

Both the test tube and SDS-gel are powerful platforms for the study of protein-protein interaction but, ultimately, functional relationships between protein binding partners must be understood within the living cell. The advent of fluorescent protein (FP) color mutants derived from the jellyfish Aequorea victoria GFP has made Förster Resonance Energy Transfer (FRET) microscopy a practical tool for the study of protein interaction in a live cell. FRET is a non-radiative process that occurs when the energy of an excited donor fluorophore is transferred to an acceptor fluorophore by dipole-dipole coupling (Förster 1949); in this process, donor fluorescence is quenched and acceptor emission is increased or “sensitized.” For energy transfer to occur, both the donor and acceptor fluorophore must be in close proximity. The efficiency of energy transfer inversely correlates with the distance between fluorophores, increasing as the gap between fluorophores decreases, and can be used to calculate the fluorophore distance, being most accurately measured from 2 to 8 nm. Thus, when an appropriate donor and acceptor FRET pair is used to respectively tag target molecules of interest, both temporal and spatial information can be quantitatively measured below the limit of resolution on a light microscope by virtue of the efficiency of energy transfer from the donor to the acceptor fluorophore. Based on the physics that define FRET, it is natural to assume that FRET is a definitive indicator of protein-protein interaction; yet, it is extremely important to recognize that FRET measurements are rulings of proxy for the interaction of the tagged molecules and, accordingly, in vitro studies remain a critical component for any rationale that drives a FRET-based experiment. But when protein associations are grounded in a supporting body of experimental evidence, FRET becomes a powerful ruler for the study of molecular interaction within a living cell.
Thus far, the preponderance of empirical evidence indicates that the FPs, when expressed on their own or when tagged to a protein of interest, do not induce measurable toxic effects on cells and, quite remarkably, rarely interfere with protein function. While FPs have been placed on either the amino- or carboxy-termini of innumerable proteins, it cannot be overstated that an assessment of tagged-protein function relative to the wild-type protein must be measured experimentally, with the best barometer being rescue of the knockout phenotype. Also, full advantage should be taken of any available structural information to intelligently design the tagged protein: it is of no use to tag an amino-terminus if it is known to be buried within the protein structure or interact with another protein especially when placement of the FP within a flexible loop can be a successful alternative (Janetopoulos, Jin et al. 2001).
In pioneering live-cell FRET experiments, the cyan (CFP) and yellow fluorescent protein (YFP) variants were used to create biosensors to monitor rapid changes in concentration of second messengers such as calcium and cAMP in response to stimuli (REFs). Here CFP and YFP were linked between a specific polypeptide sequence that would change conformation and, thus, the distance between the FRET pair upon binding to the second messenger. While keeping the ratio of the FRET pair fixed at 1:1 greatly simplifies FRET measurements, FRET has been used successfully for the study of transient protein-protein interaction in signal transduction cascades in living cells by tagging individual interacting proteins with either a donor or acceptor FP (Tolar, Sohn et al. 2005; Xu, Meier-Schellersheim et al. 2005; Sohn, Tolar et al. 2006). When well controlled, protein complex formation and dissociation between tagged proteins of interest can be studied at the subcellular level (Schaufele et al., and Wallrabe and Barroso in (Periasamy and Day 2005).
This Unit describes the acceptor-sensitized emission FRET method using a confocal microscope for image acquisition and is intended to complement the acceptor photobleaching FRET method previously described in Unit 19.5 (Wouters and Bastiaens 2001); although, an example of acceptor photobleaching is provided to demonstrate the consistency between the methods on the samples presented below. In contrast to acceptor photobleaching, which is an end-point assay that destroys the acceptor fluorophore, the sensitized emission method is amenable for FRET measurements in live cells and can be used to measure changes in FRET efficiency over time. The purpose of the Unit is to provide a basic starting point for understanding and performing the sensitized emission method with a simple teaching tool for live cell imaging. References that discuss the vagaries of acquiring and analyzing FRET between individually tagged molecules are provided.
Familiarity with DNA transfection techniques, cell culturing and molecular cloning protocols are assumed for this Unit, as is a general understanding of epi-fluorescence microscopy. The authors suggest several excellent web primers (micro.magnet.fsu.edu/; www.olympusmicro.com/; www.microscopyu.com/) for imaging techniques and principles. A step-by-step protocol is provided for cell culturing, and image acquisition using live cells using the Zeiss LSM 510, a laser-scanning microscope equipped with two photomultiplier tubes (PMTs) for detecting emission through selected bandpass filters. Data analysis was performed using the FRET analysis software package provided by Zeiss. While the chapter will be written in a generalized manner referring to “Donor” and “Acceptor” fluorophores, specific examples will be provided using newly developed FRET calibration tools composed of a CFP and YFP variant (Cerulean and Venus respectively) coupled with different lengths of amino acids (Koushik, Chen et al. 2006); these constructs are readily available by written request from the original authors. In addition, the Cerulean-Venus fusion constructs can serve as excellent teaching tools for laboratories and imaging facilities that have FRET-neophytes interested in applying the technique toward their research questions.
Materials
Phosphate buffered saline with (PBS++) and without (PBS−−) Ca2+ and Mg2+ (Invitrogen, Carlsbad, CA, USA: http://www.invitrogen.com/)
Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA)
Fetal bovine serum (FBS; HyClone: http://www.hyclone.com/)
Kanamycin (Sigma-Aldrich, St. Louis, MO, USA) – for plasmid selection in bacteria
G418 (Sigma-Aldrich, St. Louis, MO, USA) – for selection in mammalian cell
6 well culture plates (Nunc, Thermo Fisher Scientific, Rochester, NY, USA)
Plasmid DNA isolation kit (Quiagen Plasmid Mini Kit, Quiagen GmbH, Hilden, Germany)
DNA Transfection kit (Lipofectamin 2000, Invitrogen, Carlsbad, CA, USA)
Zeiss LSM 510 META laser scanning confocal microscope or equivalent
-
Chambers for live-cell imaging
Our facility uses several types:
Coverglass-bottomed Lab-Tek II multi-well chambers (Nunc: http://www.nuncbrand.com). These chambers have open tops to conveniently add reagents. The small well size limits reagent volume.
35 mm coverglass-bottomed dishes (MatTek: http://www.glass-bottom-dishes.com/)
FCS2 environmental chamber (Bioptechs: http://www.bioptechs.com/). The FCS2 is a closed coverglass system that allows profusion of reagents.
-
Microscope environmental chamber to regulate temperature and CO2
The LSM 510 is equipped with a CTI stage and lens heating system from Pecon: http://www.pe-con.de/. Another universal environmental system used in our facility is the LiveCell, (Pathology Devices: http://www.pathologydevices.com/).
-
Cells of interest
HEK 293 (cat. no. CRL-1573, ATCC; http://www.atcc.org/)
Untransfected cells to assess background autofluorescence
-
Cells transfected with constructs to express:
FRET pair – Donor fused to Acceptor
Donor-only – to assess donor spectral bleedthrough
Acceptor-only – to assess acceptor spectral bleedthrough
-
DNA plasmid constructs
The “FRET pair” constructs (pC5V, pC17V, and pC32V) consist of the Cerulean (C; Rizzo et al., 2004) and Venus (V; Nagai et al., 2002) FP variants fused to one another with 5, 17, or 32 amino acid linkers between them (Koushik et al., 2006). Each construct respectively decreases in FRET efficiency as the linker distance increases.
The “Donor-only” constructs (pC5A, pC17A, and pC32A) are comprised of the Cerulean protein (C) fused with the indicated amino acid linker distances to Amber (A), a Venus mutant that folds properly, but is not activated by either donor or acceptor excitation lasers, and is incapable of acting as a FRET acceptor (Koushik, Chen et al. 2006).
The “Acceptor-only” construct (pV1) consists of one Venus moiety.
NOTE: CFP and YFP variants comprise the most widely used FRET pair in live cells. Alternative fluorophore considerations are discussed in the following references: (Periasamy and Day 2005; Shaner, Steinbach et al. 2005; Giepmans, Adams et al. 2006).
Cell Culture
-
1.
Grow HEK 293 cells in DMEM supplemented with 10% FBS at 37 °C and 5% CO2 in T25 (25 cm2) tissue culture dishes.
Use 8–10 ml of DMEM per flask.
The doubling time for HEK 293 cells is 20–24 hrs.
-
2.
Maintain growth in log phase.
Split confluent culture 1:5 to 1:6 every 2–3 days without using trypsin/EDTA. Ad 2 ml pre-warmed PBS−− and incubate for 2–3 mins. HEK 293 cells can then be detached by either tapping the flask or pipetting the PBS−− up and down several times. Seed out cells at about 2.5–3.0 × 105 cells/25 cm2.
Transfection
-
3.
Prepare each plasmid construct (pC5V, pC5A, pC17V, pC17A, pC32V, pC32A, and pV1) for transfection using the Quiagen Plasmid Mini Kit kit.
-
4.
Transfect cells in 6 well plates using the Lipofectamine transfection method according to the manufacturer’s protocol.
HEK 293 cell cultures should be no more than 60% confluent on the day of transfection. HEK 293 should be transfected 48h after seeding to allow for a firm attachment; this ensures a higher fraction of cells to survive the transfection.
While we created stably transfected cell lines for the experiments below, transiently transfected cells may be used as well (see Koushik et al., 2006).
-
5.
Incubate cells at 37 °C for 1 hr then exchange the transfection medium to DMEM plus 10% FBS.
-
6.
After 24 hours, exchange the growth medium to selection medium: DMEM plus 10% FBS supplemented with 0.5 mg/ml G418.
-
7.
Maintain and refresh the selection medium every 3 days.
-
8.
Once single clones appear, pick and propagate clones in selection media until a sufficient density of cells is reached for freezing.
Preparation for imaging
-
9.
24 to 48 hrs before imaging, release cells from culture dishes with PBS−−, wash, count and plate 2.0 – 4.0 × 104 cells per well in a 4 well LabTek II chamber.
-
10.
30 mins prior to imaging, gently wash cells by aspiration with at least three 1 mL changes of PBS warmed to 37°C and maintain cells at 37°C in an incubator.
Microscope Setup
NOTE: Laser line and filter choice will vary from system to system and, depend on the fluorophore. We describe the setup best suited for the LSM 510 in our facility using the Cerulean and Venus probes.
-
11.
Assemble stage and lens heating apparatuses and set to 37°C at least one hour before imaging.
Warm up is essential to minimize focus drift.
-
12.
Turn on the argon laser at least one hour before imaging.
Warm up is essential to minimize power fluctuation.
-
13.
Choose “Multi-track” imaging to acquire two tracks by laser line-switching.
Line-switching is the best option for time-sensitive imaging of live specimens. While the overall scan time is the same for both line- and frame-switching modes, corresponding line positions in Ch2 and Ch3 are closer together in time (milliseconds vs seconds in frame-switching mode).
-
14.
For both Track 1 and 2 select the 458/514 HFT (excitation) and the 545 NFT (emission) beamsplitter.
The 545 NFT directs emitted wavelengths shorter than 545 nm to the “Channel 2” PMT (Ch2) and those greater to the “Channel 3” PMT (Ch3)).
-
15.
Set up Track 1 as follows: activate the 458 nm laser line, select the 470 – 500 nm bandpass emission filter for Ch2 to collect the Donor signal, and the 530 nm longpass filter for Ch3 to collect the uncorrected FRET signal.
Peak emission for Cerulean and Venus is ~477 and ~528 nm respectively.
Spectral crosstalk in the form of direct excitation of the Acceptor by the Donor laser can be minimized if a shorter wavelength laser (405, 430 or 440 nm) is available. The 430 and 440 nm lasers are closer to the excitation maximum of Cerulean and, thus, would increase signal. The 405 nm laser would eliminate spectral crosstalk. (See Rheenen et al., 2004 for a comparison between the 430 and 458 laser for confocal FRET imaging).
-
16.
Set up Track 2 as follows: activate the 514 nm laser line and select the 530 nm longpass filter for Ch3 to collect the acceptor signal.
We are limited for filter selection for Ch3 on our system. A bandpass filter would be more appropriate for Venus emission as it is unnecessary to collect emissions past 625 nm.
The Cerulean spectral bleedthrough into the FRET channel cannot be eliminated (compare emission spectra in Rizzo, Springer et al. 2004 to Nagai et al., 2002); however, as a general consideration, a bandpass filter might help minimize Donor crosstalk into the FRET channel (see Schaufele et al. in Periasamy and Day, 2005).
-
17.
Set the image size for 512 × 512 pixels with a 12 bit pixel depth (best for quantitative measurements); set the “Scan Speed” for 9 (see step 18), and average four times (see step 18).
Acceptor Sensitized Emission Image Acquisition
-
18.
Under “Palette” activate the “Range Indicator” and image cells “Continuously” expressing the FRET-pair using the 40× 1.3 NA PLAN Neofluar oil-immersion objective lens and a 4 times zoom and establish a working range for fluorescence intensity for the Donor, FRET and Acceptor channels (see Fig. 1 for the specific settings used in these experiments).
Considerations:
Do not use the full dynamic range – allow for overhead in the Donor emission channel (Track1, Ch2) for intensity increases if photobleaching experiments are planned (see below).
Laser power – Set as low as possible to minimize phototoxicity and photobleaching.
Scan Speed (controls the pixel dwell time) – The signal intensity can be increased with longer dwell times, but photobleaching may occur.
Amplifier Offset (controls the minimum intensity limit) – Set so that there are no zero values for any pixels in the image.
Detector Gain (controls the voltage applied to the PMT and, thus, its sensitivity) – It is best to keep this setting below 750 (Zeiss recommendation) because higher values do not necessarily increase signal over noise. Increasing laser power or dwell time at the cost of photobleaching can compensate for an unreasonably high Detector Gain setting. Note that in Multi-track mode the Detector Gain setting will be the same for Ch3 for both Track 1 (uncorrected FRET image) and Track 2 (Acceptor image). We found that the N-FRET efficiency calculations were most consistent by manipulating the microscope settings to roughly equalize the average image intensity for the Donor, uncorrected FRET, and Acceptor images.
Amplifier Gain (amplifies all intensities whether resulting from noise or true signal) – Do not change from 1.
Averaging – Apply averaging if it can be afforded with respect to time and photobleaching. Averaging decreases the noise in the image.
Unwanted photobleaching – A simple time-lapse can assess the level of photobleaching incurred by the above settings. Signal was quite high for the samples shown here; thus, laser power was extremely low and photobleaching was inconsequential.
-
19.
Once a balanced image with an appropriate dynamic range is established, select “No Palette” to return to the normal imaging mode and acquire an image (Fig. 1A).
-
20.
Replace cells expressing the FRET-pair with cells expressing the Donor-only construct and acquire an image with the exact same conditions (Fig. 1B).
For convenience, it is helpful to place control cells in wells next to the experimental cells in the same multi-well chamber. If doable, some experimenters find it advantageous to seed a single well with cells expressing the FRET-pair and the Donor- and Acceptor-only controls.
Collect a range of intensity values that will correspond to the values obtained for the Donor signal in the FRET-pair images. It is best to use the same intensity range for correcting the FRET image.
It cannot be overemphasized that experimental and control cells must be acquired with the same imaging settings. Changes in settings are not necessarily linear, and will, therefore, skew efficiency calculations.
-
21.
Replace cells expressing the Donor-only construct with the Acceptor-only construct and acquire an image with the exact same conditions and following the considerations above in step 20 (Fig. 1C).
Figure 1.

An example acquisition of the acceptor sensitized emission method using cells expressing the FRET-pair C32V (A), the Donor-only C32A (B), and the Acceptor only V1 (C) is shown. See Fig. 3 for statistical results. The Donor panel (Track 1, Ch2) is the Cerulean intensity from resulting 458 nm laser excitation collected through a 470 – 500 nm bandpass filter. The un-FRET panel (Track 1, Ch3) is the uncorrected FRET intensity resulting from 458 nm laser excitation collected through a 530 nm longpass filter. The Acceptor panel (Track 2, Ch3) is the Venus intensity resulting from 514 nm laser excitation collected through a 530 nm longpass filter. Each set was imaged using the conditions described in the protocol above. The 458 and 514 nm laser power was 4% and 0.3% respectively. The N-FRET image calculated in the LSM software package is shown with the scale bar in (A). Note the spectral bleedthrough in the control images: In (B), Donor-only spectral bleedthrough is observed in the uncorrected FRET channel after excitation with the Donor laser (458 nm), while no crosstalk is detected in the Acceptor channel by direct excitation with the Acceptor laser (514 nm). In (C), Acceptor-only crosstalk is detected in the uncorrected FRET channel after excitation with the Donor laser. For these experiments the resulting Donor-only signal in the Donor channel after excitation with the Acceptor laser is negligible, but keep in mind that it may be necessary to subtract this crosstalk for other FRET pairs.
Acceptor Photobleaching Image Acquisition
-
22.
Return cells expressing the FRET-pair to the stage and acquire an image with same settings established above. Using the “Edit Bleach” function, assign a region around a cell or cells of interest to be bleached, set the function to acquire several images before and after bleaching, and bleach the Acceptor fluorophore with full power using the 514 laser line in the “TimeSeries” function (Fig. 2A and 2B).
HEK 293 cells are adherent, making this technique applicable to these live cells.
The number iterations required to achieve complete Acceptor photobleaching is determined empirically.
Figure 2.

An example of the acceptor photobleaching method using cells expressing the FRET pair C17V. (A) The pre-bleach image is shown. (B) The post-bleach image shows the bottom cell bleached after 200 iterations and the expected increase in Donor fluorescence after photobleaching.
Results and Data Analysis
FRET images collected for sensitized emission and photobleaching were analyzed using the N-FRET (Xia and Liu 2001) and photobleaching algorithms in the Zeiss LSM software package. A step-by-step procedure for operating the Zeiss graphical user interface can be at the Carl Zeiss ftp site with valid software license.
An example of experimental and control data for the sensitized emission method is shown in Fig. 1. Fig. 2 shows an example of how donor fluorescence intensity increases after a typical acceptor photobleaching. Data were collected on three separate days for both methods and average N-FRET values and FRET efficiencies are shown in Fig. 3. Both techniques provided comparable results. As expected, the FRET efficiencies as measured by photobleaching decreased as the amino acid linker length increased between the Cerulean and Venus moieties: C5V ~45%, C17V ~35%, and C32V ~26%, all within approximately 5% error of the values measured byKoushik et al. (2006). Similarly, N-FRET values decreased with increasing linker length: C5V ~0.47, C17V ~0.35, and C32V ~0.26.
Figure 3.
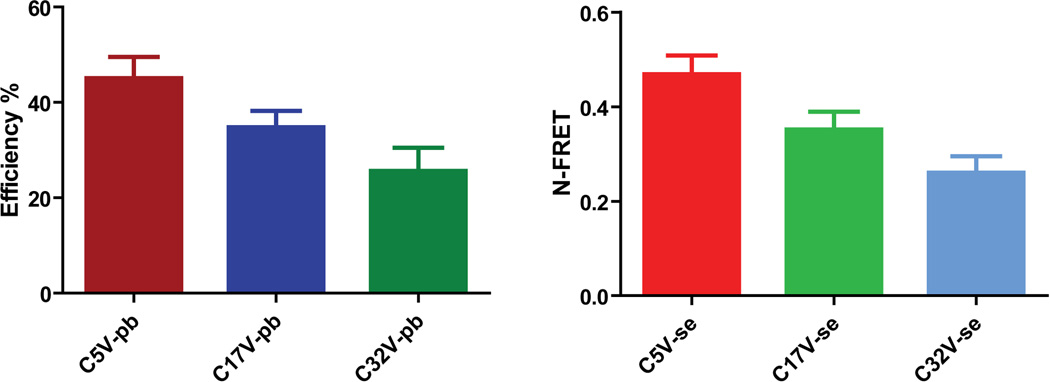
Shown is the average FRET efficiency (%) by Acceptor photobleaching (pb) and N-FRET values calculated in the LSM software for the sensitized emission method (se). Specific values and SD for photobleaching are: C5V-pb: 45.1% +/− 4.42%, n=13; C17V-pb: 34.8% +/− 3.47%, n=11; C32V-pb: 25.6% +/− 4.89%, n=17. Specific values and SD for N-FRET are: C5V-se: 0.469 +/− 0.0396, n=39; C17V-se: 0.352 +/− 0.0372, n=43; C32V-se: 0.260 +/− 0.0351, n=51.
COMENTARY
A number of methodologies are currently practiced to measure FRET between proximal fluorophores, which include ratio imaging, acceptor photobleaching, acceptor-sensitized emission, spectral FRET (a methodology that takes advantage of confocal systems equipped with spectral detectors and can be applied to the three aforementioned methods) and fluorescence lifetime imaging (FLIM). Detailed descriptions of these techniques and their sources are found in an excellent FRET compendium edited by Periassamy and Day (2006), and accessible discussions of the theory and mathematics behind these methodologies are presented in the marvelous text by Lakowicz (1999).
It is important to note that before intensity values are applied to the methods presented below, corrections for background fluorescence and autofluorescence must be made. Background values are corrected for in the Zeiss FRET macro. Autofluorescence is assessed in cells not expressing either Donor or Acceptor fluorophores.
Another consideration is adjustments to “flat-field.” If a fluorescent object having equal levels of fluorophore throughout its area yields and image with greater intensity on one side that decreases across to the other, then a flat-field correction is required. This circumstance can easily be assessed in the LSM software by measuring the intensity over a line through the image object using the “Profile” function. Flat-field error can be caused by imperfections in the specimen chamber or a misaligned system. Correction requires a reference – usually a coverslip coated with fluorophore. The current LSM software package does not have a convenient flat-field correction function but can be done with the math functions provided in the software. Alternatively, flat-field errors can be corrected for in other image processing software such as MetaMorph or through an excellent open source alternative, ImageJ (http://rsbweb.nih.gov/ij/).
Ratio Imaging
Ratio imaging is perhaps the simplest of FRET methodologies. In live cells, dynamic changes in FRET due to an experimentally induced change in fluorophore distance can be measured as a ratio of the Donor channel signal divided by the FRET channel signal, where an increase in the ratio indicates a loss in FRET (see Xu, Brzostowski et al. 2006 for an LSM 510-based spectral unmixing method). The method is not useful to compare the ratio of Donor to FRET channel signal between cells. Ratio imaging works most reliably when the Donor and Acceptor are present at a fixed ratio, either being part of the same molecule as in many biosensors (Miyawaki, Llopis et al. 1997; Nagai, Miyazaki et al. 2000; Lissandron, Terrin et al. 2005) or are tightly linked (Janetopoulos, Jin et al. 2001; Periasamy and Day 2005; Tolar, Sohn et al. 2005; Xu, Meier-Schellersheim et al. 2005; Sohn, Tolar et al. 2006) since results could be affected artificially by changes in local concentration of the fluorophores.
Acceptor Photobleaching
Acceptor photobleaching is also a relatively simple technique, requiring fewer controls than sensitized emission measurements, but the method is slow (the cost being the time for photobleaching) and is an end-point assay, making measurements of FRET change over time in live cells impossible. Only the donor is considered in the calculation for the efficiency (E) of energy transfer:
| (1) |
where FD is the fluorescence intensity of the dequenched Donor in cells expressing the FRET-pair. In other words, FD is measured in the Donor emission channel by excitation with the Donor laser after complete Acceptor photobleaching with the Acceptor laser. FDA is the fluorescence intensity of the quenched Donor in the Donor emission channel for cells expressing the FRET-pair. Simply, Donor fluorescence is quenched in the presence of an Acceptor. Acceptor photobleaching eliminates Donor quenching, thus allowing the loss of energy to be measured.
Acceptor-Sensitized Emission
The sensitized emission (also known as the three cube) method is the most complicated of the FRET measurement techniques with respect to the level of control, attention to equipment, filter choice and post-image acquisition processing. The bane of the method is the unavoidable Donor and Acceptor spectral bleedthrough that must be subtracted from each image channel to accurately access the level of FRET taking place between fluorophores.
Among other factors, FRET between two fluorophores is contingent upon overlap of the Donor’s emission and the Acceptor’s excitation spectrum, and it is this spectral proximity that gives rise to unwanted spectral bleedthrough. Fig. 4 depicts the spectral overlap between the cyan fluorescent protein (CFP) and its Acceptor, yellow fluorescent protein (YFP). As seen by the spectra when using an arc-lamp illumination source, an excitation bandpass filter can be easily selected for CFP that will not directly excite YFP, thus eliminating this spectral bleedthrough. Unfortunately for most confocal systems, the selection of monochromatic laser lines is limited to 458 nm (as in the protocol above) for adequate CFP excitation, thus requiring subtraction of the Acceptor fluorescence in the FRET channel due to direct excitation by the Donor excitation laser line. Despite this drawback, having both the Donor and Acceptor excitation lines come from the same laser head lessens problems that occur from power fluctuations. The latest confocal microscope models can now be equipped at relatively low cost with a 405, 430 or 440 nm laser line and the future likely holds the use of super-continuum white light lasers, where specific wavelengths can be selected by the user. Wisely chosen emission filters (or emission wavelength on systems using diffraction gratings) can eliminate any potential back-bleedthrough of Acceptor emission into the Donor channel from excitation by both the Donor and Acceptor lasers, but because of CFP’s broad emission spectra, significant Donor spectral bleedthrough into the Acceptor channel will always be an issue with this FRET pair (Fig. 4).
Figure 4.

Shown are the excitation (dashed) and emission (solid) spectra of the Donor CFP (blue) and Acceptor YFP (yellow); also shown are Donor and Acceptor excitation laser lines (458 and 514 nm; note the direct excitation of YFP by 458 nm), and Donor and Acceptor bandpass and longpass emission filter ranges respectively (hatched boxes in nm). The spectral overlap integral of the Donor emission and Acceptor excitation is shown in light green and the Donor spectral bleedthrough into the Acceptor channel is shown in light yellow.
Because the FRET channel contains unwanted signal due to spectral bleedthrough, these intensities must be subtracted out to obtain the true FRET signal. Nearly all methods follow the principle described by the following formula to derive the signal due to the energy transfer from the Donor to Acceptor fluorophore:
| (2) |
where Fc is the corrected FRET image after the raw FRET image (Ff) is subtracted by the Donor spectral bleedthrough (DSBT) and the Acceptor spectral bleedthrough (ASBT). The DSBT is the signal in the FRET channel that results from the direct excitation of the Donor by the Donor laser. The contribution of the DSBT is calculated indirectly by measuring the signal from a Donor-only sample excited by the Donor laser in the Donor channel (Dd; note that the abbreviations follow (Gordon, Berry et al. 1998) as used in the Zeiss manual) and in the FRET channel (Fd). The DSBT is calculated from the ratio
| (3) |
where Df is the signal from the FRET-pair excited by the Donor laser in the Donor channel. Similarly, the contribution of the ASBT is calculated by measuring the signal from an Acceptor-only sample excited by the Acceptor laser in Acceptor channel (Aa) and in the FRET channel (Fa). The ASBT is calculated from the ratio
| (4) |
where Af is the signal from the FRET-pair excited by the Acceptor laser in the Acceptor (equivalent to the FRET) channel. After substitution, the equation described by (Youvan, Silva et al. 1997) is derived
| (5) |
Eq. (5) has been shown to inadequately detect FRET under conditions where the concentration of Donor and Acceptor fluorophores can vary. There are several methods that normalize for Donor and Acceptor concentration: The FRETN method byGordon et al. (1998) is represented by
| (6) |
(Xia and Liu 2001) further normalize for Donor and Acceptor concentration by
| (7) |
which, according to the authors, takes into consideration FRET efficiency between Donor and Acceptor and is a function of the ratio of complexes to total Donor and Acceptor concentrations. The Zeiss FRET macro will report corrected FRET values for Fc, FRETN and N-FRET. Be aware that these are not calculations of FRET efficiency (E), but rather, are methods to detect or “index” the FRET signal after subtracting bleedthrough components.
Unwanted, gradual photobleaching of Donor and Acceptor fluorophore during a time-lapse acquisition affects the relationship between the intensity of the Donor and Acceptor and its relationship to the concentration of the molecules that are tagged by the fluorophores. Recently, (Zal and Gascoigne 2004) developed a way to correct for photobleaching during time-lapse acquisitions, which is incorporated into a method to calculate FRET efficiency. It is a calculation of FRET efficiency that most ideally and comparatively describes the interaction of fluorophores between system to system as this number should, within reasonable error, be the same. Unfortunately for confocal systems, technical obstacles such as multiple PMTs for image detection in the Donor, Acceptor and FRET channel, two excitation laser lines, and optics makes efficiency calculations using a sensitized emission-based method challenging. Despite the difficulty, algorithms developed by Periasamy and colleagues (2005) and van Rheenen, Langeslag et al. (2004) have provided a means to measure E with a confocal microscope. Furthermore, Tolar and colleagues (2005) fused the methods by Zal and Gascoigne (2004) and van Rheenen et al. (2004) to measure FRET efficiency between fluorophores tagged to membrane associated proteins in live B cells to measure B cell receptor-activated pathways using the Zeiss LSM 510.
FLIM
Several fine sources for the fundamentals of fluorescence lifetime imaging are Lakowicz (1999), (Becker 2005) and Redford and Clegg (in Periasamy and Day, 2005). The fluorescence lifetime (τ) is an intrinsic property of the excited fluorophore and is the most direct way to measure FRET. The advantage of lifetime measurements is that they are independent of the concentration of the fluorophores. The decay of a single molecule from the excited state is an exponential function and can be described by
| (8) |
where (τ) is the decay time for the fluorophore. However, exiting the excited state is not only accomplished by the emission of photons as other non-radiative pathways, including energy transfer, can return the fluorophore to the ground state. Therefore, if τ0 is the lifetime of a fluorophore without FRET and τ is the lifetime in the presence of FRET, the efficiency of energy transfer can be measured directly by
| (9) |
Fluorescence lifetime can be measured two ways: directly, using a so-called time-domain system, which requires an expensive, specialized picosecond-pulsed laser and the resulting fluorescence usually measured with a PMT; or indirectly, with a frequency-domain system that employs a modulated illumination source (laser or light diode) and detects fluorescence with a modulated image intensifier coupled to a CCD. While frequency-domain systems are generally less sensitive, they offer the advantage of allowing fast image acquisition and thus are more amenable for live cell imaging.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. P. Tolar for critically reading this manuscript and for the excellent technical assistance of Amy Liu. The Intramural Research Program of the NIH, NIAID, supported this research.
References
- Becker W. Advanced Time-Correlated Single Photon Counting Techniques. Berlin: Springer; 2005. [Google Scholar]
- Förster T. Experimental and theoretical investigation of the intermolecular transfer of electronic excitation energy. Z. Naturforsch. A. 1949;4:7. [Google Scholar]
- Giepmans BN, Adams SR, et al. The fluorescent toolbox for assessing protein location and function. Science. 2006;312(5771):217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Gordon GW, Berry G, et al. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998;74(5):2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos C, Jin T, et al. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 2001;291(5512):2408–2411. doi: 10.1126/science.1055835. [DOI] [PubMed] [Google Scholar]
- Koushik SV, Chen H, et al. Cerulean, Venus, and VenusY67C FRET reference standards. Biophys J. 2006;91(12):L99–L101. doi: 10.1529/biophysj.106.096206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissandron V, Terrin A, et al. Improvement of a FRET-based indicator for cAMP by linker design and stabilization of donor-acceptor interaction. J Mol Biol. 2005;354(3):546–555. doi: 10.1016/j.jmb.2005.09.089. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388(6645):882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Miyazaki M, et al. A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat Biotechnol. 2000;18(3):313–316. doi: 10.1038/73767. [DOI] [PubMed] [Google Scholar]
- Periasamy A, Day RN, editors. Molecular Imaging: FRET Microscopy and Spectroscopy. Oxford: Oxford University Press; 2005. [Google Scholar]
- Rizzo MA, Springer GH, et al. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22(4):445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, et al. A guide to choosing fluorescent proteins. Nat Methods. 2005;2(12):905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Sohn HW, Tolar P, et al. Fluorescence resonance energy transfer in living cells reveals dynamic membrane changes in the initiation of B cell signaling. Proc Natl Acad Sci U S A. 2006;103(21):8143–8148. doi: 10.1073/pnas.0509858103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar P, Sohn HW, et al. The initiation of antigen-induced B cell antigen receptor signaling viewed in living cells by fluorescence resonance energy transfer. Nat Immunol. 2005;6(11):1168–1176. doi: 10.1038/ni1262. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Langeslag M, et al. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86(4):2517–2529. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters FS, Bastiaens PIH. Imaging Protein-Protein Interactions by Fluorescence Energy Transfer (FRET) Microscopy. John Wiley & Sons, Inc.; 2001. [DOI] [PubMed] [Google Scholar]
- Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81(4):2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Brzostowski JA, et al. Using quantitative fluorescence microscopy and FRET imaging to measure spatiotemporal signaling events in single living cells. Methods Mol Biol. 2006;346:281–296. doi: 10.1385/1-59745-144-4:281. [DOI] [PubMed] [Google Scholar]
- Xu X, Meier-Schellersheim M, et al. Quantitative imaging of single live cells reveals spatiotemporal dynamics of multistep signaling events of chemoattractant gradient sensing in Dictyostelium. Mol Biol Cell. 2005;16(2):676–688. doi: 10.1091/mbc.E04-07-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youvan DC, Silva CM, et al. Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnology et alia. 1997;3:18. [Google Scholar]
- Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86(6):3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]