

Abstract
Muscle stiffness is a major clinical feature in Duchenne muscular dystrophy (DMD). DMD is the most common lethal inherited muscle-wasting disease in boys, and it is caused by the lack of the dystrophin protein. We recently showed that the extensor digitorum longus (EDL) muscle of mdx mice (a DMD mouse model) exhibits disease-associated muscle stiffness. Truncated micro- and mini-dystrophins are the leading candidates for DMD gene therapy. Unfortunately, it has never been clear whether these truncated genes can mitigate muscle stiffness. To address this question, we examined the passive properties of the EDL muscle in transgenic mdx mice that expressed a representative mini- or micro-gene (ΔH2-R15, ΔR2-15/ΔR18-23/ΔC, or ΔR4-23/ΔC). The passive properties were measured at the ages of 6 and 20 mo and compared with those of age-matched wild-type and mdx mice. Despite significant truncation of the gene, surprisingly, the elastic and viscous properties were completely restored to the wild-type level in every transgenic strain we examined. Our results demonstrated for the first time that truncated dystrophin genes may effectively treat muscle stiffness in DMD.
Keywords: Duchenne muscular dystrophy, micro-dystrophin, mini-dystrophin, passive force, muscle force
muscle weakness and stiffness are major clinical symptoms in Duchenne muscular dystrophy (DMD) (6, 7, 12). DMD is one of the most common lethal inherited muscle-wasting diseases in boys (31). It affects one in every 3,500 male births (13). Patients show difficulties in walking and climbing at the age of 2 to 5 yr old. By the age of 12, patients are confined to wheelchairs. Patients die in their early 20s due to respiratory and/or cardiac failure. Presently, there is no cure for DMD.
DMD is caused by the lack of the dystrophin protein (23). Dystrophin is a subsarcolemma cytoskeletal protein with four domains, including the NH2-terminal, rod, cysteine-rich (CR), and COOH-terminal domains (2). The rod domain comprises 24 spectrin-like repeats and four hinges. The NH2-terminal domain and a part of the rod domain bind to F-actin cytoskeleton protein, and the CR domain binds to dystroglycan transmembrane protein that connects to the extracellular matrix (2). Dystrophin is thought to stabilize the sarcolemma against mechanical stress imposed by muscle contraction (16, 30, 33). In the absence of dystrophin, muscle contraction induces cell membrane damage (35). Eventually, injured myofibers undergo necrosis and are replaced by adipose and fibrotic tissues (9, 26, 34).
Muscle weakness in DMD has been well studied by measuring muscle force generated during active contraction. However, muscle stiffness has not been clearly defined until recently (6, 7). We recently examined the passive properties (elasticity and viscosity) of the extensor digitorum longus (EDL) muscle in the mdx mouse, a widely used DMD mouse model (20). Compared with wild-type EDL muscle, the mdx EDL muscle developed more resistant force (passive stress) against stretch (strain). Furthermore, the stress relaxation rate was significantly increased.
Restoration of dystrophin expression with gene replacement may effectively treat DMD. Despite a promising modality, DMD gene therapy has been challenged by the massive size of the dystrophin gene. The full-length dystrophin cDNA far exceeds the packaging capacity of adeno-associated virus (AAV), the only vector that can lead to whole body muscle transduction (10). Several different strategies have been employed to overcome this size hurdle. Among these, a very appealing approach is the development of the truncated dystrophin genes. The intention is to generate smaller synthetic genes that can still protect muscle. This possibility has been suggested by studying Becker muscular dystrophy (BMD), a mild variant of DMD. Some patients with BMD carry large in-frame rod-domain truncations, yet they show very mild muscular dystrophy (14). Two types of truncated dystrophin genes have been pursued. The micro-genes are less than 4 kb and can fit into a single AAV vector. The mini-genes are less than 8 kb and they can be delivered by dual AAV vectors (11). The ΔR2-15/ΔR18-23/ΔC micro-gene, ΔR4-23/ΔC micro-gene, and ΔH2-R15 mini-gene are some of the representative candidates (Fig. 1) (22, 28, 29).
Fig. 1.
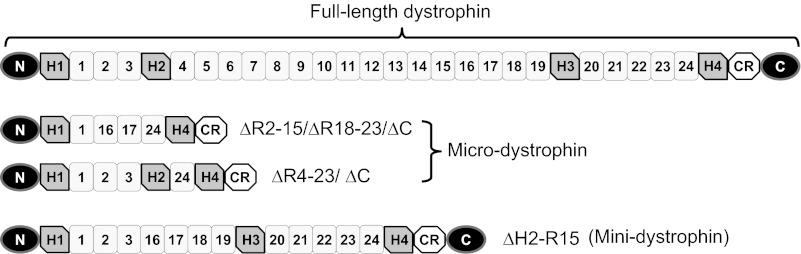
A schematic outline of the full-length dystrophin, micro-dystrophin, and mini-dystrophin genes used in the study. N, dystrophin NH2-terminal domain; H, hinge; CR, cysteine-rich domain; C, dystrophin COOH-terminal domain. Schematic outlines are not drawn to scale.
Studies from many laboratories, including ours, have provided ample evidence that, despite a significant truncation of the gene size and differences in the composition, muscle contractility is significantly enhanced by these truncated mini- and micro-genes in young adult mdx mice (22, 28, 29). However, it has been rarely evaluated whether these truncated genes can improve muscle-specific force in aged mdx mice (18). In addition, the kinetics of the contractile properties has never been studied. More importantly, it is not known whether the shorting of the dystrophin protein impacts the muscle passive property. Here, we hypothesized that 1) truncated mini- and micro-dystrophin genes can increase muscle force in aged mdx mice, 2) shortened dystrophins can restore kinetics of tetanic contraction, 3) truncated dystrophin genes can improve the elastic and viscous properties of the mdx muscle, and 4) the size and composition of a truncated dystrophin gene may influence its protection on the active and passive properties of the mdx muscle.
To test these hypotheses, we measured the active and the passive properties of the EDL muscle in transgenic mdx mice expressing either one of the two micro-genes (ΔR2-15/ΔR18-23/ΔC, or ΔR4-23/ΔC) or the mini-gene (ΔH2-R15). The specific tetanic force was restored to the wild-type level in all transgenic mdx mice at 6 and 20 mo of age. In support of our hypothesis, the elastic and viscous properties were also completely restored to those of the wild-type at 6 and 20 mo of age in every transgenic mdx strain we examined. Our data suggest that the truncated dystrophin genes not only restored active muscle contraction, but they also effectively corrected muscle stiffness.
MATERIALS AND METHODS
Experimental mice.
All animal experiments were approved by the Animal Care and Use Committee of the University of Missouri (no. 6980) and were in accordance with NIH guidelines. BL10 (wild-type) and mdx mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Skeletal muscle-specific transgenic mdx mice were generated at the University of Missouri Transgenic Animal Core. Detailed information on these mice has been published before (25, 28). Specifically, we have used three independent strains that expressed the ΔR2-15/ΔR18-23/ΔC micro-gene, ΔR4-23/ΔC micro-gene, and ΔH2-R15 mini-gene under transcriptional regulation of the muscle-specific human α-skeletal actin promoter (Fig. 1). Beside the gene size, there are several other important structural features in these minimized genes (Fig. 1). First, the ΔH2-R15 mini-gene contains the COOH-terminal domain, but the two micro-genes do not. Interestingly, a previous study suggests that the COOH-terminal domain is dispensable for muscle force production (8). Second, the ΔH2-R15 mini-gene and the ΔR2-15/ΔR18-23/ΔC micro-gene contain the R16/17 neuronal nitric oxide synthase (nNOS)-recruiting domain, but the ΔR4-23/ΔC micro-gene does not (25). Third, the ΔH2-R15 mini-gene and the ΔR4-23/ΔC micro-gene contain three hinges in the rod domain, but the ΔR2-15/ΔR18-23/ΔC micro-gene only carries two hinges. Finally, hinge 2 has been recently shown to compromise function of the truncated dystrophin genes (1). Hinge 2 is included in the ΔR4-23/ΔC micro-gene but not in the other two synthetic genes. Experimental transgenic mice were backcrossed with mdx mice for at least six generations. All experimental mice were housed in a specific pathogen-free facility. Only male mice were used in the study. The sample size for muscle physiology assay is provided in Table 1 or indicated in the figure legends.
Table 1.
Morphometric properties of experimental animals
| Strain | Age, mo | N | Body Weight, g | EDL Weight, mg | EDL CSA, mm2 | Lo, mm |
|---|---|---|---|---|---|---|
| BL10 | 6 | 10 | 32.03 ± 0.57 | 13.90 ± 0.77 | 2.12 ± 0.12 | 14.09 ± 0.04 |
| 20 | 10 | 37.44 ± 0.64 | 13.00 ± 0.18 | 2.12 ± 0.03 | 13.14 ± 0.04 | |
| mdx | 6 | 13 | 35.44 ± 0.42 | 16.73 ± 0.42† | 2.57 ± 0.07† | 13.93 ± 0.05 |
| 20 | 19 | 31.12 ± 0.56* | 15.95 ± 0.33† | 2.42 ± 0.05† | 14.13 ± 0.10† | |
| ΔR2-15/ΔR18-23/ΔC | 6 | 10 | 34.89 ± 1.74 | 13.34 ± 0.38 | 2.01 ± 0.06 | 13.17 ± 0.03 |
| 20 | 9 | 41.92 ± 1.20a | 12.49 ± 0.61 | 2.01 ± 0.10 | 13.32 ± 0.06 | |
| ΔR4-23/ΔC | 6 | 10 | 32.33 ± 0.48 | 12.51 ± 0.41 | 1.99 ± 0.06 | 13.46 ± 0.03 |
| 20 | 13 | 35.18 ± 1.24‡ | 12.12 ± 0.40 | 1.97 ± 0.06 | 13.19 ± 0.06 | |
| ΔH2-R15 | 6 | 9 | 32.51 ± 0.79 | 12.77 ± 0.22 | 2.08 ± 0.03 | 13.19 ± 0.04 |
| 20 | 9 | 32.72 ± 1.40§ | 12.26 ± 0.28 | 1.94 ± 0.05 | 13.54 ± 0.09 |
Applicable values are means ± SE.
mdx mice are significantly different from age-matched BL10 mice.
mdx mice are significantly different from all other age-matched mouse strains.
ΔR4-23/ΔC mice are significantly different from age-matched mdx mice.
ΔH2-R15 mice are significantly different from age-matched BL10 mice.
ΔR2-15/ΔR18-23/ΔC mice are significantly different from all other age-matched mouse strains. EDL, extensor digitorum longus; CSA, cross-sectional area; Lo, optimal length.
Histology and immunostaining.
Morphological examinations were performed on the intact EDL muscle that was not subjected to muscle physiology assay. Hematoxylin and eosin (HE) staining was used to study general histopathology. Central nucleation and the myofiber size were determined from five random microscopic fields of an HE-stained muscle section. The myofiber size was determined using the Feret's minimum diameter method. Fibrosis was examined by Masson trichrome staining and the hydroxyproline assay (see below for detail) (3, 27). Macrophage (rat anti-mouse F4/80; 1:200; Catalag Laboratories, Burlingame, CA) and neutrophil (rat anti-mouse Ly6-G; 1:800; BD Pharmingen, San Diego, CA) infiltration were determined by immunohistochemical staining using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA). Immunostaining for dystrophin was performed using two different monoclonal antibodies including a human dystrophin NH2 terminus-specific antibody (Dys-3, 1:20; Vector Laboratories) and a dystrophin COOH terminus-specific antibody (Dys-2, 1:20; Vector Laboratories).
EDL muscle preparation.
Experimental mice were anesthetized via intraperitoneal injection of a cocktail containing 25 mg/ml ketamine, 2.5 mg/ml xylazine, and 0.5 mg/ml acepromazine at 2.5 μl/g body wt. The EDL muscle was gently dissected and mounted to an intact muscle test system (Aurora Scientific, Aurora, ON, Canada) as previously described (20, 21). Briefly, the EDL muscle was exposed. The proximal and distal EDL tendons were tied at the muscle tendon junction. The proximal end of the EDL muscle was secured to a dual-mode servomotor transducer, and the distal end was attached to a fixed post using a 4–0 suture (SofSilk USSC Sutures, Norwalk, CT). Subsequently, the EDL muscle was submerged in a 30°C jacketed organ bath containing oxygenated (95% O2-5% CO2) Ringer's buffer.
Evaluation of active muscle contraction.
After 10-min equilibration, the optimal length (Lo) of the EDL muscle was determined by the isometric twitch method. Briefly, twitch stimulation was applied while the muscle was strained at different lengths. The length that yielded the highest twitch force is defined as the Lo, and this length was measured with an electronic digital caliper (F0.01 mm; Control Company, Friendswood, TX). The maximum isometric tetanic force (Po) was measured at 150 Hz. Time to peak tension (TPT) and half-relaxation time (1/2 RT) were determined from the maximum isometric tetanic contraction (19). Data were recorded and analyzed using the Lab View-based DMC and DMA programs (Version 3.12, Aurora Scientific).
Evaluation of the passive properties.
The elastic property of the EDL muscle was determined at Lo. The EDL muscle was passively strained from 100% Lo to 160% Lo using a six-step passive stretch protocol in the absence of electrical stimulation (20). At each step, the EDL muscle was stretched in an increment of 10% Lo at a rate of 2 cm/s. The stress-strain response was graphed. The viscous property was determined by measuring the stress-relaxation rate (SRR) while the muscle was stretched and held for 1.5 s (until passive stress reached plateau) at 110% Lo (20). The SRR was calculated using the equation (Stress1 − Stress2)/(Time2 − Time1). Specifically, the SRR was determined from the following time frames: the peak to 0.1 s postpeak, 0.1 to 0.2 s postpeak, 0.2 to 0.5 s postpeak, 0.5 to 1 s postpeak, and 1 to 1.5 s postpeak. The SRR values from all five time frames were plotted in a single bar graph. Data were recorded and analyzed using Lab View-based software (Aurora Scientific). At the end of each experiment, the tendon was removed, and the EDL muscle weight was determined. The muscle cross-sectional area (CSA) was calculated as described before (4, 20).
Quantification of the hydroxyproline content.
The hydroxyproline content was determined as previously described (20). Briefly, the proximal and distal tendons were removed from the EDL muscle. The muscle was then lyophilized overnight, and the dry weight was measured. The sample was hydrolyzed in 1 ml 6 N HCl for 3 h at 115°C. After neutralization with 10 N NaOH (to the final pH of ∼7.5), the muscle lysate was oxidized with chlormatine-T. The hydroxyproline content was quantified by measuring the color absorbance at 558 nm. The hydroxyproline concentration was determined from a standard curve calculated from a linear dilution of l-hydroxyproline (trans-4-Hydroxy-l-proline, cat. no. 56250; Sigma-Aldrich, Saint Louis, MO).
Statistical analysis.
Data are presented as means ± SE. Statistical analysis was performed using the SPSS software (IBM, Armonk, NY). Statistical significance among multiple groups was determined by one-way ANOVA followed by Bonferroni post hoc analysis. Difference was considered significant when P < 0.05.
RESULTS
Body weight and anatomic properties of the EDL muscle.
We examined male transgenic mdx mice expressing either one of the two micro-genes (ΔR2-15/ΔR18-23/ΔC or ΔR4-23/ΔC) or the mini-gene (ΔH2-R15) at the ages of 6 (young) and 20 (old) mo (Fig. 1). No significant difference in the body weight was noticed at 6 mo among all groups (Table 1). Interestingly, at 20 mo of age, the body weight was normalized in the ΔR4-23/ΔC transgenic mice but not in other two transgenic strains (Table 1). As we previously reported, EDL muscle weight and CSA were significantly increased in mdx compared with that of BL10 mice in both ages (Table 1) (20). Importantly, the anatomic parameters of the mdx EDL muscle were completely restored to those of the BL10 EDL muscle in all transgenic mdx mice at both time points (Table 1).
EDL muscle histology.
Dystrophin immunostaining showed the expected pattern (Fig. 2, A and B). The mdx EDL muscle was clearly dystrophic at both ages. It showed great variations in the fiber size, significantly elevated numbers of centrally nucleated myofibers, fibrosis, and inflammation (Figs. 2 and 3). All three strains of transgenic mdx mice showed HE staining similar to that of BL10 at both ages (Fig. 2, A and B). They displayed uniform myofiber size, minimal central nucleation, and similar distribution of myofiber size to those of BL10 (Figs. 2 and 3). There was also nominal fibrosis and inflammatory cell infiltration in transgenic mouse muscle (Fig. 2, A and B).
Fig. 2.

Characterization of the histopathology of the EDL muscle in BL10, mdx, and transgenic mdx mice at 6 (A) and 20 (B) mo of age. Representative hematoxylin and eosin (HE), Masson trichrome (MTC), macrophage, and neutrophil staining as well as dystrophin immunofluorescence photomicrographs are shown for each strain. For dystrophin immunostaining, 2 antibodies were used including a human dystrophin NH2 terminus-specific antibody (N-dys) and a pan-species dystrophin COOH terminus-specific antibody (C-dys). Arrow, macrophage; arrowhead, neutrophil; asterisks, to mark the same myofiber in serial. Scale bar = 100 μm for all images.
Fig. 3.

Quantitative characterization of extensor digitorum longus (EDL) muscle histopathology. A and B: 6 mo old. C and D: 20 mo old. The percentage of the myofiber with centrally localized nuclei in all 5 mouse strains is shown in A and C. Distribution of myofiber cross-sectional area in BL10, mdx, ΔR2-15/ΔR18-23/ΔC, and ΔH2-R15 is shown in B and D. †mdx mice are significantly different from all other age-matched mouse strains.
Characterization of the isometric tetanic force of the EDL muscle.
The specific isometric tetanic force was restored to the control level in young adult transgenic mdx mice as we reported before (Fig. 5A) (22, 25, 28). To determine whether the truncated dystrophin genes can result in long-term muscle force preservation, we measured tetanic force at 20 mo of age (Fig. 5B). Despite the differences in transgene configuration in three different transgenic strains (Fig. 1), the specific isometric tetanic force was fully restored in every transgenic strain in aged mice (Fig. 5B).
Fig. 5.
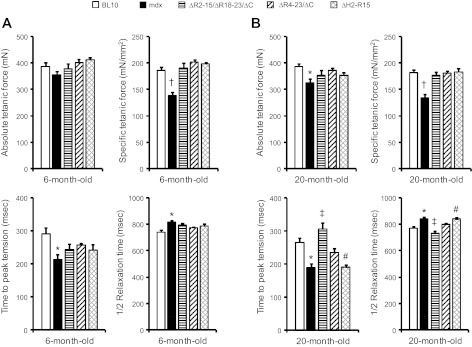
Assessment of maximum isometric tetanic force properties of the EDL muscle. The absolute isometric tetanic force, specific isometric tetanic force, time to maximum force, and half relaxation time of the EDL muscle were compared between BL10, mdx, and all 3 transgenic mdx mice at 6 (A) and 20 (B) mo of age. Time to peak tension and half relaxation time are determined from maximal tetanic stimulation. *mdx mice are significantly different from age-matched BL10 mice. †mdx mice are significantly different from all other age-matched mice strains. ‡ΔR2-15/ΔR18-23/ΔC transgenic mice are significantly different from age-matched mdx, ΔR4-23/ΔC, and ΔH2-R15 transgenic mdx mice. #ΔH2-R15 transgenic mdx mice are significantly different from age-matched BL10 mice.
To further investigate the kinetics properties of the isometric tetanic force, we examined the TPT and the 1/2 RT (Fig. 5). Consistent with our previous study (19), the mdx EDL muscle reached the peak tension significantly faster than BL10. However, it took longer time for mdx muscle to relax (Fig. 5). In 6-mo-old transgenic mdx mice, both TPT and 1/2 RT showed a trend of normalization although not statistically significant (Fig. 5A). Some differences were noticed in 20-mo-old transgenic mdx mice (Fig. 5B). The ΔR2-15/ΔR18-23/ΔC transgenic line showed a TPT significantly slower, whereas 1/2 RT was significantly faster than those of mdx and other two transgenic lines (Fig. 5B). Surprisingly, the ΔH2-R15 strain of transgenic mice showed a quite unique pattern. The TPT and 1/2 RT were not improved in this strain at 20 mo of age (Fig. 5B).
Characterization of the EDL muscle passive properties.
The stress-strain profile was examined. Similar to our previous reports (19, 20), mdx muscles exhibited increase in stiffness at both ages. The resistant force developed at the peak stress (130% Lo strain) was significantly higher than that of BL10. Furthermore, the resistant force developed after peak stress decreased rapidly in mdx, indicating muscle failure (Fig. 6A) (20). The increased muscle stiffness in the mdx muscle was brought to the wild-type level in all three transgenic strains (Fig. 6A).
Fig. 6.
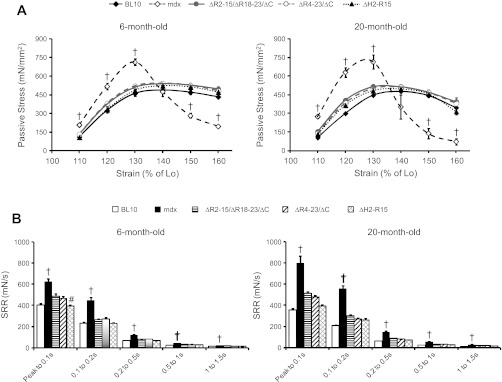
Characterization of the EDL muscle passive properties in transgenic mdx mice. Comparison of the stress-strain relationship (A) and stress relaxation rate (SRR) (B) between BL10, mdx, and all 3 transgenic mdx mice was determined at 6 and 20 mo old. †mdx mice are significantly different from all other age-matched mouse strains. #ΔH2-R15 transgenic mdx mice are significantly different from age-matched ΔR2-15/ΔR18-23/ΔC transgenic mdx mice. Lo, optimal length.
The viscous property of the mdx muscle was significantly altered compared with that of BL10 (19, 20). In particular, the mdx muscle had a significantly higher SRR (Fig. 6B). The SRR was corrected in every transgenic strain (Fig. 6B).
Fibrosis quantification with the hydroxyproline assay.
We have previously shown that the amount of fibrosis positively correlated with the level of muscle stiffness (20). To further understand the mechanism(s) of the improved passive properties in transgenic mice, we quantified the hydroxyproline content. Consistent with the morphological data (Fig. 2), the hydroxyproline content appeared lower in transgenic mdx mice compared with that of mdx mice at 6 mo of age (Fig. 4). However, to our surprise, multigroup analysis did not reveal significant improvement over mdx in 6-mo-old transgenic mdx mice (Fig. 4). The fibrosis content was significantly lower than that of age-matched mdx mice in 20-mo-old transgenic mdx mice (Fig. 4). Nevertheless, it was still significantly higher than that of age-matched BL10 mice (Fig. 4).
Fig. 4.

Quantification of the hydroxyproline (HP) content in the EDL muscle. The hydroxyproline content in the EDL muscle was compared between BL10, mdx, and 3 transgenic mdx strains at 6 mo of age (A) and 20 mo of age (B). *mdx mice are significantly different from age-matched BL10 mice. #ΔR4-23/ΔC transgenic mdx mice are significantly different from age-matched mdx mice. †mdx mice are significantly different from all other age-matched mouse strains. ‡BL10 mice are significantly different from age-matched ΔR4-23/ΔC and ΔH2-R15 transgenic mdx mice. n = 5 for 20-mo-old ΔR2-15/ΔR18-23/ΔC transgenic mdx mice; n = 7 for all other groups.
DISCUSSION
In this study, we tested hypotheses that shortened dystrophins can increase muscle-specific force in aged mdx mice, improve the kinetics of the contractile force, and restore the passive properties of dystrophin-deficient muscle in a transgene-dependent manner. Specifically, we compared active force and passive mechanic properties of the EDL muscle in 6- and 20-mo-old BL10, mdx, and three strains of mini/micro-dystrophin transgenic mdx mice that each expressed one of the three truncated dystrophin genes including the ΔR2-15/ΔR18-23/ΔC micro-gene, ΔR4-23/ΔC micro-gene, and ΔH2-R15 mini-gene (Fig. 1). To correlate physiology study results, we have also examined the anatomical and histological properties of the EDL muscle in these mouse strains at the 6- and 20-mo ages (Fig. 2, Table 1).
We first measured the force generated under tetanic contraction. Previous studies suggest that transgenic overexpression of any one of above three mini-/micro-dystrophin genes can correct force deficit in young adult mdx mice (25, 29). Consistent with the published results, we also found that specific tetanic force was completely restored to that of the wild-type level in all three strains of transgenic mdx mice at 6 mo of age (Fig. 5A). Because young adult mdx mice do not show characteristic signs of muscular dystrophy and only aged mdx mice display clinical disease similar to that of human patients (10), we examined muscle force in aged mice (Fig. 5B). Encouragingly, all transgenic mdx mice showed muscle force similar to that of BL10 mice at 20 mo of age (Fig. 5B). These results confirm our initial hypothesis and suggest that the truncated synthetic dystrophin genes may lead to long-term preservation of muscle contractility.
TPT and 1/2 RT were compromised in the mdx EDL muscle (Fig. 5) (19). We have initially hypothesized that truncated dystrophins can correct these kinetic parameters. Surprisingly, this hypothesis was disapproved by our experimental results (Fig. 5). TPT and 1/2 RT showed a trend of improvement but were not completely restored to the control level in 6-mo-old transgenic mdx mice (Fig. 5A). Furthermore, 20-mo-old transgenic mdx mice showed a variety of different patterns from no correction (such as the ΔH2-R15 strain) to the normal level (such as the ΔR2-15/ΔR18-23/ΔC strain) (Fig. 5B). Muscle contraction and relaxation is essentially directed by the release of calcium from the sarcoplasmic reticulum and active uptake of calcium back to the sarcoplasmic reticulum, respectively. Intracellular calcium concentration thus plays a pivotal role in determining the kinetic profile of muscle contraction. Shortened TPT and elongated 1/2 RT are consistent with chronic calcium overload in mdx muscle (reviewed in Refs. 15 and 24). We speculate that the lack of full correction of TPT and 1/2 RT may suggest that overexpression of truncated dystrophins may have not fully restored calcium homeostasis in transgenic mdx muscle. Future study on the sarcoplasmic reticulum calcium uptake ATP-dependent pump and the myosin ATPase activity may help to uncover the mechanisms underlying our observations (36).
The main question we intended to address in this study is whether the truncated dystrophin genes can correct muscle stiffness. To this end, we have proposed two hypotheses at the beginning of our study. On one hand, we hypothesized that truncated dystrophin proteins can improve the passive properties of the mdx muscle. On the other hand, we hypothesized that the difference in the size and composition of the truncated dystrophin protein may lead to different levels of recovery. To test these two hypotheses, we examined the elastic and viscous properties of the EDL muscle. We chose the EDL muscle as the target muscle because we have previously shown that mdx EDL muscle is stiffer than that of BL10 (19, 20). Our results demonstrated that transgenic mice had the same stress-strain curve as that of BL10 mice at either 6- or 20-mo time points. Furthermore, the SRR was also significantly improved in all transgenic lines (Fig. 6B). Based on these results, we concluded that truncated dystrophins were fully capable of rectifying abnormal elastic and viscous properties of the mdx muscle. In addition, the length and the composition (such as the presence or absence of the COOH-terminal domain, nNOS binding domain, and hinge 2 and the number of hinges in the rod domain) of the shortened dystrophin gene did not significantly affect the restoration of the passive properties in mdx skeletal muscle.
We have previously noted that muscle stiffness correlated with fibrosis (20). For this reason, we also quantified the hydroxyproline content. Despite the remarkable improvement of the elastic and viscous properties in transgenic mice at both 6- and 20-mo time points (Fig. 6), the hydroxyproline content in transgenic mdx mice was not restored to the level of BL10 mice (Fig. 4, A and B). Nevertheless, expression of the truncated dystrophin genes has significantly prevented muscle fibrosis at the age of 20 mo (Fig. 4B). Our results suggest that the total amount of the hydroxyproline content cannot fully explain muscle stiffness seen in DMD murine model. Other factors (such as the type of the collagen and the pattern of the cross-linking) should also be considered (5, 17, 32, 37). Future studies may help clarify this issue.
Collectively, our results here suggest that truncated dystrophin genes used in this study have greatly reduced histological lesions and resulted in long-term restoration of muscle-specific force in both young adult and very old mdx mice (Figs. 2–5). More importantly, these therapeutic candidate genes have effectively recovered the passive properties of the mdx EDL muscle in a transgene-independent manner. Our results provide strong support to further develop the mini- and micro-dystrophin genes for DMD gene therapy. Nevertheless, it should be emphasized that the results described in this study were from transgenic mice. Further studies using AAV-gene mediated therapy is warranted to truly test therapeutic effect in a more clinical relevant context.
GRANTS
This work was supported by grants from the National Institutes of Health (AR-49419 to D. Duan), Muscular Dystrophy Association (D. Duan) and NIH training grant T90DK70105 (C. Hakim).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: C.H.H. and D.D. conception and design of research; C.H.H. performed experiments; C.H.H. and D.D. analyzed data; C.H.H. and D.D. interpreted results of experiments; C.H.H. prepared figures; C.H.H. drafted manuscript; C.H.H. and D.D. edited and revised manuscript; C.H.H. and D.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Marianne Abdo, Yongping Yue, Keqing Zhang, and Juveria Nayeem for excellent technical assistance. We also thank Drs. Elizabeth S. Crister and Natalia Karasseva at the University of Missouri Transgenic Animal Core for help with generating the founder transgenic mice.
REFERENCES
- 1. Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet 6: e1000958, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82: 291–329, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J, Duan D. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice. Mol Ther 17: 253–261, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burgeson RE. The collagens of skin. Curr Probl Dermatol 17: 61–75, 1987 [PubMed] [Google Scholar]
- 6. Cornu C, Goubel F, Fardeau M. Muscle and joint elastic properties during elbow flexion in Duchenne muscular dystrophy. J Physiol 533: 605–616, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cornu C, Goubel F, Fardeau M. Stiffness of knee extensors in Duchenne muscular dystrophy. Muscle Nerve 21: 1772–1774, 1998 [DOI] [PubMed] [Google Scholar]
- 8. Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol 150: 1399–1410, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De la Porte S, Morin S, Koenig J. Characteristics of skeletal muscle in mdx mutant mice. Int Rev Cytol 191: 99–148, 1999 [DOI] [PubMed] [Google Scholar]
- 10. Duan D. Duchenne muscular dystrophy gene therapy: Lost in translation? Res Rep Biol 2011: 31–42, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duan D. From the smallest virus to the biggest gene: marching towards gene therapy for Duchenne muscular dystrophy. Discov Med 6: 103–108, 2006 [PMC free article] [PubMed] [Google Scholar]
- 12. Dubowitz V. Deformities in Duchenne dystrophy. Neuromuscul Disord 20: 282, 2010 [DOI] [PubMed] [Google Scholar]
- 13. Emery AE. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscul Disord 1: 19–29, 1991 [DOI] [PubMed] [Google Scholar]
- 14. England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343: 180–182, 1990 [DOI] [PubMed] [Google Scholar]
- 15. Gailly P. New aspects of calcium signaling in skeletal muscle cells: implications in Duchenne muscular dystrophy. Biochim Biophys Acta 1600: 38–44, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Garcia-Pelagio KP, Bloch RJ, Ortega A, Gonzalez-Serratos H. Biomechanics of the sarcolemma and costameres in single skeletal muscle fibers from normal and dystrophin-null mice. J Muscle Res Cell Motil 31: 323–336, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gosselin LE, Adams C, Cotter TA, McCormick RJ, Thomas DP. Effect of exercise training on passive stiffness in locomotor skeletal muscle: role of extracellular matrix. J Appl Physiol 85: 1011–1016, 1998 [DOI] [PubMed] [Google Scholar]
- 18. Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther 16: 657–664, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hakim CH, Duan D. Gender differences in contractile and passive properties of mdx extensor digitorum longus muscle. Muscle Nerve 45: 250–256, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hakim CH, Grange RW, Duan D. The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J Appl Physiol 110: 1656–1663, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hakim CH, Li D, Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol Biol 709: 75–89, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 8: 253–261, 2002 [DOI] [PubMed] [Google Scholar]
- 23. Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928, 1987 [DOI] [PubMed] [Google Scholar]
- 24. Hopf FW, Turner PR, Steinhardt RA. Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem 45: 429–464, 2007 [DOI] [PubMed] [Google Scholar]
- 25. Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 119: 624–635, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lefaucheur JP, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat Rec 242: 70–76, 1995 [DOI] [PubMed] [Google Scholar]
- 27. Li D, Shin JH, Duan D. iNOS ablation does not improve specific force of the extensor digitorum longus muscle in dystrophin-deficient mdx4cv mice. PLoS One 6: e21618, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li D, Yue Y, Lai Y, Hakim CH, Duan D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J Pathol 223: 88–98, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu M, Yue Y, Harper SQ, Grange RW, Chamberlain JS, Duan D. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther 11: 245–256, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lynch GS, Rafael JA, Chamberlain JS, Faulkner JA. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am J Physiol Cell Physiol 279: C1290–C1294, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. Postgrad Med J 85: 622–630, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Mays PK, Bishop JE, Laurent GJ. Age-related changes in the proportion of types I and III collagen. Mech Ageing Dev 45: 203–212, 1988 [DOI] [PubMed] [Google Scholar]
- 33. Pasternak C, Wong S, Elson EL. Mechanical function of dystrophin in muscle cells. J Cell Biol 128: 355–361, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci 129: 97–105, 1995 [DOI] [PubMed] [Google Scholar]
- 35. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90: 3710–3714, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stephenson DG, Lamb GD, Stephenson GM. Events of the excitation-contraction-relaxation (E-C-R) cycle in fast- and slow-twitch mammalian muscle fibres relevant to muscle fatigue. Acta Physiol Scand 162: 229–245, 1998 [DOI] [PubMed] [Google Scholar]
- 37. Viidik A. Elasticity and tensile strength of the anterior cruciate ligament in rabbits as influenced by training. Acta Physiol Scand 74: 372–380, 1968 [DOI] [PubMed] [Google Scholar]