NPR-A−/− mice. Baseline permeability, as assessed by measuring TER across PMVEC monolayers, was similar in PMVEC isolated from NPR-A+/+ and NPR-A−/− mice (711 ± 128 vs. 665 ± 113 Ω for NPR-A+/+ and NPR-A−/− cells, respectively; P = 0.19). TER fell to a similar degree in response to thrombin (Fig. 9). When the change in resistance in all experiments was normalized to TER at the time of thrombin administration, maximal TER response to thrombin in PMVECs treated with ANP was ∼85% of that seen in cells treated with saline vehicle. This was true for PMVEC isolated from both NPR-A+/+ and NPR-A−/− mice.
Fig. 9.

ANP protects against agonist-induced permeability of endothelial cells isolated from both NPR-A+/+ and NPR-A−/− mice. Resistance across monolayers of PMVEC isolated from NPR-A+/+ and NPR-A−/− mice was assessed following exposure to thrombin (2 U/ml) and/or ANP (1 μM) (or corresponding vehicles). Values are means ± SD; n = 17–19. *P < 0.05 vs. PMVEC treated with thrombin.
DISCUSSION
Numerous studies have demonstrated the ability of ANP and to a lesser extent, BNP, to protect against acute lung injury in intact animals subjected to a variety of lung injuries (3a, 7, 19, 20, 22, 31, 34, 39, 43, 47, 49). As early as 1987, Inomata et al. (20) demonstrated that ANP could ameliorate pulmonary edema formation in isolated pig lungs exposed to arachidonic acid. They concluded that the protective effect of ANP was receptor mediated because the concentration of ANP needed to blunt edema formation was similar to the dissociation constant of ANP binding to lung homogenates. However, the receptor responsible for mediating the protective effect of ANP was not identified, and few studies to date have examined the roles of the various NPRs in modulating pulmonary vascular permeability.
Previous studies showing that the permeability of systemic and PVECs in vitro can be reduced by the administration of cell-permeant analogs of cGMP (10, 35, 43, 46) suggested a role for NPR-A in mediating the inhibitory effect of ANP on lung edema formation. However, most of these studies were done with cells isolated from larger conduit vessels. In a previous study of rat PMVEC, we saw no significant inhibitory effect of 8-bromo-cGMP on thrombin-induced permeability (25). Conversely, cGMP analogs have been reported to blunt H2O2-induced permeability in bovine PMVEC (38), and studies in isolated mouse lungs found that the NPR-A antagonist, antanin, protected against pulmonary edema induced by ischemia-reperfusion (9), suggesting that NPR-A signaling may worsen lung injury in this model. Thus the role of cGMP and NPR-A in mediating the protective effect of ANP on agonist-induced pulmonary endothelial permeability remains unclear.
In the present study, we sought to better understand the role of NPR-A signaling by examining acute lung injury in mice with disrupted expression of the gene for NPR-A. Loss of functional NPR-A signaling was demonstrated in knockout mice by the lack of NPR-A transcript expression in lung and isolated PMVEC, increased cardiac mass characteristic of the NPR-A knockout phenotype (37), and loss of an increase in intracellular cGMP synthesis in response to ANP. In response to saline alone, we found no difference in lung edema formation, as assessed by lung W/D weight ratio and protein and cell concentration of BAL fluid between mice with two, one, or zero copies of a functional NPR-A gene. However, in response to intravenous administration of the thrombin receptor agonist, TRAP, and intratracheal administration of LPS, lung W/D weight was greater in NPR-A knockout mice than in wild-type or heterozygote mice. LPS-induced increases in BAL cell counts were also greater in mice with disrupted expression of NPR-A.
These findings were similar to previous reports showing that LPS and Staphylococcus aureus induce greater lung injury and edema formation in ANP knockout mice than in wild-type mice (5, 50). In those studies, pretreatment with ANP attenuated LPS- or staphylococcus-induced lung injury, demonstrating that ANP-deficient mice could be rescued by administration of exogenous ANP. However, the receptor by which ANP confers its protective effects against agonist-induced lung injury was not examined. Initially, our findings demonstrating agonist-induced lung edema was worse in NPR-A-deficient mice suggested that ANP may be signaling via NPR-A. However, several phenotypic differences between NPR-A wild-type and knockout mice may contribute to differences in lung injury. For example, NPR-A is expressed in alveolar epithelial cells, and NPR-A knockout mice could have greater lung W/D weight due to impaired resorption of alveolar fluid rather than increased permeability. Previous studies have shown that NPR-A knockout mice have greater plasma volume and higher pulmonary arterial pressures than wild-type mice (26, 27, 37, 51). NPR-A knockout mice also lose their ability to increase hematocrit in response to ANP (42). These differences could affect lung W/D weight ratios because hemoglobin contributes to lung dry weight. Indeed, we found no difference in TRAP-induced pulmonary edema between NPR-A+/+ and NPR-A−/− mice when lung edema formation was measured using bloodless lung W/D weight. We also found no significant difference in LPS-induced increases in BAL protein concentration between NPR-A+/+ and NPR-A−/− mice, although BAL cell counts were greater in NPR-A−/− mice. Taken together, it is unclear if disrupted NPR-A expression predisposes to greater lung injury in response to the agents used in this study. However, future studies that examine the role of NPRs in ameliorating acute lung injury will need to consider the effects of these receptors on blood volume and vascular tone.
To further examine the role of NPR-A in mediating the protective effects of ANP on acute lung injury, we administered ANP to mice with intact and disrupted NPR-A signaling during administration of TRAP or LPS. ANP infusion completely abolished TRAP-induced increases in lung W/D weight in NPR-A knockout and wild-type mice. There was also a trend toward ANP inhibition of LPS-induced lung edema in the NPR-A knockout mice. This could have been due to the acute effects of ANP on reduction in plasma volume. However, in light of previous studies demonstrating that the effects of ANP on decreasing plasma volume and intravascular pressure are mediated by NPR-A (17, 42), it is unlikely that the protective effect of ANP on lung edema formation observed in NPR-A knockout mice in the present study was due to a reduction in hydrostatic forces. This hypothesis was supported by our studies showing that ANP was equally effective at blunting thrombin-induced increases in permeability in PMVEC isolated from NPR-A knockout and wild-type mice in vitro. We also found a significant effect of ANP on reducing BAL protein and cell count in LPS-injured animals. This effect was at least as effective in NPR-A−/− mice as it was in NPR-A+/+ mice. Our findings suggest that ANP has an in vivo inhibitory effect on TRAP- and LPS-induced increases in acute lung injury that cannot be explained by effects on hydrostatic forces and is mediated via NPR-A independent pathway.
The ability of ANP to protect against agonist-induced lung injury in the absence of NPR-A strongly suggests that it is acting via NPR-C. Although it is possible that the protective effects of ANP in NPR-A knockout mice could be mediated through NPR-B, this seems unlikely for several reasons: 1) the binding affinity of this receptor for ANP is at least three orders of magnitude less than that for NPR-C (28); 2) the elevation in circulating ANP levels achieved by the dose of ANP used in our experiments was less than twofold above baseline; 3) we found no evidence of increased NPR-B or NPR-C expression in whole lung homogenates and lung microvascular cell cultures obtained from NPR-A knockout mice; and 4) ANP produced no discernible increase in intracellular cGMP levels in PMVEC isolated from NPR-A knockout mice. Furthermore, in earlier studies using rat PMVEC, we found only a mild protective effect of C-type natriuretic peptide, the primary ligand for NPR-B, despite significant increases in intracellular cGMP concentrations (25).
To further test the hypothesis that the protective effects of ANP in NPR-A knockout mice are mediated by NPR-C, we performed additional experiments with the NPR-C-specific ligand, cANF. Infusion of cANF blocked TRAP-induced increases in lung W/D ratio in NPR-A+/+ and NPR-A−/− mice and decreased BAL fluid cell count and protein concentration in NPR-A+/+ and NPR-A−/− mice with LPS-induced lung injury. The finding that the protective effect of cANF was similar in both NPR-A genotypes suggested that NPR-C played an important role in protecting against agonist-induced lung injury, even in genetically unaltered mice. These findings strongly suggest that NPR-C plays an important role in mediating the protective effects of ANP on agonist-induced lung injury. Findings from our study add to those of an earlier study by Hempel et al. (16) in which cANF attenuated isoproterenol-induced increases in the permeability of rat coronary artery endothelial cells. In that study, cANF also inhibited isoproterenol-induced increases in endothelial cAMP levels. Both effects of cANF were about one-half that of the same molar concentration of ANP and blocked by pertussis toxin, suggesting that they were mediated via a NPR-C-Gi-protein-coupled mechanism. Taken together, these findings suggest that ANP can act via NPR-C to blunt agonist-induced endothelial barrier dysfunction and explain the efficacy of ANP in protecting against acute lung injury in NPR-A knockout mice.
The signal transduction pathway by which NPR-C may mediate pulmonary endothelial barrier function is unclear. NPR-C lacks an intracellular guanylate cyclase domain, but several investigators have reported evidence of biological activity, including inhibition of adenylate cyclase and activation of phospholipase C (2, 36). Previous studies describing a NPR-C/cAMP-mediated pathway for inhibition of vascular smooth muscle proliferation and hypertrophy support the hypothesis that some biological effects of ANP are mediated via NPR-C (15, 30). Recent studies suggest that the protective effects of ANP on pulmonary vascular permeability may be mediated by the small Rho GTPases, including Rac and its cytoskeletal effector p21 activated kinase (5, 6, 25, 49). ANP has been shown to be a novel endogenous activator of Rac1 in human microvascular endothelial cells (14). Activity of the Rho GTPases is mediated by cAMP via exchange protein directly activated by cAMP (18). Thus it is possible that the effects of ANP on PMVEC permeability are mediated via NPR-C modulation of intracellular cAMP levels.
The role of NPR-C in protecting against pulmonary endothelial barrier dysfunction may be particularly important, considering that it is the most abundant NPR in the lung (24). Furthermore, pulmonary expression of NPR-C and ANP binding to NPR-C in the lung are markedly decreased during exposure to chronic hypoxia (23, 29), suggesting that pulmonary NPR-C may be downregulated in hypoxic lung diseases. Thus, a decrease in pulmonary NPR-C expression during the development of acute lung injury could impair the ability of ANP to protect against lung edema formation.
A potential role for ANP in the treatment of human lung injury has not been established. The protective effect of ANP on LPS-induced lung injury in the present study was achieved with an approximate doubling of plasma ANP levels, well within the physiological range of plasma ANP levels and the eightfold increase reported in patients with ARDS (11). Clinical trials of ANP in patients with acute lung injury are limited. Mikata et al. (34) found a significant increase in arterial Po2/fraction of inspired O2 and thoracic compliance and a decrease in lung injury score and shunt fraction in response to ANP in patients with ARDS, whereas Bindels et al. (4) saw no change in extravascular lung water index or in pulmonary gas exchange. However, in the latter study, the dose of ANP and duration of infusion were considerably smaller than in the former. Further studies are needed to better elucidate the timing of administration and dose of ANP or BNP needed to protect against acute lung injury. In the meantime, the present study suggests that the protective effects of ANP on pulmonary endothelial barrier function are not mediated solely by NPR-A and cGMP-dependent mechanisms. Further studies that examine the role of NPR-C in pulmonary endothelial barrier function may provide novel insights into the pathogenesis and treatment of acute lung injury.
GRANTS
This material is the result of work supported with resources and the use of facilities at the Providence VA Medical Center and supported with National Heart, Lung, and Blood Institute Grants HL-088328 and HL-088328-03S1 (J. R. Klinger), VA Merit Review and HL67795 Grants (E. O. Harrington), and T32 HL094300 (supported K. L. Grinnell).
DISCLAIMER
The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.R.K. and E.O.H. conception and design of research; J.R.K., S.G., K.L.G., J.T.M., and E.O.H. analyzed data; J.R.K., S.-W.T., J.T.M., and E.O.H. interpreted results of experiments; J.R.K. and K.L.G. drafted manuscript; J.R.K. and E.O.H. edited and revised manuscript; J.R.K., S.-W.T., S.G., K.L.G., and E.O.H. approved final version of manuscript; S.-W.T. and S.G. performed experiments; K.L.G. and E.O.H. prepared figures.
ACKNOWLEDGMENTS
Some of these results were presented at the 2010 and 2012 American Thoracic Society International Meetings and were published in abstract form (23a, 23b).
REFERENCES
- 1. Almeida FA, Suzuki M, Maack T. Atrial natriuretic factor increases hematocrit and decreases plasma volume in nephrectomized rats. Life Sci 39: 1193–1199, 1986 [DOI] [PubMed] [Google Scholar]
- 2. Anand-Srivastava MB, Sehl PD, Lowe DG. Cytoplasmic domain of natriuretic peptide receptor-C inhibits adenylyl cyclase. Involvement of a pertussis toxin-sensitive G protein. J Biol Chem 271: 19324–19329, 1996 [DOI] [PubMed] [Google Scholar]
- 3. Atkinson B, Lim Y, Zervoglos M. Isolation of Mouse Lung Endothelial Cells (MLEC)/Mouse Heart Endothelial Cells (MHEC) (Online) Boston, MA: Brigham and Women's Hospital; http://vrd.bwh.harvard.edu/core_facilities/mlec.html [October 2004]. [Google Scholar]
- 3a. Baron DA, Lofton CE, Newman WH, Currie MG. Atriopeptin inhibition of thrombin-mediated changes in the morphology and permeability of endothelial monolayers. Proc Natl Acad Sci U S A 86: 3394–3398, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bindels AJ, van der Hoeven JG, Groeneveld PH, Frolich M, Meinders AE. Atrial natriuretic peptide infusion and nitric oxide inhalation in patients with acute respiratory distress syndrome. Crit Care 5: 151–157, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birukova AA, Xing J, Fu P, Yakubov B, Dubrovskyi O, Fortune JA, Klibanov AM, Birukov KG. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol 299: L652–L663, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol 215: 715–724, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchan KW, Martin W. Modulation of barrier function of bovine aortic and pulmonary artery endothelial cells: dissociation from cytosolic calcium content. Br J Pharmacol 107: 932–938, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Curry FR. Atrial natriuretic peptide: an essential physiological regulator of transvascular fluid, protein transport, and plasma volume. J Clin Invest 115: 1458–1461, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dodd-o JM, Hristopoulos ML, Kibler K, Gutkowska J, Mukaddam-Daher S, Gonzalez A, Welsh-Servinsky LE, Pearse DB. The role of natriuretic peptide receptor-A signaling in unilateral lung ischemia-reperfusion injury in the intact mouse. Am J Physiol Lung Cell Mol Physiol 294: L714–L723, 2008 [DOI] [PubMed] [Google Scholar]
- 10. Draijer R, Atsma DE, van der Laarse A, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res 76: 199–208, 1995 [DOI] [PubMed] [Google Scholar]
- 11. Eison HB, Rosen MJ, Phillips RA, Krakoff LR. Determinants of atrial natriuretic factor in the adult respiratory distress syndrome. Chest 94: 1040–1045, 1988 [DOI] [PubMed] [Google Scholar]
- 12. Fluckiger JP, Waeber B, Matsueda G, Delaloye B, Nussberger J, Brunner HR. Effect of atriopeptin III on hematocrit and volemia of nephrectomized rats. Am J Physiol Heart Circ Physiol 251: H880–H883, 1986 [DOI] [PubMed] [Google Scholar]
- 13. Fukuda N, Folkesson HG, Matthay MA. elationship of interstitial fluid volume to alveolar fluid clearance in mice: ventilated vs. in situ studies. J Appl Physiol 89:672–679, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Furst R, Brueckl C, Kuebler WM, Zahler S, Krotz F, Gorlach A, Vollmar AM, Kiemer AK. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res 96: 43–53, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Hashim S, Li Y, Anand-Srivastava MB. Small cytoplasmic domain peptides of natriuretic peptide receptor-C attenuate cell proliferation through Giα protein/MAP kinase/PI3-kinase/AKT pathways. Am J Physiol Heart Circ Physiol 291: H3144–H3153, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Hempel A, Noll T, Bach C, Piper HM, Willenbrock R, Hohnel K, Haller H, Luft FC. Atrial natriuretic peptide clearance receptor participates in modulating endothelial permeability. Am J Physiol Heart Circ Physiol 275: H1818–H1825, 1998 [DOI] [PubMed] [Google Scholar]
- 17. Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A 99: 7142–7147, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol 577: 5–15, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imamura T, Ohnuma N, Iwasa F, Furuya M, Hayashi Y, Inomata N, Ishihara T, Noguchi T. Protective effect of alpha-human atrial natriuretic polypeptide (alpha-hANP) on chemical-induced pulmonary edema. Life Sci 42: 403–414, 1988 [DOI] [PubMed] [Google Scholar]
- 20. Inomata N, Ohnuma N, Furuya M, Hayashi Y, Kanai Y, Ishihara T, Noguchi T, Matsuo H. Alpha-human atrial natriuretic peptide (alpha-hANP) prevents pulmonary edema induced by arachidonic acid treatment in isolated perfused lung from guinea pig. Jpn J Pharmacol 44: 211–214, 1987 [DOI] [PubMed] [Google Scholar]
- 21. Irwin DC, Rhodes J, Baker DC, Nelson SE, Tucker A. Atrial natriuretic peptide blockade exacerbates high altitude pulmonary edema in endotoxin-primed rats. High Alt Med Biol 2: 349–360, 2001 [DOI] [PubMed] [Google Scholar]
- 22. Irwin DC, Tissot van Patot MC, Tucker A, Bowen R. Direct ANP inhibition of hypoxia-induced inflammatory pathways in pulmonary microvascular and macrovascular endothelial monolayers. Am J Physiol Lung Cell Mol Physiol 288: L849–L859, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Klinger JR, Arnal F, Warburton RR, Ou LC, Hill NS. Downregulation of pulmonary atrial natriuretic peptide receptors in rats exposed to chronic hypoxia. J Appl Physiol 77: 1309–1316, 1994 [DOI] [PubMed] [Google Scholar]
- 23a. Klinger JR, Green S, Grinnell K, Tsai S-W, Harrington EO. Gene-targeted disruption of NPR-A worsens agonist-induced lung injury in mice (Abstract). Am J Respir Crit Care Med 183: A1973, 2011 [Google Scholar]
- 23b. Klinger JR, Green S, Tsai S-W, Harrington EO. Natriuretic peptide receptor-C specific ligand cANF blunts agonist-induced acute lung injury (Abstract). Am J Respir Crit Care Med 185: A5517, 2012 [Google Scholar]
- 24. Klinger JR, Moalli R, Warburton RR, Wrenn DS, Hill NS. C-receptor ligand blocks pulmonary clearance of atrial natriuretic peptide in isolated rat lungs. Proc Soc Exp Biol Med 201: 154–158, 1992 [DOI] [PubMed] [Google Scholar]
- 25. Klinger JR, Warburton R, Carino GP, Murray J, Murphy C, Napier M, Harrington EO. Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp Cell Res 312: 401–410, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Klinger JR, Warburton RR, Pietras L, Oliver P, Fox J, Smithies O, Hill NS. Targeted disruption of the gene for natriuretic peptide receptor-A worsens hypoxia-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 282: H58–H65, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Klinger JR, Warburton RR, Pietras LA, Smithies O, Swift R, Hill NS. Genetic disruption of atrial natriuretic peptide causes pulmonary hypertension in normoxic and hypoxic mice. Am J Physiol Lung Cell Mol Physiol 276: L868–L874, 1999 [DOI] [PubMed] [Google Scholar]
- 28. Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H, Goeddel DV. Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science 252: 120–123, 1991 [DOI] [PubMed] [Google Scholar]
- 29. Li H, Oparil S, Meng QC, Elton TS, Chen YF. Selective downregulation of ANP clearance-receptor gene expression in lung of rats adapted to hypoxia. Am J Physiol Lung Cell Mol Physiol 268: L328–L335, 1995 [DOI] [PubMed] [Google Scholar]
- 30. Li Y, Hashim S, Anand-Srivastava MB. Intracellular peptides of natriuretic peptide receptor-C inhibit vascular hypertrophy via Gq alpha/MAP kinase signaling pathways. Cardiovasc Res 72: 464–472, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Lofton CE, Baron DA, Heffner JE, Currie MG, Newman WH. Atrial natriuretic peptide inhibits oxidant-induced increases in endothelial permeability. J Mol Cell Cardiol 23: 919–927, 1991 [DOI] [PubMed] [Google Scholar]
- 32. Maack T, Suzuki M, Almeida FA, Nussenzveig D, Scarborough RM, McEnroe GA, Lewicki JA. Physiological role of silent receptors of atrial natriuretic factor. Science 238: 675–678, 1987 [DOI] [PubMed] [Google Scholar]
- 33. Mitaka C, Hirata Y, Habuka K, Narumi Y, Yokoyama K, Makita K, Imai T. Atrial natriuretic peptide improves pulmonary gas exchange by reducing extravascular lung water in canine model with oleic acid-induced pulmonary edema. Crit Care Med 30: 1570–1575, 2002 [DOI] [PubMed] [Google Scholar]
- 34. Mitaka C, Hirata Y, Nagura T, Tsunoda Y, Amaha K. Beneficial effect of atrial natriuretic peptide on pulmonary gas exchange in patients with acute lung injury. Chest 114: 223–228, 1998 [DOI] [PubMed] [Google Scholar]
- 35. Moldobaeva A, Welsh-Servinsky LE, Shimoda LA, Stephens RS, Verin AD, Tuder RM, Pearse DB. Role of protein kinase G in barrier-protective effects of cGMP in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 290: L919–L930, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Murthy KS, Makhlouf GM. Identification of the G protein-activating domain of the natriuretic peptide clearance receptor (NPR-C). J Biol Chem 274: 17587–17592, 1999 [DOI] [PubMed] [Google Scholar]
- 37. Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A 94: 14730–14735, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pearse DB, Shimoda LA, Verin AD, Bogatcheva N, Moon C, Ronnett GV, Welsh LE, Becker PM. Effect of cGMP on lung microvascular endothelial barrier dysfunction following hydrogen peroxide. Endothelium 10: 309–317, 2003 [DOI] [PubMed] [Google Scholar]
- 39. Pedram A, Razandi M, Levin ER. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J Biol Chem 277: 44385–44398, 2002 [DOI] [PubMed] [Google Scholar]
- 40. Perreault T, Gutkowska J. Role of atrial natriuretic factor in lung physiology and pathology. Am J Respir Crit Care Med 151: 226–242, 1995 [DOI] [PubMed] [Google Scholar]
- 41. Potter LR, Hunter T. Guanylyl cyclase-linked natriuretic peptide receptors: structure and regulation. J Biol Chem 276: 6057–6060, 2001 [DOI] [PubMed] [Google Scholar]
- 42. Sabrane K, Kruse MN, Fabritz L, Zetsche B, Mitko D, Skryabin BV, Zwiener M, Baba HA, Yanagisawa M, Kuhn M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest 115: 1666–1674, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suttorp N, Hippenstiel S, Fuhrmann M, Krull M, Podzuweit T. Role of nitric oxide and phosphodiesterase isoenzyme II for reduction of endothelial hyperpermeability. Am J Physiol Cell Physiol 270: C778–C785, 1996 [DOI] [PubMed] [Google Scholar]
- 44. Suzuki T, Yamazaki T, Yazaki Y. The role of the natriuretic peptides in the cardiovascular system. Cardiovasc Res 51: 489–494, 2001 [DOI] [PubMed] [Google Scholar]
- 45. Wakabayashi G, Ueda M, Aikawa N, Naruse M, Abe O. Release of ANP and its physiological role in pulmonary injury due to HCl. Am J Physiol Regul Integr Comp Physiol 258: R690–R696, 1990 [DOI] [PubMed] [Google Scholar]
- 46. Westendorp RG, Draijer R, Meinders AE, van Hinsbergh VW. Cyclic-GMP-mediated decrease in permeability of human umbilical and pulmonary artery endothelial cell monolayers. J Vasc Res 31: 42–51, 1994 [DOI] [PubMed] [Google Scholar]
- 47. Westendorp RG, Roos AN, van der Hoeven HG, Tjiong MY, Simons R, Frolich M, Souverijn JH, Meinders AE. Atrial natriuretic peptide improves pulmonary gas exchange in subjects exposed to hypoxia. Am Rev Respir Dis 148: 304–309, 1993 [DOI] [PubMed] [Google Scholar]
- 48. Wijeyaratne CN, Moult PJ. The effect of alpha human atrial natriuretic peptide on plasma volume and vascular permeability in normotensive subjects. J Clin Endocrinol Metab 76: 343–346, 1993 [DOI] [PubMed] [Google Scholar]
- 49. Xing J, Birukova AA. ANP attenuates inflammatory signaling and Rho pathway of lung endothelial permeability induced by LPS and TNF alpha. Microvasc Res 79: 56–62, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xing J, Moldobaeva N, Birukova AA. Atrial natriuretic peptide protects against Staphylococcus aureus-induced lung injury and endothelial barrier dysfunction. J Appl Physiol 110: 213–224, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao L, Long L, Morrell NW, Wilkins MR. NPR-A-Deficient mice show increased susceptibility to hypoxia-induced pulmonary hypertension. Circulation 99: 605–607, 1999 [DOI] [PubMed] [Google Scholar]