

Abstract
Aims/hypothesis
Endoplasmic reticulum (ER) stress has been recognised as a common pathway in the development of obesity-associated insulin resistance. Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of insulin signalling and is localised on the ER membrane. The aim of the study was to investigate the cross-talk between ER stress and PTP1B.
Methods
Leptin-deficient obese (ob/ob), Ptp1b (also known as Ptpn1) knockout and C57BL/6J mice were subjected to a high-fat or normal-chow diet for 20 weeks. ER stress was induced in cultured myotubes by treatment with tunicamycin. Immunohistochemistry and western blotting were used to assess proteins involved in the ER stress response. Myotube glucose uptake was determined by measuring 2-deoxy[3H]glucose incorporation.
Results
A high-fat diet induced ER stress and PTP1B expression in the muscle tissue of mice and these responses were attenuated by treatment with the ER chaperone tauroursodeoxycholic acid (TUDCA). Cultured myotubes exhibited increased levels of PTP1B in response to tunicamycin treatment. Silencing of Ptp1b with small interfering RNA (siRNA) or overexpression of Ptp1b with adenovirus construct failed to alter the levels of ER stress. Ptp1b knockout mice did not differ from the wild-type mice in the extent of tunicamycin-induced upregulation of glucose-regulated protein-78. However, tunicamycin-induced phosphorylation of eukaryotic initiation factor 2α and c-Jun NH2-terminal kinase-2 were significantly attenuated in the Ptp1b knockout mice. Treatment with TUDCA or silencing of PTP1B reversed tunicamycin-induced blunted myotube glucose uptake.
Conclusions/interpretation
Our data suggest that PTP1B is activated by ER stress and is required for full-range activation of ER stress pathways in mediating insulin resistance in the skeletal muscle.
Keywords: Diabetes, Endoplasmic reticulum stress, Insulin resistance, Obesity, Protein tyrosine phosphatase 1B, Tunicamycin
Introduction
Obesity is a major risk factor for the development of metabolic syndrome and insulin resistance and is growing at pandemic proportions [1]. Obesity and insulin resistance predispose to cardiovascular disease, which is the leading cause of morbidity and mortality in the western world [2]. Insulin resistance is characterised by impairment in glucose uptake by skeletal muscle and the failure of the body to suppress hepatic gluconeogenesis and is observed years before the development of full-blown diabetes. Insulin resistance by itself, without overt diabetes, has been shown to be an independent risk factor for cardiovascular disease. Thus, obesity-associated insulin resistance provides a potential therapeutic avenue to pre-empt future complications. Although diet and exercise can alleviate insulin resistance, compliance with these lifestyle-change measures is difficult. Drugs such as thiazolidinediones and metformin, although they are good insulin-sensitising agents, are associated with a variety of adverse effects [3]. Therefore, identifying and characterising pathways leading to insulin resistance would help in designing strategies to treat this problem.
Although the exact molecular mechanisms of obesity-associated insulin resistance are not fully understood, several postulates have been proposed, including alterations in adipokines (leptin, resistin, adiponectin) [4], elevated levels of circulating pro-inflammatory cytokines (such as tumour necrosis factor-α) [5], glucotoxicity mediated by reactive oxygen species and glycated proteins [6] and lipotoxicity consequent to increased circulating levels of NEFA [4, 7], all of which can lead to impaired insulin signalling and hence insulin resistance. Insulin mediates its metabolic action by binding to the insulin receptor tyrosine kinase and activating the receptor. Activated insulin receptor phosphorylates the downstream docking protein IRS, which subsequently through the activation of the phosphatidylinositol 3-kinase and Akt pathway leads to the translocation of GLUT4 vesicles to the cell surface resulting in cellular glucose uptake [8]. Several studies have reported that protein tyrosine phosphatase 1B (PTP1B) is a hydrolase that targets the tyrosine-phosphorylated insulin receptor β and IRS-1 and thereby functions as a negative regulator of insulin signalling [9, 10]. Deletion of PTP1B results in improved insulin sensitivity and glucose tolerance suggesting that PTP1B is a potential target for therapeutic intervention in insulin-resistant conditions [11–13]. Several PTP1B inhibitors have been proposed for the management of diabetes mellitus [14].
In mammalian cells, PTP1B is tethered to the endoplasmic reticulum (ER) via its C-terminal targeting sequence [15]. The ER is the cellular organelle responsible for the folding and assembly of secretory proteins. Conditions such as obesity overwhelm the capacity of the ER leading to the accumulation of misfolded/unfolded proteins, a condition termed ER stress. In response to ER stress, three ER-localised transmembrane signal transducers are activated to initiate adaptive responses called unfolded protein response (UPR): inositol-requiring enzyme (IRE1), double-stranded RNA-activated protein kinase-like ER kinase (PERK) and activating transcription factor 6 (ATF6) [16]. Emerging evidence suggests that ER stress may have a pivotal role in the pathophysiology of obesity, insulin resistance and type 2 diabetes [17, 18]. Accordingly, interventions that suppress ER stress can improve diabetes and associated comorbidities [19, 20]. Because PTP1B appears to be closely associated with the ER, in terms of both cellular localisation and function, this study was undertaken to determine the cross-talk between ER stress and PTP1B and its role in the regulation of skeletal muscle glucose uptake.
Methods
Materials
2-Deoxy-D-[3H]glucose was from Sigma (St Louis, MO, USA). Tauroursodeoxycholic acid (TUDCA) and tunicamycin were from Calbiochem (Darmstadt, Germany). PTP1B adenovirus was from Vector Biolabs (Philadelphia, PA, USA). Ptp1b small interfering RNA (siRNA) sequences, non-target siRNA sequences and DharmaFECT transfection reagent were from Thermo Scientific (Rockford, IL, USA). Antibodies against glucose-regulated protein 78 (GRP78), CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), phosphorylated eukaryotic initiation factor 2α (phospho-eIF2α), phosphorylated c-Jun NH2-terminal kinase (phospho-JNK), eIF2α, JNK, phospho-Akt, Akt, phosphorylated IRS-1 (phospho-IRS-1; Tyr1222 and Ser307) and IRS-1 and LumiGLO Reagent were from Cell Signaling Technology (Boston, MA, USA). Antibodies against PTP1B were from Millipore (Billerica, MA, USA). Anti-rabbit IgG-FITC antibody and anti-mouse IgG-tetramethyl rhodamine isothiocyanate (TRITC) antibody were from Sigma-Aldrich (St Louis, MO, USA). DMEM, FBS and horse serum were from Gibco/Invitrogen (Carlsbad, CA, USA).
Animals
The experimental procedures described in this study were approved by the University of Wyoming Animal Use and Care Committee (Laramie, WY, USA). All mice were maintained with free access to food and water and were housed in the School of Pharmacy Animal Facility at the University of Wyoming. ob/ob mice and C57BL6 mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Ptp1b whole body knockout mice were kindly provided by M. L. Tremblay (McGill Cancer Center, Quebec, QC, Canada).
In brief, 7-week-old adult male homozygous B6.V-lep<ob>/J (ob/ob) and age- and sex-matched C57BL/6J lean mice were randomly assigned to receive either TUDCA (50 mgkg/day, orally) in 0.1% carboxymethylcellulose (CMC) or the vehicle (0.1% CMC) for a period of 5 weeks. This dose of TUDCA was selected based on previous studies by us [20] and others [21]. At the end of the 5-week treatment, mice were killed and gastrocnemius muscle and livers were collected. Tissues were homogenised in radioimmunoprecipitation assay (RIPA) lysis buffer (Upstate, Lake Placid, NY, USA) using a PowerGen Homogenizer 125 (Fisher Scientific, Hampton, NH, USA) and subsequently sonicated using Sonic Dismembrator 100 (Fisher Scientific). The homogenates were centrifuged at 14,000 g for 15 min and the soluble fraction was used for protein analysis by western blot as described below.
For diet-induced obesity experiments 5-week-old male mice were randomly assigned to low-fat (10% of total calories) or high-fat (45% of total calories) diets for a period of 20 weeks. At the end of treatment, mice were killed and proteins from the gastrocnemius muscle were extracted as described above. For in-vivo ER stress induction male adult mice were given an i.p. injection of 1 mg/kg tunicamycin or vehicle. After 24 h mice were killed and gastrocnemius muscles collected. Proteins for western blot analysis were isolated as described above.
Cell culture and differentiation
The mouse muscle myoblast cell line C2C12 (American Type Culture Collection, Rockville, MD, USA) was maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin under a humidified atmosphere of 5% CO2 in air. After confluence, the culture medium was changed to DMEM with 2% horse serum to initiate myogenic differentiation [22]. After differentiation, myotubes were starved of serum for 12 h and then were subjected to the various treatments described below. Adenoviral infection and siRNA treatment (described below) were performed in conjunction with serum starvation.
Treatment of cells
ER stress was induced in C2C12 myotubes by treating them with tunicamycin (0.1–2 μg/ml for experiments described in Fig. 2, 0.1 μg/ml for subsequent experiments) or palmitic acid (0.2–0.8 mmol/l). Palmitic acid was prepared by conjugating it with bovine serum albumin as previously reported [23].
Fig. 2.
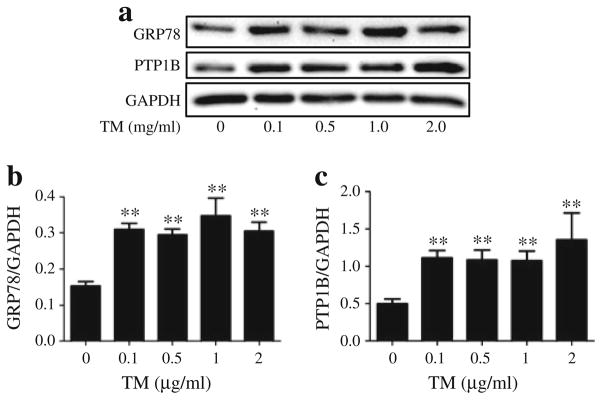
Tunicamycin (TM)-induced ER stress and PTP1B in cultured myotubes. Representative western blots (a) and densitometric analysis (mean ± SEM, n=4) of GRP78 (b) and PTP1B (c) protein levels in C2C12 myotubes, incubated with 0.1, 0.5, 1 and 2 μg/ml of TM for 24 h. **p<0.01 compared with non-treated control
Transfection of cells with Ptp1b siRNA
A total of 25 nmol/l of Ptp1b siRNA or the same amount of non-target siRNA were transfected for 48 h into C2C12 myotubes using DharmaFECT transfection reagent according to the manufacturer’s instructions.
Infection of cells with PTP1B adenovirus
PTP1B adenoviruses (Vector Biolabs, Philadelphia, PA, USA) were produced in HEK-293 cells with subsequent purification using Adeno-XTM virus purification kit (Clontech, Mountain View, CA, USA) according to the manufacturer’s instructions. The purified PTP1B adenoviruses were transfected to myotubes in complete medium without antibiotics. Cells transfected with vector adenovirus not expressing Ptp1b were used as a control.
Western blot analysis
Cells were lysed in RIPA lysis buffer followed by sonication and centrifugation at 14,000 g for 15 min. A 50-μg sample of lysates was separated using 10% SDS-PAGE. Subsequently, proteins were transferred to nitrocellulose membranes, which were incubated in the primary antibody against specific proteins. Following treatment with anti-rabbit or anti-mouse IgG horseradish-peroxidase-linked antibody immunoreactive bands were detected and quantified by enhanced chemiluminescence autoradiography using a molecular imager Gel Doc XR + System (Bio Rad, Hercules, CA, USA). All protein levels were normalised to GAPDH levels; phospho-eIF2α, -JNK, -Akt, and -IRS-1 (Tyr1222 and Ser307) were normalised to corresponding total protein levels. Average values for control (untreated) group were used for normalisation between different blots, when acquired ratios for controls were substantially different among the blots.
RNA isolation and real-time RT-PCR
Total RNA was isolated from myotubes using RNeasy Mini Kit (QIAGEN, Hilden, Germany) followed by genomic DNA digestion with RNase-Free DNase Set (QIAGEN). Reverse transcription was performed from 1 μg of total RNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. The synthesised cDNAs were amplified by PCR in the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using iQ SYBR Green Supermix (Bio-Rad). All the primers used were from Integrated DNA Technologies (Coralville, IA, USA) (see electronic supplementary material [ESM] Table 1). Results were normalised to 18S ribosome RNA as endogenous control. Relative expression levels of mRNAs were calculated using the 2−ΔΔCt method [24].
Immunocytochemistry
C2C12 cultured myoblasts were grown on chamber slides. After the treatment, the cells were fixed with 4% paraformaldehyde and permeabilised with 0.25% Triton X-100. The fixed cells were probed with anti-PTP1B and anti-CHOP antibodies and incubated with secondary anti-rabbit antibody conjugated with FITC and anti-mouse antibody conjugated with TRITC. Following nuclear staining with DAPI, the cells were observed under a Zeiss 710 confocal microscope.
Glucose uptake assay
2-Deoxy[3H]glucose-uptake assay was performed as previously described [25]. Briefly, cells were incubated in Krebs–Ringer phosphate HEPES buffer for 30 min at 37°C in the presence of 2-deoxy[3H]glucose (7,400 Bq) and 50 nmol/l of insulin. Cells were then lysed in PBS containing 0.2 mol/l NaOH, and glucose uptake was assessed by scintillation counting. The counts were adjusted by the protein content.
Statistical analysis
Data were presented as mean ± SEM. Statistical analysis was performed with ANOVA using GraphPad Prism 5.04 (GraphPad Software, La Jolla, CA, USA). A p value of less than 0.05 was considered to be statistically significant.
Results
Obesity-associated elevated PTP1B in skeletal muscle is attenuated by ER chaperone
Leptin-deficient ob/ob mice exhibited significantly higher PTP1B protein levels in gas-trocnemius muscle compared with the age- and sex-matched lean control mice (Fig. 1a, b). Oral supplementation with a well-established ER chaperone, TUDCA, resulted in a near complete inhibition of this elevated PTP1B level in the obese mice. In contrast, TUDCA did not have any effect on the PTP1B levels in the lean mice. Obese mice also exhibited robust levels of ER stress marker protein GRP78 (Fig. 1a, c) compared with their lean counterparts. As anticipated, the TUDCA treatment resulted in an attenuation of elevated GRP78 level in obese mice. Similar inhibition of PTP1B abundance and ER stress by TUDCA was observed in liver tissue samples (ESM Fig. 1). To further confirm these findings we used a diet-induced obesity model. Mice fed a high-fat diet exhibited significantly higher levels of PTP1B proteins in muscle tissue (Fig. 1d). Furthermore high-fat diet resulted in elevated levels of ER stress in the skeletal muscle, as demonstrated by GRP78 levels and eIF2α phosphorylation (Fig. 1d).
Fig. 1.

Obesity-induced ER stress leads to increased PTP1B protein levels. Representative western blots (a) and densitometric analysis (mean ± SEM, n=3) of PTP1B (b) and GRP78 (c) protein levels in gastrocnemius muscle of ob/ob and C57BL/6J lean mice receiving TUDCA or vehicle for 5 weeks. **p<0.01 compared with lean mice, ††p<0.01 compared with ob/ob mice. (d) Representative western blots of PTP1B, GRP78 and phospho- and total-eIF2α (p-eIF2α and p-eIF2α, respectively) protein levels in gastrocnemius muscle of C57BL/6J mice on a normal diet (ND) or high-fat diet (HFD)
ER stressor tunicamycin induces PTP1B production in cultured myotubes
To understand the molecular mechanisms involved in the cross-talk between PTP1B and ER stress, quiescent cultured C2C12 myotubes were challenged with the ER stressor tunicamycin for 24 h, followed by western blot analysis of levels of PTP1B and GRP78. Tunicamycin blocks the synthesis of N-linked glycoproteins to induce UPR. Western blot analysis revealed upregulation of GRP78 levels in the myotubes even at the lowest concentration of the compound used (Fig. 2a, b). Furthermore, tunicamycin treatment resulted in elevation of PTP1B levels indicating that ER stress induces PTP1B production (Fig. 2a, c). As we did not observe any further increase in the amount of ER stress or PTP1B levels with higher concentrations of tunicamycin, a concentration of 0.1 μg/ml was used for further experiments. Immunocytochemical analysis of C2C12 myoblasts was consistent with the aforementioned western blot analysis. In untreated myoblasts we observed some basal levels of PTP1B with no CHOP (Fig. 3). Tunicamycin treatment caused a significant elevation in both PTP1B and CHOP immunoreactivity, further substantiating the finding that ER stress augments PTP1B protein levels. Interestingly, PTP1B was localised primarily in the cytoplasm whereas CHOP was limited to the nucleus. To further confirm our findings we studied the effect of another ER stressor, palmitic acid, the predominant unsaturated fatty acid component present in the high-fat diet (ESM Fig. 2). Both ER-stress and PTP1B protein levels were elevated in cells treated with palmitic acid.
Fig. 3.
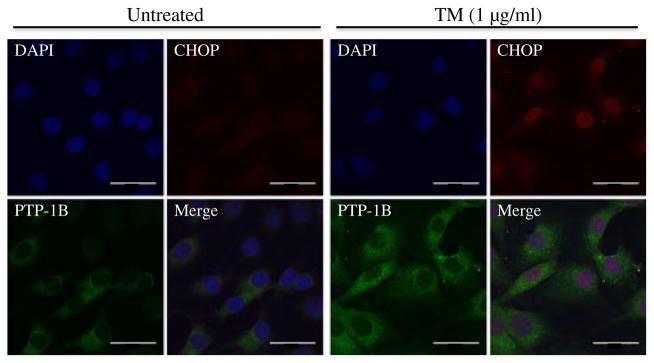
Tunicamycin (TM)-induced localisation of PTP1B and CHOP in cultured myotubes. Immunocytochemistry staining of PTP1B and CHOP proteins in C2C12 myoblasts treated with TM for 24 h. PTP1B is shown as green, CHOP is shown as red, the nucleus is stained with DAPI (blue), while purple suggests colocalisation. Scale bars, 1 μm
To determine the temporal ER stress and PTP1B responses, we incubated myotubes with tunicamycin for different time periods. We observed increases in the levels of ER stress markers GRP78 and CHOP in response to tunicamycin by about 6 h (Fig. 4a–d). In contrast, increase in PTP1B occurred only after 24 h of incubation with tunicamycin. Further, to explore the potential mechanism of the tunicamycin-induced increase in protein levels, we used real-time RT-PCR to assess mRNA levels in myotubes treated with tunicamycin for various lengths of time. We observed increases in Grp78 and Chop (also known as Ddit3) mRNA levels already after 2 h of tunicamycin treatment (Fig. 4e–g). However, increase in Ptp1b (also known as Ptpn1) mRNA expression occurred only after 24 h of incubation, consistent with the pattern of protein levels.
Fig. 4.

Time course of tunicamycin (TM)-induced ER stress and PTP1B expression in C2C12 myotubes. Representative western blots (a) and den-sitometric analysis (mean ± SEM, n=3) of PTP1B (b), GRP78 (c) and CHOP (d) protein levels in C2C12 myotubes incubated with TM for 6, 12, 24 and 48 h. (e–g) Real time RT-PCR analysis (mean ± SEM, n=4) of mRNA content of Ptp1b (e), Grp78 (f) and Chop (g) in in C2C12 myotubes incubated with TM for 2, 6, 12, 24 and 48 h. *p<0.05 compared with non-treated control
Tunicamycin-associated elevated PTP1B in cultured myotubes is attenuated by ER chaperone
To further investigate the cross-talk between PTP1B and ER stress, tunicamycin-induced cultured myotubes were treated with the ER chaperone TUDCA. As expected, TUDCA attenuated the degree of ER stress caused by addition of tunicamycin (Fig. 5). Furthermore, cells treated with TUDCA had lower protein levels of GRP78 and a lower level of phospho-eIF2α than the cells treated with tunicamycin alone (Fig. 5c, d). Consistent with our in-vivo findings, TUDCA treatment also decreased tunicamycin-induced upregulation of PTP1B (Fig. 5b). Neither PTP1B nor ER stress levels were altered by TUDCA in cells that had not been treated with tunicamycin.
Fig. 5.

Effect of TUDCA on tunicamycin (TM)-induced PTP1B protein levels in myotubes. Representative western blots (a) and densitometric analysis (mean ± SEM, n=3) of PTP1B (b), GRP78 (c) and phospho- and total eIF2α (p-eIF2α and t-eIF2α, respectively) (d) in C2C12 myotubes treated with tunicamycin in presence of 1 mmol/l TUDCA for 24 h. **p<0.01 compared with non-treated control cells; ††p<0.01 compared with TM-treated cells
Silencing of Ptp1b does not inhibit ER stress
Our in-vivo and in-vitro data demonstrate that attenuating ER stress using the ER chaperone TUDCA attenuates PTP1B protein levels. However, it is not clear whether the converse is true. To test this, Ptp1b was silenced using siRNA technique in cultured myotubes, following which the effect of tunicamycin on markers of ER stress was evaluated. As shown in Fig. 6a, siRNA mediated-silencing of Ptp1b successfully abrogated tunicamycin-induced increase in the protein levels of PTP1B. Interestingly, silencing of Ptp1b failed to alter the induction of ER stress markers GRP78 and CHOP by tunicamycin (Fig. 6c, d). In contrast, silencing of Ptp1b blocked tunicamycin-induced phosphorylation of eIF2α and JNK1/2 (Fig. 6e, f), suggesting a potential role for PTP1B in the activation of these branches of the ER stress response. Furthermore, in cells overexpressing Ptp1b (by means of the adenoviral transgene delivery system expressing wild-type recombinant PTP1B), we failed to observe any upregulation of GRP78 (Fig. 7c) or CHOP (data not shown). Nevertheless, myotubes that overexpressed Ptp1b had higher protein levels of GRP78 upon stimulation with tunicamycin. To confirm our in vitro findings, we used intra-peritoneal injection of tunicamycin (1 mg/kg) to induce in vivo ER stress response in mice lacking PTP1B (Fig. 7d–h). In response to tunicamycin injection C57BL/6J, Ptp1b+/− and Ptp1b−/− male adult mice exhibited significantly higher protein levels of GRP78 in the skeletal muscle (Fig. 7d, e). In contrast, we did not observe any differences in the extent of GRP78 induction in skeletal muscle by tunicamycin between wild-type and Ptp1b knockout mice (Fig. 7f), further demonstrating that PTP1B is not required for its activation. However, the induction of other ER stress-associated signalling pathways–phosphorylation of eIF2α and JNK1/2, was slightly lower in Ptp1b heterozygotic mice and almost undetectable in Ptp1b knockout mice upon tunicamycin injection, which is consistent with in vitro data (Fig. 7g, h).
Fig. 6.

Effect of Ptp1b knockdown on tunicamycin (TM)-induced ER stress in myotubes. Representative western blots (a) and densitometric analysis (mean ± SEM, n=3) of PTP-1B (b), GRP78 (c), CHOP (d), phospho and total-eIF2α (p-eIF2α and t-eIF2α, respectively) (e), phospho- and total-JNK1/2 (p-JNK and t-JNK, respectively) (f) in C2C12 myotubes transfected with 25 nmol/l of Ptp1b siRNA and incubated with TM for 24 h. **p<0.01 compared with non-target siRNA control; ††p<0.01 compared with Ptp1b siRNA-treated cells
Fig. 7.

Effect of Ptp1b overexpression and knockout on tunicamycin (TM)-induced ER stress. Representative western blots (a) and densitometric analysis (mean ± SEM, n=4) of PTP1B (b) and GRP78 (c) protein levels in C2C12 myotubes transfected with adenovirus expressing wild-type recombinant PTP1B (Ad-PTP1B) and treated with TM for 24 h. **p<0.01 compared with control; ††p<0.01 compared with Ad-PTP1B cells. Representative western blots (d) and densitometric analysis (mean ± SEM, n=3) of PTP1B (e), GRP78 (f), phospho (p) to total (t)eIF2α (g) and phospho (p) to total (t)JNK1/2 (h) in gastrocnemius muscle of C57BL/6J, Ptp1b+/− and Ptp1b−/− male adult mice, 24 h after i.p. injection of 1 mg/kg TM or vehicle
Ptp1b silencing alters ER stress-mediated impairment of glucose uptake
To assess the cellular consequence of this cross-talk, we assessed the glucose uptake in cultured differentiated myotubes. As expected, insulin stimulation for 30 min resulted in a greater than twofold increase in cellular glucose uptake compared with unstimulated cells (Fig. 8). The insulin-stimulated glucose uptake was abrogated by pretreating the cells with tunicamycin, which in turn was rescued by pretreating the cells with TUDCA (Fig. 8a). Furthermore, silencing of Ptp1b rescued tunicamycin-induced downregulation of glucose uptake in cultured myotubes (Fig. 8b). To further understand the mechanism involved in ER stress-induced insulin resistance, we evaluated the effect of tunicamycin on insulin-stimulated phosphorylation of Akt and IRS-1 in cultured myotubes. Tunicamycin abrogated the threonine phosphorylation of Akt and tyrosine phosphorylation of IRS-1 in response to insulin stimulation (Fig. 8c, d). Interestingly, this blunting of tunicamycin-induced insulin signalling was rescued by treatment with TUDCA or by silencing of Ptp1b (ESM Fig. 3). In contrast, a robust increase in the phosphorylation of IRS-1 at the Ser307 residue (which inhibits insulin signalling) was seen when the cells were treated with tunicamycin (Fig. 8c, d). This tunicamycin-induced elevation of IRS-1 phosphoserine was nullified by TUDCA and by Ptp1b siRNA (ESM Fig. 3).
Fig. 8.

The role of PTP1B and ER stress in tunicamycin (TM)-induced insulin resistance. C2C12 myotubes were incubated with TM and treated with TUDCA (a, c) or Ptp1b siRNA (b, d) for 24 h (48 h for siRNA experiment), and stimulated with insulin (50 nmol/l, 30 min). (a, b) 2-Deoxy[3H]glucose uptake assay (mean ± SEM, n=3). **p< 0.01 compared with corresponding no-insulin conditions; ††p<0.01 compared with insulin-stimulated cells; ‡‡p<0.01 compared with TM + insulin treated cells. (c, d) Western blot analysis of phospho-and total Akt (p-Akt and t-Akt, respectively) and phospho- and total-IRS-1 (p-IRS-1 (Tyr1222 and Ser307 and t-IRS, respectively) protein levels. Representative blots are shown
Discussion
Accumulating evidence demonstrates that ER stress represents a common pathway to link obesity, insulin resistance and type 2 diabetes [17, 18]. The activation of multiple branches of the UPR response in the pathogenesis of obesity and diabetes has been previously described in liver and white adipose tissue [19, 26, 27]. However, the role of ER stress in the development of diet-induced insulin resistance in skeletal muscle is more controversial, with some authors showing increased ER stress in diabetic patients [28] and others indicating that UPR is not involved in the impairment of glucose tolerance induced by a fat-rich diet [29]. In our study we focused on PTP1B as a candidate molecule linking ER stress and insulin sensitivity in muscle cells. PTP is a well-known negative regulator of insulin signalling and can be a link between ER stress and the development of insulin resistance [30]. In the present study we examined the effect of the ER stress inducer tunicamycin on UPR, PTP1B levels and insulin sensitivity in skeletal muscle cells.
Our data demonstrate that levels of PTP1B are elevated in obesity, along with ER stress, and that countering the ER stress with an ER chaperone downregulated this obesity-associated elevated PTP1B level. Our in vitro studies using C2C12 myotubes showed that protein mRNA levels of ER stress markers GRP78 and CHOP were both increased already after 6 h of treatment with tunicamycin. However, an increase in PTP1B levels was detected only after 24 h. These differences in temporal production suggest that tunicamycin-induced activation of ER stress precedes the upregulation of PTP1B. Consistent with our findings, Bettaieb and coworkers demonstrated an increase in GRP78 levels early after palmitic acid treatment in pancreatic beta cells, and an increase in PTP1B levels only after prolonged treatment [31]. Interestingly, tunicamycin-induced production of CHOP was observed after several hours, although CHOP protein levels and mRNA content steadily decreased with time. This bell shaped curve of the time course of CHOP production is consistent with observations by Oliveira and coworkers using human hepatoma HepG2 cells that were treated with homocysteine or dithiothreitol [32].
Previous studies have shown that the absence of PTP1B results in impaired ER stress [33]. In Ptp1b knockout mice, ER-stress-mediated JNK activation, X-box binding protein 1 (XBP-1) splicing and apoptosis were attenuated in response to challenge with the ER stressor tunicamycin [34]. Consistent with these findings, Delibegovic and coworkers found that liver-specific deletion of Ptp1b inhibited the high-fat diet-induced ER stress response in mice [35]. PTP1B contains two proline-rich domains and has the potential to interact with scaffold proteins. Indeed, recently it has been shown that the sarcoma (Src) homology 3-containing adaptor protein, non-catalytic region of tyrosine kinase (NCK), directly interacts with PTP1B [36]. Earlier Nguyen et al showed that in fibroblasts, azetidine-2-carboxylic-acid-stimulated GRP78 production was NCK independent, although the phosphorylation of ERK MAPK mediated by IRE1 was negatively regulated by NCK [37]. Such a NCK-dependent inhibitory system has been also shown to regulate PERK activation, preventing translational attenuation mediated by eIF2α phosphorylation [38]. These findings are in accordance with both our in vitro studies on cultured myotubes using siRNA-mediated silencing of Ptp1b and our in vivo studies using Ptp1b knockout mice, where tunicamycin induced normal activation of Grp78 expression but decreased phosphorylation levels of JNK2 (IRE1 pathway) and eIF2α (PERK pathway). It is likely that PTP1B contributes to ER stress signalling by regulating scaffold protein NCK, either by direct binding or by regulating its tyrosine phosphorylation state. From our data it appears that robust PTP1B activation is particularly important for the complete activation of PERK (eIF2α phosphorylation) and IRE1α (JNK2 phosphorylation) branches of UPR. Our results are consistent with those of Bettaieb and coworkers who showed that PTP1B regulates the PERK/eIF2α arm of ER stress signalling in pancreatic beta cells [31]. On the other hand Gu et al showed that PTP1B potentiates IRE1 signalling during ER stress [34].
We observed a striking consistency in our in vitro observation and in vivo data with respect to the upregulation of PTP1B in response to ER stress. However, a major shortcoming is that our in vivo studies were performed in muscle homogenate and not with isolated myotubes from the muscle. Therefore, although our in vitro data from myotubes is suggestive that the changes observed in the muscle homogenate may represent those observed in the myotubes, the likelihood that other cellular components in the muscle homogenate may also contribute to the observed response cannot be ruled out. In previous studies focusing on the role of PTP1B in insulin signalling in skeletal muscle, palmitic acid was used to induce Ptp1b and insulin resistance [39, 40]. However, it has been shown recently that ER stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells [41, 42]. Palmitate is more likely to increase Ptp1b in muscle cells by inducing inflammation pathways [43]. In the present study, we have focused on cross-talk between ER stress and PTP1B in obesity, and the role of this cross-talk in development of insulin resistance in skeletal muscle, which is why we chose to use tunicamycin, a potent and specific inducer of ER stress. Our observations that TUDCA was able to reverse the effect of tunicamycin on glucose uptake in cultured myotubes are consistent with the findings of Hage Hassan and coworkers [41]. What is interesting is that silencing of Ptp1b was able to reconcile the tunicamycin-induced blunted glucose uptake back to normal levels, indicating that PTP1B mediates ER stress-induced insulin resistance. Our results indicate that tunicamycin-induced insulin resistance in myotubes is a result of inhibition of threonine phosphorylation of Akt and tyrosine phosphorylation of IRS-1, which is reversed by TUDCA or PTP1B siRNA treatment. Interestingly, tunicamycin also increases phosphorylation of the Ser307 residue of IRS-1, which has a negative effect on insulin signalling. These detrimental effects of tunicamycin on insulin signalling molecules were rescued by either TUDCA or by silencing Ptp1b. Furthermore, TUDCA treatment or Ptp1b silencing decreased the activation of JNK. It is well established that JNK negatively regulates insulin signalling by phosphorylating IRS-1 at the Ser307 residue [44]. It is therefore likely that both ER-dependent and -independent pathways may contribute to the induction of PTP1B in obesity, which eventually leads to an insulin-resistant state in skeletal muscle cells.
Collectively, our data demonstrate that there is a close cross-talk between ER stress response and PTP1B in regulating cellular glucose uptake, which may have implications in the pathophysiology of insulin-resistant conditions. ER stress causes elevation in PTP1B levels, which may lead to insulin resistance in rodent muscle cells. On the other hand, Ptp1b knockdown results in impaired UPR response in vivo. These finding indicate that PTP1B is required for the full range of UPR responses and that a positive feedback loop between PTP1B and ER stress may exist. Taken together, it is likely that ER stress may be functioning upstream of PTP1B in the pathogenesis of obesity-associated insulin resistance.
Supplementary Material
Acknowledgments
The real-time PCR system was provided by the University of Wyoming Macromolecular Analysis Core facility.
Funding This work was supported in part by grants from NIH P20RR016474 (S. Nair, J. Ren).
Abbreviations
- ATF6
Activating transcription factor 6
- eIF2α
Eukaryotic initiation factor 2α
- ER
Endoplasmic reticulum
- C/EBP
CCAAT/enhancer binding protein
- CHOP
C/EBP homologous protein
- CMC
Carboxymethylcellulose
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GRP78
Glucose-regulated protein 78
- IRE1
Inositol-requiring kinase 1
- JNK
c-Jun NH2-terminal kinase
- MAPK
Mitogen-activated protein kinase
- NCK
Non-catalytic region of tyrosine kinase
- PERK
Double-stranded RNA-activated protein kinase-like ER kinase
- PTP1B
Protein tyrosine phosphatase 1B
- RIPA
Radioimmuoprecipitation assay
- siRNA
Small interfering RNA
- TRITC
Tetramethyl rhodamine isothiocyanate
- TUDCA
Tauroursodeoxycholic acid
- UPR
Unfolded protein response
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00125-012-2782-z) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement EP contributed to design, acquired and analysed data and drafted the article; YH contributed to design, acquired data and drafted the article; BC contributed to design, analysed data and contributed to drafting the article; JR contributed to conception and design, helped analyse the data and contributed to drafting the article; SN contributed to conception and design, analysed and interpreted data, and drafted and revised the article. All authors approved the final version to be published.
Contributor Information
E. Panzhinskiy, School of Pharmacy, University of Wyoming, College of Health Sciences, Laramie, WY 82071, USA. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY, USA
Y. Hua, School of Pharmacy, University of Wyoming, College of Health Sciences, Laramie, WY 82071, USA. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY, USA
B. Culver, School of Pharmacy, University of Wyoming, College of Health Sciences, Laramie, WY 82071, USA. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY, USA
J. Ren, School of Pharmacy, University of Wyoming, College of Health Sciences, Laramie, WY 82071, USA. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY, USA
S. Nair, Email: sreejay@uwyo.edu, School of Pharmacy, University of Wyoming, College of Health Sciences, Laramie, WY 82071, USA. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY, USA
References
- 1.James PT, Rigby N, Leach R. The obesity epidemic, metabolic syndrome and future prevention strategies. Eur J Cardiovasc Prev Rehabil. 2004;11:3–8. doi: 10.1097/01.hjr.0000114707.27531.48. [DOI] [PubMed] [Google Scholar]
- 2.Bahrami H, Bluemke DA, Kronmal R, et al. Novel metabolic risk factors for incident heart failure and their relationship with obesity: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;51:1775–1783. doi: 10.1016/j.jacc.2007.12.048. [DOI] [PubMed] [Google Scholar]
- 3.Caldwell SH, Argo CK, Al-Osaimi AM. Therapy of NAFLD: insulin sensitizing agents. J Clin Gastroenterol. 2006;40(suppl 1):S61–S66. doi: 10.1097/01.mcg.0000168647.71411.48. [DOI] [PubMed] [Google Scholar]
- 4.Halberg N, Wernstedt-Asterholm I, Scherer PE. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am. 2008;37:753–768. x–xi. doi: 10.1016/j.ecl.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishima Y, Kuyama A, Tada A, Takahashi K, Ishioka T, Kibata M. Relationship between serum tumor necrosis factor-alpha and insulin resistance in obese men with Type 2 diabetes mellitus. Diabetes Res Clin Pract. 2001;52:119–123. doi: 10.1016/s0168-8227(00)00247-3. [DOI] [PubMed] [Google Scholar]
- 6.Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci. 2011;32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Eckardt K, Taube A, Eckel J. Obesity-associated insulin resistance in skeletal muscle: role of lipid accumulation and physical inactivity. Rev Endocr Metab Disord. 2011;12:163–172. doi: 10.1007/s11154-011-9168-2. [DOI] [PubMed] [Google Scholar]
- 8.Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116:1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J Biol Chem. 1996;271:19810–19816. doi: 10.1074/jbc.271.33.19810. [DOI] [PubMed] [Google Scholar]
- 10.Dadke S, Kusari J, Chernoff J. Down-regulation of insulin signaling by protein-tyrosine phosphatase 1B is mediated by an N-terminal binding region. J Biol Chem. 2000;275:23642–23647. doi: 10.1074/jbc.M001063200. [DOI] [PubMed] [Google Scholar]
- 11.Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 12.Klaman LD, Boss O, Peroni OD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bence KK. Hepatic PTP1B deficiency: the promise of a treatment for metabolic syndrome? J Clin Metab Diabetes. 2010;1:27–33. [PMC free article] [PubMed] [Google Scholar]
- 14.Thareja S, Aggarwal S, Bhardwaj TR, Kumar M. Protein tyrosine phosphatase 1B inhibitors: a molecular level legitimate approach for the management of diabetes mellitus. Med Res Rev. 2010;32:459–517. doi: 10.1002/med.20219. [DOI] [PubMed] [Google Scholar]
- 15.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 16.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 17.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54(suppl 2):S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 18.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 19.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceylan-Isik AF, Sreejayan N, Ren J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol. 2011;50:107–116. doi: 10.1016/j.yjmcc.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Ono T, Nagasue N, Kohno H, et al. Effect of tauroursodeoxycholic acid on bile flow and calcium excretion in ischemia-reperfusion injury of rat livers. J Hepatol. 1995;23:582–590. doi: 10.1016/0168-8278(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 22.Nedachi T, Kanzaki M. Regulation of glucose transporters by insulin and extracellular glucose in C2C12 myotubes. Am J Physiol Endocrinol Metab. 2006;291:E817–E828. doi: 10.1152/ajpendo.00194.2006. [DOI] [PubMed] [Google Scholar]
- 23.Wang XL, Zhang L, Youker K, et al. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes. 2006;55:2301–2310. doi: 10.2337/db05-1574. [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Kandadi MR, Rajanna PK, Unnikrishnan MK, et al. 2-(3,4-Dihydro-2H-pyrrolium-1-yl)-3oxoindan-1-olate (DHPO), a novel, synthetic small molecule that alleviates insulin resistance and lipid abnormalities. Biochem Pharmacol. 2010;79:623–631. doi: 10.1016/j.bcp.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kammoun HL, Chabanon H, Hainault I, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flamment M, Kammoun HL, Hainault I, Ferre P, Foufelle F. Endoplasmic reticulum stress: a new actor in the development of hepatic steatosis. Curr Opin Lipidol. 2010;21:239–246. doi: 10.1097/MOL.0b013e3283395e5c. [DOI] [PubMed] [Google Scholar]
- 28.Peter A, Weigert C, Staiger H, et al. Individual stearoyl-CoA desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes. 2009;58:1757–1765. doi: 10.2337/db09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deldicque L, van Proeyen K, Francaux M, Hespel P. The unfolded protein response in human skeletal muscle is not involved in the onset of glucose tolerance impairment induced by a fat-rich diet. Eur J Appl Physiol. 2011;111:1553–1558. doi: 10.1007/s00421-010-1783-1. [DOI] [PubMed] [Google Scholar]
- 30.Asante-Appiah E, Kennedy BP. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab. 2003;284:E663–670. doi: 10.1152/ajpendo.00462.2002. [DOI] [PubMed] [Google Scholar]
- 31.Bettaieb A, Liu S, Xi Y, et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;286:9225–9235. doi: 10.1074/jbc.M110.186148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliveira SJ, Pinto JP, Picarote G, et al. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS One. 2009;4:e6618. doi: 10.1371/journal.pone.0006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agouni A, Mody N, Owen C, et al. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438:369–378. doi: 10.1042/BJ20110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu F, Nguyen DT, Stuible M, Dube N, Tremblay ML, Chevet E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279:49689–49693. doi: 10.1074/jbc.C400261200. [DOI] [PubMed] [Google Scholar]
- 35.Delibegovic M, Zimmer D, Kauffman C, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–599. doi: 10.2337/db08-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu CL, Buszard B, Teng CH, et al. Dock/Nck facilitates PTP61F/PTP1B regulation of insulin signalling. Biochem J. 2011;439:151–159. doi: 10.1042/BJ20110799. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen DT, Kebache S, Fazel A, et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell. 2004;15:4248–4260. doi: 10.1091/mbc.E03-11-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kebache S, Cardin E, Nguyen DT, Chevet E, Larose L. Nck-1 antagonizes the endoplasmic reticulum stress-induced inhibition of translation. J Biol Chem. 2004;279:9662–9671. doi: 10.1074/jbc.M310535200. [DOI] [PubMed] [Google Scholar]
- 39.Bakhtiyari S, Meshkani R, Taghikhani M, Larijani B, Adeli K. Protein tyrosine phosphatase-1B (PTP-1B) knockdown improves palmitate-induced insulin resistance in C2C12 skeletal muscle cells. Lipids. 2010;45:237–244. doi: 10.1007/s11745-010-3394-3. [DOI] [PubMed] [Google Scholar]
- 40.Mohammad Taghvaei N, Meshkani R, Taghikhani M, Larijani B, Adeli K. Palmitate enhances protein tyrosine phosphatase 1B (PTP1B) gene expression at transcriptional level in C2C12 skeletal muscle cells. Inflammation. 2011;34:43–48. doi: 10.1007/s10753-010-9206-3. [DOI] [PubMed] [Google Scholar]
- 41.Hage Hassan R, Hainault I, Vilquin JT, et al. Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia. 2012;55:204–214. doi: 10.1007/s00125-011-2328-9. [DOI] [PubMed] [Google Scholar]
- 42.Rieusset J, Chauvin MA, Durand A, et al. Reduction of endoplasmic reticulum stress using chemical chaperones or Grp78 overexpression does not protect muscle cells from palmitate-induced insulin resistance. Biochem Biophys Res Commun. 2012;417:439–445. doi: 10.1016/j.bbrc.2011.11.135. [DOI] [PubMed] [Google Scholar]
- 43.Parvaneh L, Meshkani R, Bakhtiyari S, et al. Palmitate and inflammatory state additively induce the expression of PTP1B in muscle cells. Biochem Biophys Res Commun. 2010;396:467–471. doi: 10.1016/j.bbrc.2010.04.118. [DOI] [PubMed] [Google Scholar]
- 44.Sykiotis GP, Papavassiliou AG. Serine phosphorylation of insulin receptor substrate-1: a novel target for the reversal of insulin resistance. Mol Endocrinol. 2001;15:1864–1869. doi: 10.1210/mend.15.11.0725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.