

Abstract
A proposed key event in the pathogenesis of Alzheimer’s disease (AD) is the formation of neurotoxic amyloid β (Aβ) oligomers and amyloid plaques in specific brain regions that are affected by the disease. The main plaque component is the 42 amino acid isoform of Aβ (Aβ1-42), which is thought to initiate plaque formation and AD pathogenesis. Numerous isoforms of Aβ, e.g., Aβ1-42, Aβ1-40 and the 3-pyroglutamate derivate of Aβ3-42 (pGluAβ3-42), have been detected in the brains of sporadic AD (SAD) and familial AD (FAD) subjects. However, the relative importance of these isoforms in the pathogenesis of AD is not fully understood. Here, we report a detailed study using immunoprecipitation in combination with mass spectrometric analysis to determine the Aβ isoform pattern in the cerebellum, cortex and hippocampus in AD, including subjects with a mutation in the presenilin (M146V) or amyloid precursor protein (KM670/671NL) genes, SAD subjects and non-demented controls. We show that the dominating Aβ isoforms in the three different brain regions analyzed from control, SAD, and FAD are Aβ1-42, pGluAβ3-42, Aβ4-42 and Aβ1-40 of which Aβ1-42 and Aβ4-42 are the dominant isoforms in the hippocampus and the cortex in all groups analyzed, controls included. No prominent differences in Aβ isoform patterns between FAD and SAD patients were seen, underscoring the similarity in the amyloid pathology of these two disease entities.
Keywords: Alzheimer’s disease, Amyloid precursor protein, Brain, Immunoprecipitation, Mass spectrometry
Introduction
According to the amyloid cascade hypothesis, there is an imbalance in the production and clearance of amyloid beta (Aβ) leading to the formation of neurotoxic oligomers and larger assemblies of Aβ [15]. In Alzheimer’s disease (AD), Aβ forms amyloid plaques in specific regions in the brain, which has been considered a key element in the pathogenesis of AD [3, 6, 8]. Of all AD cases, the majority have the sporadic form (SAD) of unknown cause, while a minority (accounting for less than 1% of all AD cases) have the familial form (FAD) with an onset before the age of 65 years [17]. Most FAD cases are caused by autosomal dominant mutations in amyloid metabolism-associated genes. To date, more than 170 different AD-causing missense mutations have been identified in the amyloid precursor protein (APP) and presenilin 1 and 2 (PSEN1 and PSEN2, respectively) [4, 33].
Aβ is produced along the amyloidogenic pathway via cleavages of APP by β- and γ-secreatase [1]. β-Secretase is an aspartyl protease encoded by the β-site APP-cleaving enzyme 1 (BACE1) gene [18, 34, 37, 39] while γ-secretase is a protease complex consisting of at least four essential components of which the homologous presenilin 1 and 2 proteins (PSEN1 and PSEN2, respectively) constitute the active site [5]. Due to its imprecise cleavage preference, γ-secretase directly or indirectly mediates cleavage of Aβ at multiple sites between amino acids 17 and 42 [31]. This was shown by treating cells with a γ-secretase inhibitor with the result that all isoforms longer than and including Aβ1-17 were abolished while the shorter isoforms were expressed at even higher levels. In another pathway, APP is first cleaved by β-secretase followed by α-secretase, thus generating shorter isoforms, e.g., Aβ1-15 and Aβ1-16 [19, 31]. In yet another non-amyloidogenic pathway, α-secretase cleaves between amino acid 16 and 17 in the Aβ sequence generating α-sAPP followed by γ-secretase cleavages, generating a fragment called p3 (Aβ17-40/42) [7, 11, 12]. This isoform has been isolated from AD brains containing vast deposits of diffuse amyloid plaques and also from patients with Down’s syndrome, but it has not been detected in cerebrospinal fluid (CSF) [10, 20, 29].
Numerous isoforms of Aβ have previously been detected in the brains of SAD and FAD subjects [35]. However, the relative importance of these truncated isoforms in the pathogenesis of AD is not fully understood. Different studies of brain Aβ report that the most abundant isoform is either Aβ1-42 or Aβ1-40 while others have reported the 3-pyroglutamate derivate of Aβ3-42 (pGluAβ3-42) as one the most abundant isoforms [16, 24, 26, 27]. These divergent results may be due to that the different studies are studying different brain regions, different ages of the subjects and different stages of disease. Further, different protocols and techniques are used for analysing the different Aβ isoforms.
To date, no detailed mass spectrometric survey of the different Aβ isoforms present in different brain regions of SAD and FAD subjects has been performed. Proteomic studies using targeted approaches and mass spectrometry have previously led to the design of several new Alzheimer-related protein and peptide panels [40]. Here, we report the results of a study using immunoprecipitation (IP) in combination with matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) or nanoflow liquid chromatography (LC) electrospray (ESI) high resolution tandem mass spectrometry (MS/MS) to determine the Aβ isoform pattern in the cerebellum, cortex and hippocampus of FAD subjects harboring a mutation in the PSEN1 (M146V) or APP (KM670/671NL) gene, SAD subjects and non-demented controls.
Materials and methods
Patient characteristics
The brain samples from AD patients obtained from Huddinge Brain Bank at Krolinska Institute, Stockholm, fulfilled the clinical NINCDS criteria for probable AD [23] and met the neuropathological CERAD criteria for definitive AD [25]. Neuropathological controls were cases who had died from acute cardiac or malignant disease, without history of dementia, psychiatric or neurological diseases. The PSEN1 M146V mutation carriers had a history of AD traced trough four generations with an early age at onset (mid-late 30 s) of the disease and a rapid progression [14]. The Swedish APP double mutation (KM670/671NL) induces increased cleavage by β-secretase to generate more Aβ1-40 and Aβ1-42 [13, 21]. Detailed patient characteristics are given in Table 1. The study was conducted in accordance with the provisions of the Helsinki declaration and approved by the regional ethics committee.
Table 1.
Subject characteristics
| Subject ID | Diagnosis | Mutation | Age | Sex | PM | Plaque count
|
||
|---|---|---|---|---|---|---|---|---|
| Cerebellum | Hippocampus | Cortex | ||||||
| 1 | Control | NA | 80 | F | 7 | 0 | 0 | 1 |
| 2 | Control | NA | 64 | F | 5 | 0 | 0 | 0 |
| 3 | Control | NA | 81 | F | 25 | 0 | 1 | 1 |
| 4 | Control | NA | 81 | F | 3 | 0 | 2 | 4 |
| 5 | Control | NA | 80 | M | 13 | 0 | 0 | 0 |
| 6 | AD | NA | 78 | F | 12 | 0.5 | 16 | 29 |
| 7 | AD | NA | 70 | F | 12 | 0.5 | 20 | 32 |
| 8 | AD | NA | 80 | M | 12 | 1 | 24 | 35 |
| 9 | AD | NA | 83 | F | 4 | 0 | 5 | 9 |
| 10 | AD | NA | 79 | F | 7 | 0 | 5 | 10 |
| 11 | FAD | APP670/671 | 68 | M | <24 | 6 | 27 | 35 |
| 12 | FAD | APP670/671 | 66 | M | <24 | 2 | 28 | 28 |
| 13 | FAD | APP670/671 | 62 | M | 40 | 12 | 14 | 56 |
| 14 | FAD | APP670/671 | 56 | M | <24 | 3 | 24 | 36 |
| 15 | FAD | APP670/671 | 62 | F | <24 | 24 | 14 | 35 |
| 16 | FAD | PSEN1 M146 V | 48 | F | ? | 46 | 87 | 74 |
| 17 | FAD | PSEN1 M146 V | 50 | F | ? | 34 | 78 | 86 |
NA not applicable, PM postmortem, time in hours
Brain tissue
Brain tissue (cortex, hippocampus and cerebellum) from SAD (n = 5), APP670/671 (KM670/671NL, n = 5) and PSEN1 (M146V, n = 2) subjects and non-demented controls (n = 5) (Table 1) was homogenized on ice in Tris-buffered saline containing complete protease inhibitor (Roche Diagnostics GmBH, Mannheim, Germany) as described previously [30]. Briefly, formic acid (FA) was added (final concentration 70%) followed by further homogenization, sonication and centrifugation (30,000g, 1 h, +4°C). The supernatant was collected and dried in a vacuum centrifuge. The dried fraction was dissolved in 70% FA followed by centrifugation (30,000g, 1 h, +4°C). Before IP, the supernatant was neutralized using 0.5 M Tris.
Immunoprecipitation
The IP using the KingFisher magnetic particle processor (Thermo Scientific, Waltman, MA, USA) was performed as described earlier with some modifications [32]. Briefly, an aliquot (8 μL, 1 mg/mL) of the Aβ specific antibodies 6E10 and 4G8 (epitope 4–9 and epitope 18–22, respectively, Signet Laboratories, Inc., Dedham, MA, USA) was separately added to 50 μL Dynabeads M-280 (Dynal®) sheep anti-mouse according to the manufacturer’s product description. The washed beads with bound antibody (50 μL 6E10 and 50 μL 4G8) were combined and used for immunoprecipitation of the neutralized FA fraction to which Tween-20 (Bio-Rad Laboratories Inc., end concentration 0.025%) was added and incubated.
The beads/FA fraction was transferred to a KingFisher magnetic particle processor (polypropylene tubes, Thermo Scientific) for automatic washing and elution of the Aβ peptides. The collected supernatant was dried in a vacuum centrifuge and redissolved in 5 μl 0.1% FA in 20% acetonitrile. The Aβ peptides were analyzed using MALDI-TOFMS and nanoflow LC-MS/MS. All solvents used were of HPLC quality. Control experiments included IP with only beads (pre-coated with sheep anti mouse IgG from the manufacturer) and beads with coupled IgG from mouse serum.
Mass spectrometry
MALDI-TOFMS measurements were performed using an AUTOFLEX instrument (Bruker Daltonics, Bremen, Germany) operating in linear or reflector mode. Each spectrum represents an average of 1,500 shots acquired 75 at a time. The MALDI samples were prepared with the seed layer method as described previously [29] with α-cyano-4-hydroxycinnamic acid (CHCA) used as matrix. The sequence of Aβ peptides best matching the molecular mass obtained with MALDI-TOFMS was evaluated with an in-house developed software (PeptideMassCalculator).
The Aβ levels are expressed as MS signal/mg tissue, calculated from the original brain weight (40–250 mg) measured before homogenization. A MS signal/mg tissue <100 is called small, 100–500 is called minor, 500–1,000 is called intermediate and a MS signal/mg tissue>1,000 is called major.
LC-MS/MS was conducted by nanoflow liquid chromatography coupled to electrospray ionization Fourier transform ion cyclotron resonance tandem mass spectrometry (LC-ESI-FTICR-MS/MS) with an Ettan MDLC (GE Healthcare, Uppsala, Sweden) coupled to an LTQ-FT Ultra (ThermoFisher Scientific, Bremen, Germany), a hybrid linear quadrupole ion trap-Fourier transform ion cyclotron resonance mass spectrometer equipped with a 7 T magnet.
Quantification of neuropathology
Sample processing and plaque counts on all the brain tissues used in the present study were performed as described previously in detail [2]. In short, brain tissue was processed within 24–48 h of death. The right hemisphere was fixed in buffered 4% formaldehyde for 4 weeks. Blocks were embedded with paraffin, and stained using the Bielschowsky method. The number of plaques was counted within superficial layers 1–3 and deep layers 4–6 for 11 cortical areas and for 12 subcortical structures using the Olympus Video Stereological Analysis System (BICO, Copenhagen) and the GRID v2.0 software (Interactivision ApS, Silkeborg). Plaque counts are given as number of plaques per square millimeter. In the cortex neuritic plaques, while in the cerebellum and basal ganglia diffuse plaques were counted. List of the regions used is described elsewhere [2].
Results
In general, the most abundant isoforms throughout all different brain regions analyzed were Aβ1-40, Aβ1-42, Aβ4-42 and pGluAβ3-42. See Fig. 1a–c for representative mass spectra displaying the Aβ isoform pattern from the cortex, hippocampus and cerebellum from a FAD subject.
Fig. 1.

Representative mass spectra from a sporadic AD patient displaying the Aβ isoform patterns from three different brain regions; the cerebellum, hippocampus and cortex. 1 Aβ4-40 [M+2H]2+, 2 Aβ4-42 [M+2H]2+, 3 Aβ1-40 [M+2H]2+ and 4 Aβ1-42 [M+2H]2+. Asterisk represents an unidentified peak
Control brains
In the cerebellum, only one out of five subjects displayed four minor peaks corresponding to Aβ (Fig. 2). In the hippocampus, another subject displayed two major peaks corresponding to Aβ1-42 and Aβ4-42 and four small peaks corresponding to Aβ2-42, Aβ3-42, pGluAβ3-42 and Aβ5-42. The major isoforms detected in the cortex were Aβ1-42 and Aβ4-42, constituting ~80% of all Aβ isoforms detected (Fig. 6a). In one of the subjects, no peaks corresponding to Aβ isoforms were detected. Notably, Aβ1-40 was not detected in the cortex, hippocampus or cerebellum.
Fig. 2.

Chart displaying the distribution of Aβ isoforms in the cerebellum, hippocampus and cortex of control subjects. A light blue box a small peak, a blue box minor peak, a dark blue/white box an intermediate peak and a dark blue/red box a major peak. Plaque count = lesions/m2
Fig. 6.
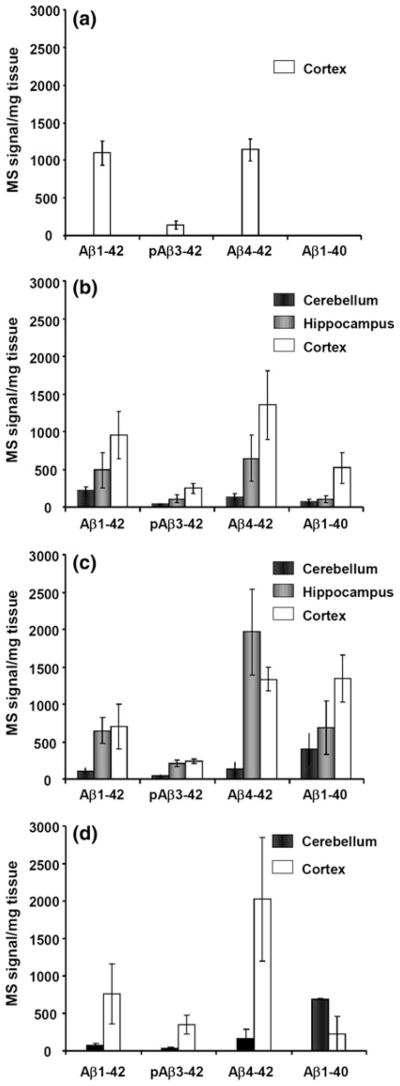
The MS signal/mg tissue is displayed for a controls, b SAD, c APP670/671 and d PSEN1 patients. The error bars represent one standard of the mean. It should be noted that the ratio between the different isoforms detected in the mass spectrum cannot be interpreted as a direct reflection of their absolute or relative abundance in the brain since the ionization efficiency might be different for the different isoforms and since different isoforms are more hydrophobic and less soluble than others
SAD brains
All SAD subjects, except one, displayed various Aβ isoforms in all different brain regions analyzed (Fig. 3). In the cerebellum, all of the Aβ isoforms detected were minor except Aβ1-42 and Aβ4-42, which were small in three of the subjects. In the hippocampus, Aβ4-42 and Aβ1-42 were intermediate while Aβ1-40 and pGluAβ3-42 were minor (Fig. 6b). In the cortex, Aβ4-42 and Aβ1-42 constituted ~60% of all Aβ isoforms detected while Aβ1-40, constituting ~18% of all Aβ isoforms detected, were intermediate and pGluAβ3-42 was minor (Fig. 6b). In the three brain regions analyzed, the MS signal/mg tissue of Aβ4-42 and Aβ1-42 was higher compared to Aβ1-40 (Fig. 6b).
Fig. 3.

Chart displaying the distribution of Aβ isoforms in the cerebellum, hippocampus and cortex of SAD patients. A light blue box a small peak, a blue box minor peak, a dark blue/white box an intermediate peak and a dark blue/red box a major peak. Plaque count = lesions/m2
APP670/671 brains
In the cortex, most of the subjects displayed major or intermediate peaks corresponding to Aβ4-40, Aβ4-42, Aβ1-40 and Aβ1-42, of which the dominant Aβ isoforms were Aβ4-42 and Aβ1-40, constituting ~54% of all Aβ isoforms detected (Fig. 4). The hippocampus from two of the subjects was not available, but the Aβ isoform pattern from the remaining three showed distinct peaks corresponding to Aβ4-42, Aβ1-40, Aβ1-42 and pGluAβ3-42. Aβ4-42 was the most abundant isoform in the hippocampus while Aβ1-40 was the most abundant isoform in the cerebellum (Fig. 6c). Aβ1-42 and pGluAβ3-42 were equally abundant in the hippocampus and cortex. Interestingly, one of the subjects (subject 12, see Fig. 4) also had peaks corresponding to Aβ1-39, Aβ1-38 and Aβ1-37 in all brain regions. These isoforms were not detected in any other subjects.
Fig. 4.

Chart displaying the distribution of Aβ isoforms in the cerebellum, hippocampus and cortex of APP670/671 patients. A light blue box a small peak, a blue box minor peak, a dark blue/white box an intermediate peak and a dark blue/red box a major peak. Plaque count = lesions/m2
PSEN1 brains
The hippocampi were not available from the PSEN1 M146V subjects. The remaining brain regions displayed numerous Aβ isoforms with Aβ1-42, pGluAβ3-42 Aβ4-42 and Aβ1-40 as the dominant isoforms (Fig. 5). Aβ4-42 was the dominant isoform in the cortex constituting ~48% of all Aβ isoforms detected. This isoform was ~9 times more abundant compared to Aβ1-40 and ~3 times more abundant than Aβ1-42 (Fig. 6d). In the cerebellum, Aβ1-40 was the most abundant isoform, ~9 times more abundant compared to Aβ1-42 and ~4 times more abundant compared to Aβ4-42.
Fig. 5.

Chart displaying the distribution of Aβ isoforms in the cerebellum, hippocampus and cortex of PSEN1 patients. A light blue box a small peak, a blue box minor peak, a dark blue/white box an intermediate peak and a dark blue/red box a major peak. Plaque count = lesions/m2
Correlation of Aβ isoform results with plaque counts
In general, the detected Aβ isoform signals corresponded well to neuropathological plaque counts. The one control with major Aβ peaks in the hippocampus also had high plaque counts in this brain region (subject 4, Fig. 2). Another control with no detectable Aβ in the mass spectra had no plaques in the brain (subject 5, Fig. 2). With regards to the AD brains, patients with many plaques all had major or intermediate Aβ peaks in the mass spectra of the corresponding brain region (Figs. 3, 4, 5).
Discussion
Here, we describe a detailed study of Aβ and its isoforms in different brain regions from different SAD, FAD and control subjects. Using 70% FA in the extraction protocol, we extracted most isoforms of Aβ present in the brain tissue. Overall, for all diagnoses, controls included, the cortex contained the most Aβ isoforms followed by hippocampus and cerebellum. This is in agreement with the view that plaque pathology begins with Aβ deposits in the neocortex, extending to the hippocampus and finally to the cerebellum [36]. Aβ deposition was frequently detected in the cortices of the controls, but was rarely seen in the cerebellum and hippocampus. Previous studies have shown that some elderly people with no AD symptoms have Aβ deposits [22, 26], which is in agreement with the present study.
In the cortex of APP670/671 subjects, Aβ1-40 was the most abundant isoform (accounting for ~27% of all Aβ isoforms detected), whereas it was the third most abundant isoform in the cortex from SAD subjects and absent in the cortex from control subjects. Of the two PSEN1 subjects, one did not have any Aβ1-40. Thus, Aβ1-40 retention in the brain might be an AD-specific phenomenon rarely seen in controls.
The major Aβ isoforms detected in the different brain regions analyzed from control, SAD, and FAD subjects were Aβ1-42, pGluAβ3-42, Aβ4-42 and Aβ1-40 with the exception that Aβ1-40 was not detected in the brain regions from controls. These isoforms also displayed an additional peak, which had the mass of the peptide plus 16 Da, corresponding to the oxidized form. In the cortex, most of the SAD and FAD patients also displayed minor or small peaks corresponding to pGluAβ11-42 and pGluAβ3-40 while pGluAβ3-40 was absent in the cortex from control subjects. Studies have suggested that pGluAβ3-42 has a higher aggregation propensity and shows increased toxicity compared to Aβ1-42 and that pGluAβ appears in early stages of AD. Further, it has also been suggested that Aβ4-42 has faster aggregation kinetics than the intact Aβ1-42 and that N-terminal deletions generally enhance aggregation of Aβ into neurotoxic, β-sheet fibrils in a manner that may initiate and/or nucleate the pathological deposition of Aβ [28]. Even though there are suggestions (e.g., neprilysin and plasmin), the enzymes responsible for the N-terminal cleavages remain to be verified [1]. Yet another minor peak detected in the hippocampus and cortex from most of the patients corresponded to the mass of Aβ1-43. This isoform has recently been identified to be presented in plaque cored sodium dodecyl sulfate preparations from both SAD as well as FAD cases [38].
Aβ17-40/42 (p3) was not detected in any of the brain regions analyzed. Earlier reports have shown its presence in AD brains containing vast deposits of diffuse amyloid plaques and also from patients with Down’s syndrome [10, 20]. One can argue that this study did not analyze diffuse amyloid plaques and/or the soluble fractions of the brain homogenate. However, p3 is a hydrophobic peptide that should be present in plaques extracted using FA. Importantly, we have previously shown that synthetic p3 can be retrieved from CSF upon spiking and that it can be analyzed using the MS method employed in the present work [29]. It has previously been suggested that the p3 fragment actually might be Aβ11-42 [9], which is closer in mass to 3 kDa (3,334 Da and 3,153 Da for Aβ11-42 and Aβ11-40, respectively) than the weight of p3 (2,578 Da and 2,394 Da for Aβ17-42 and Aβ17-40, respectively). Notably, many studies that have detected and analyzed the p3 fragment have used antibodies reactive to epitopes within the amino acid sequence 17–42 of Aβ followed of tryptic digestion. However, if the antibody is not end-specific for the N-terminus of p3, it also recognizes Aβ11-42. Tryptic digestion of this peptide will generate a Aβ17-28 fragment, thus the N-terminus will start after the proposed α-secretase cleavage site. Altogether, the data presented here suggest that p3, if existing at all, is a minor Aβ isoform.
We recently identified a set of 18N- and C-terminally truncated Aβ peptides in CSF along with additionally 11 APP/Aβ peptides starting N-terminally of the β-secretase site using IP-MS. These shorter isoforms, e.g., Aβ1-16/17, were not detected in this study. This might be due to that either they are not present in the brain due to their more hydrophilic nature or the method used is not sensitive enough for detecting these shorter isoforms.
It should be noted that the ratio between the different isoforms detected in the mass spectrum cannot be interpreted as a direct reflection of their absolute or relative abundance in the brain since the ionization efficiency might be different for the different isoforms and since different isoforms are more hydrophobic and less soluble than others. Further, the different Aβ isoforms may differ in protein interaction capacity, including affinity to antibodies, and active or passive transport from brain tissue into CSF. Altogether, there are several non-quantitative aspects of IP-MS.
The brain tissue was processed within 24–48 h of death. The different lag times could potentially introduce artefacts in the mass spectrometric analysis such as proteolytic degradation and/or oxidation of the isoforms. However, oxidation of the isoforms can be monitored in the mass spectrometric analysis; the mass of the peptide plus 16 Da and the degree of oxidation of methionine were not different between the different subjects. Further, no new Aβ isoforms were detected when comparing the Aβ isoform pattern from brains which had been processed within a short period of time or 48 h. However, degradation can still be a factor to have in account.
The identification of the different isoforms is based on the average mass of the peak detected which is matched to the calculated theoretical mass and some of the isoforms might be false positive. However, all isoforms presented showed high mass accuracy and corresponded well to the calculated peptide masses (100 parts per million deviation or less). Further, the identities of the Aβ1-42, Aβ1-40, Aβ4-42 and Aβ4-40 peaks were confirmed using LC-ESI-FTICR-MS/MS (data not shown).
In conclusion, we here show that the dominating Aβ isoforms in AD brains are Aβ4-42 and Aβ1-40 and Aβ1-42. These peptides are also frequently detected in the cortex of controls. Their presence in hippocampus and cerebellum is, however, firmly associated with AD. An AD-like brain Aβ isoform pattern was seen in one of the five controls. No prominent differences in Aβ isoform patterns between FAD and SAD patients were seen, underscoring the similarity in the amyloid pathology of these two disease entities.
Acknowledgments
This work was supported by grants from the Swedish Research Council (projects 2006-6227, 2006-2740 and 2006-3505), the Alzheimer’s Association (NIRG-08-90356), cNEUPRO, the Royal Swedish Academy of Sciences, the Sahlgrenska University Hospital, the Inga-Britt and Arne Lundberg Research Foundation, the Göteborg Medical Society, the Swedish Medical Society, Swedish Brain Power, Stiftelsen Gamla Tjänarinnor, Gun och Bertil Stohnes stiftelse, Åhlén-stiftelsen, Alzheimer Foundation, Sweden, U.S. PHS K08 AG-22228, AG-O5131 and AG-023185.
Footnotes
Conflict of interest statement The authors declare that they have no conflict of interest.
Contributor Information
Erik Portelius, Email: erik.portelius@neuro.gu.se, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 431 80 Mölndal, Sweden.
Nenad Bogdanovic, Alzheimer Disease Research Center, Karolinska Institute, Huddinge University Hospital, Stockholm, Sweden.
Mikael K. Gustavsson, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 431 80 Mölndal, Sweden
Inga Volkmann, Alzheimer Disease Research Center, Karolinska Institute, Huddinge University Hospital, Stockholm, Sweden.
Gunnar Brinkmalm, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 431 80 Mölndal, Sweden.
Henrik Zetterberg, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 431 80 Mölndal, Sweden.
Bengt Winblad, Alzheimer Disease Research Center, Karolinska Institute, Huddinge University Hospital, Stockholm, Sweden.
Kaj Blennow, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 431 80 Mölndal, Sweden.
References
- 1.Andreasson U, Portelius E, Andersson ME, Blennow K, Zetterberg H. Aspects of beta-amyloid as a biomarker for Alzheimer’s disease. Biomark Med. 2007;1:59–78. doi: 10.2217/17520363.1.1.59. [DOI] [PubMed] [Google Scholar]
- 2.Bogdanovic N, Corder E, Lannfelt L, Winblad B. APOE polymorphism and clinical duration determine regional neuropathology in Swedish APP(670, 671) mutation carriers: implications for late-onset Alzheimer’s disease. J Cell Mol Med. 2002;6:199–214. doi: 10.1111/j.1582-4934.2002.tb00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 4.Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- 5.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 6.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 8.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 9.Gouras GK, Xu H, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S. Generation and regulation of beta-amyloid peptide variants by neurons. J Neurochem. 1998;71:1920–1925. doi: 10.1046/j.1471-4159.1998.71051920.x. [DOI] [PubMed] [Google Scholar]
- 10.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney M, Little SP, Ball MJ. Chemical characterization of A beta 17–42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J Biol Chem. 1994;269:10987–10990. [PubMed] [Google Scholar]
- 11.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 12.Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ. beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem. 1993;268:3021–3024. [PubMed] [Google Scholar]
- 13.Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- 14.Haltia M, Viitanen M, Sulkava R, Ala-Hurula V, Poyhonen M, Goldfarb L, Brown P, Levy E, Houlden H, Crook R, et al. Chromosome 14-encoded Alzheimer’s disease: genetic and clinicopathological description. Ann Neurol. 1994;36:362–367. doi: 10.1002/ana.410360307. [DOI] [PubMed] [Google Scholar]
- 15.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 16.Harigaya Y, Saido TC, Eckman CB, Prada CM, Shoji M, Younkin SG. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem Biophys Res Commun. 2000;276:422–427. doi: 10.1006/bbrc.2000.3490. [DOI] [PubMed] [Google Scholar]
- 17.Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry. 2003;74:1206–1209. doi: 10.1136/jnnp.74.9.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 19.Jager S, Leuchtenberger S, Martin A, Czirr E, Wesselowski J, Dieckmann M, Waldron E, Korth C, Koo EH, Heneka M, Weggen S, Pietrzik CU. Alpha-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Abeta generation. J Neurochem. 2009;111(6):1369–1382. doi: 10.1111/j.1471-4159.2009.06420.x. [DOI] [PubMed] [Google Scholar]
- 20.Lalowski M, Golabek A, Lemere CA, Selkoe DJ, Wisniewski HM, Beavis RC, Frangione B, Wisniewski T. The “non-amyloidogenic” p3 fragment (amyloid beta17-42) is a major constituent of Down’s syndrome cerebellar preamyloid. J Biol Chem. 1996;271:33623–33631. doi: 10.1074/jbc.271.52.33623. [DOI] [PubMed] [Google Scholar]
- 21.Lannfelt L, Bogdanovic N, Appelgren H, Axelman K, Lilius L, Hansson G, Schenk D, Hardy J, Winblad B. Amyloid precursor protein mutation causes Alzheimer’s disease in a Swedish family. Neurosci Lett. 1994;168:254–256. doi: 10.1016/0304-3940(94)90463-4. [DOI] [PubMed] [Google Scholar]
- 22.Mackenzie IR, McLachlan RS, Kubu CS, Miller LA. Prospective neuropsychological assessment of nondemented patients with biopsy proven senile plaques. Neurology. 1996;46:425–429. doi: 10.1212/wnl.46.2.425. [DOI] [PubMed] [Google Scholar]
- 23.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 24.Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005;44:10810–10821. doi: 10.1021/bi0508237. [DOI] [PubMed] [Google Scholar]
- 25.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 26.Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, Silberring J, Gandy SE, Winblad B, Greengard P, et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA. 1994;91:8378–8382. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, Giliberto L, Armirotti A, D’Arrigo C, Bachi A, Cattaneo A, Canale C, Torrassa S, Saido TC, Markesbery W, Gambetti P, Tabaton M. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- 28.Pike CJ, Overman MJ, Cotman CW. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J Biol Chem. 1995;270:23895–23898. doi: 10.1074/jbc.270.41.23895. [DOI] [PubMed] [Google Scholar]
- 29.Portelius E, Tran AJ, Andreasson U, Persson R, Brinkmalm G, Zetterberg H, Blennow K, Westman-Brinkmalm A. Characterization of amyloid beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J Proteome Res. 2007;6:4433–4439. doi: 10.1021/pr0703627. [DOI] [PubMed] [Google Scholar]
- 30.Portelius E, Brinkmalm G, Tran AJ, Zetterberg H, Westman-Brinkmalm A, Blennow K. Identification of novel APP/Abeta isoforms in human cerebrospinal fluid. Neurodegener Dis. 2009;6:87–94. doi: 10.1159/000203774. [DOI] [PubMed] [Google Scholar]
- 31.Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, Westman-Brinkmalm A, Zetterberg H, Simon AJ, Blennow K. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Portelius E, Zhang B, Gustavsson MK, Brinkmalm G, Westman-Brinkmalm A, Zetterberg H, Lee VM, Trojanowski JQ, Blennow K. Effects of gamma-secretase inhibition on the amyloid beta isoform pattern in a mouse model of Alzheimer’s disease. Neurodegener Dis. 2009;6:258–262. doi: 10.1159/000264639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, Kelleher RJ., III The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 35.Tekirian TL. Commentary: A beta N-terminal isoforms: critical contributors in the course of AD pathophysiology. J Alzheimers Dis. 2001;3:241–248. doi: 10.3233/jad-2001-3209. [DOI] [PubMed] [Google Scholar]
- 36.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 37.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 38.Welander H, Franberg J, Graff C, Sundstrom E, Winblad B, Tjernberg LO. Abeta43 is more frequent than Abeta40 in amyloid plaque cores from Alzheimer disease brains. J Neurochem. 2009;110:697–706. doi: 10.1111/j.1471-4159.2009.06170.x. [DOI] [PubMed] [Google Scholar]
- 39.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 40.Zellner M, Veitinger M, Umlauf E. The role of proteomics in dementia and Alzheimer’s disease. Acta Neuropathol. 2009;118:181–195. doi: 10.1007/s00401-009-0502-7. [DOI] [PubMed] [Google Scholar]