

Summary
The important biological roles of nitric oxide (NO) have prompted the development of analytical techniques capable of sensitive and selective detection of NO. Electrochemical sensing, more than any other NO-detection method, embodies the parameters necessary for quantifying NO in challenging physiological environments such as blood and the brain. Herein, we provide a broad overview of the field of electrochemical NO sensors, including design, fabrication, and analytical performance characteristics. Both electrochemical sensors and biological applications are detailed.
Introduction
Few diatomic molecules have received as much attention as nitric oxide (NO). Although well known as a potent environmental pollutant, it was not until 1987 that Furchgott, Ignarro, and Murad separately identified NO as the endothelium-derived relaxation factor,1–3 a discovery that would ultimately lead to their shared Nobel prize in physiology in 1998. In the years since this discovery, many scientists have continued to unravel the roles of NO in physiology. When produced endogenously from L-arginine by a family of enzymes called nitric oxide synthases (NOSs),4 NO has been found to be active in the cardiovascular,5 nervous,6 and immune7, 8 system, and in the wound-healing process.9 Exogenously released NO has been shown to elicit diverse biological responses such as reduced microbial viability10 and decreased platelet activation.11
Widespread interest in NO and its biological roles has generated demand for analytical techniques capable of its measurement and quantification. Such technology is not straightforward due to NO’s widely varying concentration. In the human body, the effect of NO is dependent on its concentration, ranging from sub-nanomolar to micromolar levels.12, 13 To complicate matters further, NO has a short half-life (typically <10 s) in biological milieu due to its reactivity with oxygen, thiols, free radicals and hemes.14 Effective NO detection schemes thus require a wide dynamic range, adequate sensitivity, and fast response time. Furthermore, the method must be highly selective toward NO over interfering species, which is often challenging due to the overwhelming complexity of biological systems.
The majority of analytical approaches for measuring NO may be categorized into spectroscopic and electrochemical methods. Most spectroscopic NO detection methods involve either indirect measurement of byproducts of reactions between NO and other chemical species (i.e., Griess reaction and chemiluminescence); or, direct measurement of adducts formed between NO and metal complexes (absorbance), fluorescent dyes (fluorescence), or spin traps (electron paramagnetic resonance spectroscopy).15 Some spectroscopic methods offer high sensitivity and selectivity for NO. For example, fluorescence NO detection is widely used for intracellular imaging of NO, enabling NO measurement at concentrations as low as 2 pM.16 However, most spectroscopic methods present obstacles for in vivo NO detection due to the complex instrumentation that is difficult to miniaturize. Conversely, electrochemical NO sensors allow for direct NO analysis with attractive analytical performance (i.e., sensitivity, selectivity, response time, sensor size, and inexpensive fabrication and operation).
Electrochemical Detection of Nitric Oxide
Electrode materials
The materials used to construct an electrochemical NO sensor play a pivotal role in the sensitivity and quality of the ensuing analytical measurement. Materials most often chosen as the working electrode include platinum (Pt) and its alloys,7, 17 carbon fiber,18 glassy carbon (GC),19 and gold (Au).20 By varying the composition and surface characteristics of the electrode material, the sensitivity, selectivity, signal stability, and required oxidation or reduction potential become tunable to varying extent. For example, Meyerhoff’s group found that platinum electrodes could be made more stable and sensitive to NO via platinization, a process where platinum black particles are electrochemically formed on the electrode surface, increasing the roughness and effective surface area.21 By platinizing the platinum electrode of the NO sensor, 10-fold gains in both the NO detection limit and sensitivity were achieved. The authors surmised that the source of this performance enhancement was a concomitant increase in electron-transfer kinetics with a decrease in the potential required to drive the oxidation of NO.
Electroactive biological interferences
Of the various examples of electrochemical NO sensors intended for biological applications, few have been tested against more than a handful of applicable biological interferences. The extent to which a particular interfering species influences an NO measurement depends on the type of sensor, the applied potential, the characteristics of the permselective membrane (i.e., surface charge, porosity, hydrophobicity, and thickness), and the intended biological location of the analysis. For example, interference from gaseous oxygen is only a concern if NO is being measured via electroreduction, since the reduction potential for NO and oxygen are similarly negative. Predicting likely interfering species is further complicated by the dependence of NO and interfering species concentrations on a multitude of outside stimuli (e.g., disease, injury, age, nutrition, and prior medical history). The most commonly encountered interfering species in biological milieu and their typical biological concentration ranges are listed in Table 1.22–25 Nitrite is of particular concern due to its high concentration and similar size and oxidation potential to NO, making it difficult to discriminate against. Additionally, nitrite is a stable byproduct of the auto-oxidation of NO by endogenous oxygen and oxyhemoglobin, resulting in a direct dependence between the two species. Carbon monoxide (CO) is equally problematic because of its similarities to NO in size, hydrophobicity, oxidation potential, and physiological roles.26 Recent studies have made it apparent that NO and CO regulate each other through various physiological processes.27 As a result, attempts to exclude CO using a NO selective membrane often fail. Providing selectivity to NO sensors over all of the above-mentioned interfering species is clearly challenging. The discussion of strategies for conferring selectivity to NO sensors is a major thrust of our tutorial.
Table 1.
Possible interfering species and their physiological concentrations during the electrochemical NO measurements.
| compound | concentration range | specimen | ref. |
|---|---|---|---|
| nitrite | <20 m Ma | blood (plasma) | 25 |
| ascorbic acid | 34 – 114 mM | blood (plasma) | 22 |
| uric acid | 150 – 470 mM | blood (serum) | 22 |
| acetaminophen | 66 – 199 mMb | blood (serum or plasma) | 22 |
| carbon monoxide | 0.5 – 1.5 mMa | mouse kidney | 24 |
| dopamine | <2.0 nM | blood (plasma) | 22 |
| norepinephrine | 0.35 – 2.96 nM | blood (plasma) | 22 |
| serotonin | 0.28 – 1.14 mM | whole blood | 22 |
| DOPACc | 5.88 – 23.10 nM | blood (plasma) | 23 |
| 5-HIAAd | 18.31 – 65.91 nM | blood (serum) | 22 |
Basal concentration
Therapeutic concentration
DOPAC, 3,4-dihydroxyphe-nylacetic acid
5-HIAA, 5-hydroxyindole-3-acetic acid
Sensor classification
A wide variety of sensor designs have been developed and adapted for use in the measurement of NO in solution. While the construction of these devices varies widely, sensors are typically composed of a surface capable of the electro-oxidation or –reduction of NO, and a mechanism for discriminating against electroactive interferences. A permselective membrane is commonly employed for this purpose. In general, sensor styles may be categorized as follows; 1) Shibuki-style, 2) solid permselective, and 3) solid catalytic (Figure 1).
Figure 1.
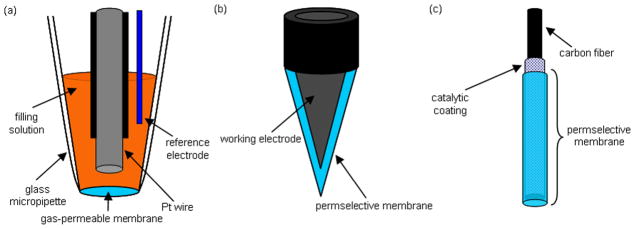
Schematic diagrams of (a) Shibuki-style, (b) solid permselective, and (c) solid catalytic NO sensors.
Shibuki-style NO sensors are modified versions of the initial oxygen (O2) sensor first reported by Leland Clark in 1956.28 This sensor comprises an electrolyte-filled micropipette into which both platinum working and silver reference wires are placed, and covered with a thin gas-permeable rubber membrane. Low molecular weight gases (e.g., NO and O2) easily diffuse through the membrane and to the electrode surface while larger species are excluded. By applying a negative or positive potential at the platinum wire electrode, electroactive species are reduced or oxidized, respectively, at the electrode surface, resulting in current of magnitude proportional to the analyte concentration. Shibuki reported the fabrication of the first NO-selective sensor in 1990, for which a positive electrode potential (i.e., electrooxidation) was used to oxidize and detect NO.29 While this type of sensor measures NO with adequate selectivity over nitrite, the sensitivity of the sensor varied over time and between sensors from 2.5–106.3 pA/nM NO, leading to unstable measurements. In addition, the sensor was not readily amenable to miniaturization (>150 μm diameter) due to the complexity of construction and the requirement of an internal filling solution.
Solid permselective NO electrodes have been developed to eliminate the need for an internal filling solution. Fabrication is accomplished by directly modifying a noble metal or carbon electrode with a typically hydrophobic membrane permeable to the analyte of interest but impermeable to other electroactive interferences.30 The simple design and construction of solid permselective electrodes allows them to be more easily miniaturized than Shibuki-style sensors. By layering multiple types of membranes on the electrode, the sensor selectivity may be tuned to discriminate over a wide variety of interferences including nitrite, dopamine, and acetaminophen, enabling unambiguous NO concentration determination in biological milieu. Nitric oxide is measured directly either by electrooxidation or electroreduction.
Solid catalytic electrodes were developed to further reduce the effect of electroactive interferences on the NO-selective electrode. While similar in construction to solid permselective electrodes, the catalytic electrodes incorporate a mediator (e.g., metalloporphyrins and metal phthalocyanines) either directly on the electrode surface or within a permselective membrane.30 By including a mediator capable of catalyzing the oxidation or reduction of NO, the magnitude of the required electrochemical potential (for NO measurement) is decreased, minimizing interference from other electroactive species. When combined with a permselective membrane, solid catalytic electrodes provide unparalleled selectivity. Similar to solid permselective electrodes, NO is measured directly either by catalytic electrooxidation or electroreduction.
Placement of a reference electrode (typically silver/silver chloride) is also an important consideration for all three types of NO sensors. Integration of the reference electrode inside the envelope created by the NO-selective membrane (internal) maintains a constant potential between the reference and working electrodes. Conversely, the use of a reference electrode external to the membrane results in an additional membrane potential. External reference may be problematic if physical or chemical changes to the membrane or biofouling (e.g., adhesion of platelets, blood proteins) occur during the measurement, thereby altering the membrane potential and potential applied to the working electrode. As shown in Figure 1a, Shibuki-style sensors typically integrate a reference electrode within the electrolyte filling solution. Solid permselective and solid catalytic electrodes may utilize an internal or external reference electrode depending on the measurement requirements.
Modes of Operation
Electrochemical NO sensors operate via the application of a potential at an electrode surface sufficiently positive or negative to electrochemically oxidize or reduce NO. The resulting transfer of electrons to or from the electrode surface is measured as a current, proportional to the concentration of NO in solution. The potential required for measuring NO for each operational mode depends on both the identity (e.g., gold, platinum, carbon) and specific surface properties (e.g., catalytic activity and surface roughness) of the electrode. The three most common modes of electrode operation are explained below.
Electroreduction of NO
While the majority of NO sensor-related publications involve the electrooxidation of NO (direct or catalytic), sensors that measure NO via its electroreduction have been reported. Depending on the electrode type and sample solution pH, NO is reduced at negative potentials ranging from −0.5 to −1.4 V (vs. Ag/AgCl).30 The reaction proceeds via a two electron reduction mechanism:
| (4) |
The primary advantage of electroreduction is the avoidance of most interfering species that are commonly troublesome at positive potentials. However, electroreductive sensors are often plagued by diminished sensitivity, oxygen interference, and dependence on pH and electrode surface characteristics. With proper optimization, recent reports indicate some utility for specialized biological analysis.31–33
The most problematic requirement for electroreductive NO sensors is the ability to operate at physiological pH. Perhaps the first example of a physiologically useful electroreductive NO sensor, Meulemans used differential pulse voltammetry to detect NO (reduction potential at −1.2 V vs. Ag/AgCl) in phosphate-buffered saline (PBS) with a sharpened carbon fiber.33 While the sensor did not respond to nitrite or oxygen, the reported detection limit for NO was poor (10 μM) and no information was given concerning interferences.
To improve the sensitivity and selectivity of electroreductive NO sensors, electrodes have been coated with transition metal complexes that catalyze the reduction of NO, thereby minimizing the magnitude of the potential required. For example, Maskus and coworkers used a chromium (Cr) complex to create a sensitive NO electrode capable of operation in aqueous buffer at pH 7.0.32 The authors modified a glassy carbon electrode with a film of [Cr(4′-vinyl-2,2′,6,2″-terpyridyl)2]3+ and a thin coating of Nafion. The resulting sensor was capable of measuring NO at sub-micromolar concentrations at −0.9 V vs. saturated calomel electrode (SCE). Neither nitrite nor O2 were found to pose significant interferences. As a demonstration of the biological utility of the sensor, the authors successfully detected stimulated NO generation from bacteria in buffer at pH 7.
Metalloproteins such as hemoglobin have also proven useful for catalyzing the electroreduction of NO. For example, Liu and colleagues fabricated an NO sensor composed of hemoglobin immobilized within polyethyleneimine (a biocatalyst stabilizer) on a pyrolytic graphite surface.31 With the hemoglobin, the required potential for NO was reduced from −1.3 V for the bare pyrolytic graphite electrode to −0.680 V (vs. SCE). Despite the more positive potential, the authors reported a 40 nM NO detection limit and linear response range from 0.1 – 8 μM in PBS (pH 7.4). Ascorbic acid was the only interferent tested, resulting in a 0.5% increase in current for equimolar concentrations of ascorbic acid in an NO solution. The selectivity of the sensor to more concerning interfering species such as nitrite and O2 was not reported.
Though less biologically applicable, a greater degree of NO sensitivity has been reported for sensors operating at non-physiological pH.34 For example, Zen et al. fabricated an NO sensor using a Nafion/lead ruthenate pyrochlore electrode with a NO detection limit of 4.8 nM at pH 1.65 (buffered). While the low pH requirement renders this sensor unsuitable for most biological applications, it highlights the analytical sensitivity attainable in the electroreduction mode. Nevertheless, the majority of published NO sensor manuscripts have employed the electrooxidative mode due to the poor limits of detection and unavoidable oxygen interference for biological application via electroreduction.
Direct Electrooxidation of NO
The electrochemical reaction of NO on metal surfaces at positive electrode potentials (typically 0.6 – 0.9 V vs. Ag/AgCl)35 proceeds via a three-electron oxidation mechanism:
| (1) |
| (2) |
| (3) |
During the third step/reaction, nitrite is electrochemically oxidized to nitrate. As a result, endogenously produced nitrite presents a significant source of electrochemical interference.36 Typically present in biological tissues at more than an order of magnitude higher concentration than NO, a successful NO electrode via electrooxidation must include a physical contingency for excluding nitrite. Other common interfering species include acetaminophen, ascorbic acid, uric acid, dopamine, and CO. Careful attention thus must be given to understand the type and concentration of such interferences when making NO measurements with bare electrodes.
Many direct oxidation NO sensors are based on the solid permselective platform, where an electroactive surface is modified directly with an NO-selective membrane (Figure 1b). The permselective membrane limits the diffusion of interfering species to the working electrode by electrostatic repulsion and/or hydrophobic character. Selectivity is also dependent on membrane thickness, where thicker membranes enable greater selectivity, but often at the expense of sensitivity to NO.
While a direct comparison of NO sensitivity and limit of detection is easily made between the sensors described below, the variety of sensor designs and biological applications (with divergent biological interferences) make it difficult to fully compare sensors in terms of selectivity over interferences. A list comparing the performance characteristics of the direct oxidation NO sensors discussed is provided in Table 2.
Table 2.
Materials and performance characteristics of selected NO sensors that operate via the direct electrooxidation of NO.
| Electrode materiala | NO-selective membraneb | E-chem modec | Diameter (μM) | Linear range (μM) | LODd (nM) | Calibration methode | Interfering species testedf | Reference |
|---|---|---|---|---|---|---|---|---|
| Au | Nation | DPV | 25 | 10 – 100 | dNO | nitrite | 20 | |
| Pt | polycarbazole | DPV | 0.01 – ~1000 | dNO | AA,DA | 38 | ||
| Pt | polystyrene | CPA | 10 | 0.1 – 1.0 | 75 | dNO | nitrite | 39 |
| Pt/Pt black | fluorinated xerogel | CPA | 20 | 0.0002 – 0.003, 0.5 – 4.0 | 0.083 | dNO | nitrite, AA, UA, DA, ammonia, CO | 40 |
| Pt/Pt black | PTFE | CPA | 250 | Sub-nM - 1 | 1 nM | dNO | CO | 24 |
| CF | Nation, o-PD | CPA, CA | 39 | 0.1 – 1.2 | 35 | dNO | nitrite, AA, DA | 41 |
CF, carbon fiber
PTFE, polytetrafluoroehtylene; o-PD, o-phenylenediamine
DPV, differential pulse voltammetry; CPA, constant potential amperometry; CA, chronoamperometry
LOD, limit of detection
dNO, dissolved NO
AA, ascorbic acid; DA, dopamine; UA, uric acid; CO, carbon monoxide
Since anionic (e.g., ascorbic acid and nitrite) and cationic (e.g., dopamine) species are significant sources of interference for electrooxidative measurements of NO in biological systems, exclusion by electrostatic repulsion is the most common method for imparting selectivity to NO sensors. Nafion, a polymeric cation exchanger (Figure 2), has been employed extensively to exclude nitrite via electrostatic repulsion from the sulfonate group present at neutral pH.20 In an early example of a permselective electrode that selects against anionic species, Bedioui and coworkers coated gold fiber and microdisk electrodes with a Nafion film. In addition to providing good selectivity for NO, the electrodes exhibited a linear dynamic range for NO from 10 – 100 μM.20 While not exceedingly sensitive to NO, the authors noted that by decreasing the thickness of the Nafion membrane, sensitivity to NO was increased at the expense of selectivity over nitrite. Unfortunately, sensors coated with Nafion still respond to cationic and neutral species such as dopamine and acetaminophen, respectively.37
Figure 2.
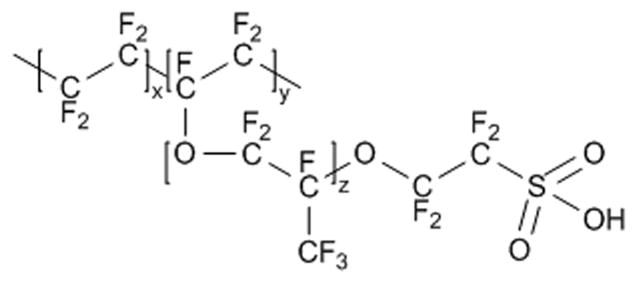
Structure of the polymeric cation exchanger Nafion.
As an alternative to Nafion, Prakash and coworkers electrochemically deposited polycarbazole on a Pt electrode, fabricating an NO sensor capable of excluding ascorbic acid.38 The use of polycarbazole, an electrically conductive polymer, increased the active surface area of the electrode, and thus enhanced NO sensitivity. Sensor response to dopamine was minimized by making differential pulse voltammetry measurements, where the oxidation peaks for NO (~0.65 V vs. Ag/AgCl) and dopamine (at 0.25 V) were monitored, allowing for their discrimination. The resulting sensor exhibited a limit of detection of 50 nM and a linear dynamic range up to 1 μM. Curiously, interference from nitrite was not tested, although the anionic nature of polycarbazole would likely enable some degree of selectivity.
Hydrophobic polymer membranes have also been employed as electrode coatings to increase both the sensitivity and selectivity of NO sensors, promoting NO diffusion over hydrophilic interfering species. For example, Kitamura et al. dip-coated polystyrene (and collodion as a protection layer) onto a Pt microcoaxial sensor (incorporating an Ag/AgCl electrode on the perimeter of the glass-insulated Pt wire).39 The detection limit of the sensor was 75 nM NO with a linear range of 0.1 – 1.0 μM, and no response to nitrite (up to 10 μM aliquots).
The ability to control the degree of hydrophobicity of an NO-selective membrane is important for maximizing sensitivity to NO and selectivity over interfering species. Utilizing multiple fluorinated alkylalkoxysilane precursors, Shin et al. demonstrated the ability to tune the hydrophobicity of NO-selective polymeric xerogel membranes, allowing for optimization of the sensor’s response to NO over electroactive interferences.40 The permeability and selectivity of the sensor to NO were maximized utilizing a 20% (heptadeca uoro-1,1,2,2-tetrahydrodecyl)trimethoxysilane (v/v, balance methyltrimethoxysilane) xerogel membrane applied to a 5 μm diameter Pt black/Pt-coated conical tungsten microelectrode (Figure 3). The limit of detection and dynamic linear range of the NO sensor were 83 pM and 0.2 nM – 4.0 μM, respectively. Furthermore, the sensor had little response to nitrite, ascorbic acid, uric acid, acetaminophen, dopamine, and ammonia. Although not commonly tested, the authors reported that the sensor also responded to carbon monoxide (CO), which might have been expected due to its physical similarity to NO.
Figure 3.

(a) SEM image of conical W-Pt-Pt black microelectrode. (b) Magnified view of electrode tip shown in (a). (c) Schematic diagram showing cutaway of W, Pt, and Pt black layers at tip of microelectrode. Reprinted with permission from reference 40. Copyright 2008 American Chemical Society.
To date, the only successful direct electrooxidation strategy for NO discrimination over CO is based on the use of a Shibuki-style dual NO/CO sensor. Due to the difficulty in designing a membrane capable of CO exclusion, Lee and Kim relied on the sensitivity ratios of NO to CO on Pt black and Pt black/tin (Sn) electrodes to determine the concentration of both NO and CO, respectively, in solution.24 The authors found that the Pt black/Sn-coated electrode was more sensitive to CO than the electrode coated with Pt black only. The addition of a polytetrafluoroethylene (PTFE) membrane provided effective discrimination against nitrite. Although no other interferences were tested, the resulting sensor exhibited adequate sensitivity to NO with a dynamic linear range from nM to μM NO. When used in vivo in mouse kidney tissues, the concentrations of CO generated were up to 8 times greater than that of NO, suggesting that without a mechanism for CO discrimination, electrochemical NO sensors would certainly have overestimated the NO concentrations. Future NO sensor work related to physiological measurements should include this contingency for CO.
A single membrane is often unable to exclude all physiological interfering species because of the diverse size and charge characteristics of common electroactive interferences. By combining multiple NO-selective membrane types, the selectivity of a sensor may be tailored for a variety of physiological environments. For example, Friedemann and coworkers deposited layers of o-phenylenediamine (o-PD) and Nafion onto carbon fiber electrodes, resulting in a sensor capable of excluding interferences by size and charge, respectively.41 Electrodes with the hybrid coating of Nafion/o-PD were found to have adequate selectivity over ascorbic acid, nitrite, and dopamine, with a detection limit of 35 nM and a linear response up to 6 μM NO. Although multiple layers may confer selectivity over a broad range of interfering species, careful study of the effects of the ensuing membrane thickness on sensor response (e.g., sensitivity) is required.
Catalytic Electrooxidation of NO
While the sensitivity and selectivity of direct electrooxidation NO sensors are dependent primarily on the permselective membrane, sensors that operate via catalytic electrooxidation utilize a redox mediator (Figure 4) to further improve analytical performance. For example, metalloporphyrins may be immobilized on the electrode surface or incorporated into a polymer coating to function as catalysts for the oxidation of NO. By increasing the electron transfer kinetics for NO oxidation, the sensitivity to NO is enhanced. The selectivity over interfering species is further improved by employing one or more permselective membranes. Table 3 lists the performance characteristics of example electrocatalytic oxidation NO sensors.
Figure 4.

Structures of redox mediators commonly incorporated into catalytic electrooxidative NO sensors: (a) Tetrakis(3-methoxy-4-hydroxyphenyl)-nickel(II) porphyrin (NiTMHPP), (b) metal N,N′-ethylenebis(salicylideneiminato) (M(Salen)), and (c) metallophthalocyanine.
Table 3.
Materials and performance characteristics of selected NO sensors that operate via the catalytic electrooxidation of NO.
| Electrode materiala | Catalytic systemb | NO-selective membranec | E-chem moded | Diameter (μM) | Linear Range (MM) | LOD (nM) | Interfering Species Tested | Reference |
|---|---|---|---|---|---|---|---|---|
| CF | poly-TMHPP-Ni | Nafion | DPV, CPA | 0.5 | <300 | 10 | nitrite | 18 |
| Pt | Mn(II)TCPPyP | Nafion | CPA, DPV | 50 | 0.3 – 2.4 | 100 | nitrite, nitrate, AA | 44 |
| CF | CoPc | Nafion | DPV | 11 | 0.2 | seratonin, DA | 46 | |
| Pt | Fe(salen) | Nafion | DPV | 15 | 0.0196 – 1.13 | 10 | nitrite | 50 |
| CF | TMHPP | Nafion, AAO, polylysine/Polypyridinium | CC | 35 – 40 | 0.5 | nitrite, DA, AA, UA, H2O2, CO | 37 |
CF, carbon fiber
TMHPP, tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin; Mn(II)TCPPyP, Mn(II)-meso-tetracarboxyphenylprophyrin; CoPc, cobalt phthalocyanine; Fe(salen), iron ethylenebis(salicylideneiminate)
AAO, ascorbic acid oxidase
DPV, differential pulse voltammetry; CPA, constant potential amperometry; CC, chronocoulometry
dNO, dissolved NO
AA, ascorbic acid; DA, dopamine; UA, uric acid; CO, carbon monoxide
Metalloporphyrins, are the most common mediator chosen for fabrication of catalytic NO sensors. Nickel (Ni), is the most frequently employed central metal ion. In their seminal report, Malinski and Taha utilized a Ni-porphyrin (Figure 4a) electropolymerized on a carbon fiber subsequently modified with a Nafion film.18 In this configuration, the sensor had a detection limit of 10 nM and linear response up to 300 μM NO. When tested in solutions containing both NO and nitrite, the authors observed only a small increase in current and no change in peak potential indicating minimal interference from nitrite due to the lower oxidation potential employed. An important advantage of utilizing carbon fiber as an electrode is its small size and wide biological applicability. Indeed, the Malinski/Taha sensor was roughly 0.5 μm in diameter, and thus able to measure NO release from single endothelial and smooth muscle cells. Despite the use of Ni porphyrins, porphyrins containing central metal ions such as iron (Fe) and manganese (Mn) have also been used to construct successful NO sensors.42–44 For example, Diab and Schuhmann coated platinum electrodes with Mn porphyrin-modified polypyrrole films, resulting in NO sensors with little interference from nitrite or ascorbic acid.44
Metallophthalocyanines represent another type of metal-ligand complex recently suggested as a catalytic coating for fabricating NO sensors (Figure 4c). Although similar in structure to metalloporphyrins, metallophthalocyanines are advantageous due to their ability to resist degradation.45 Vilakazi and Nyokong examined a variety of metal-phthalocyanine complexes, including zinc (Zn), Mn, cobalt (Co), Ni and Fe.46 Although sensors modified with the Fe complex resulted in the greatest sensitivity to NO, the strong NO-Fe-phthalocyanine interaction led to poor signal stability due to fouling caused by strong interactions with NO and its oxidation byproducts. In contrast, a carbon fiber microelectrode modified with Co-phthalocyanine and Nafion allowed successful regeneration of the electrode without fouling. Metallophthalocyanines have also been applied to other electrode materials including carbon nanotube-modified glassy carbon,47 platinum,48 and screen-printed carbon.49 For example, Silva and coworkers modified a glassy carbon electrode with single walled carbon nanotubes (SWCNT), followed by commercially available Ni-tetrasulfonated phthalocyanine and Nafion.47 The resulting sensors exhibited a two-fold enhancement in the oxidation current for NO compared to a sensor lacking SWCNT modification.
While metal-porphyrin and porphyrin-like complexes have found widespread use as catalytic coatings for NO sensors, non-porphyrin complexes have performed equally well. Mao and colleagues fabricated NO sensors by modifying electrodes with an electropolymerized salen ligand conjugated to iron, cobalt, copper, or manganese (Figure 4b), and then a Nafion film.50 Each of the metal complexes tested resulted in sensors sensitive to NO, with the Fe(salen) complex exhibiting both the lowest detection limit (10 nM) and most promising linear range (19.6 nM – 1.13 μM). As seen in Table 3, these performance characteristics were similar to the Ni-porphyrin-modified electrodes with selectivity for NO over dopamine, nitrite, and ascorbic acid. Of note, the reported selectivity over dopamine, a positively charged species at physiological pH, was the result of the electrochemical transduction mode. Indeed, the use of differential pulse voltammetry allowed for the pre-oxidation of any cationic species, thereby reducing the magnitude of their interference.
As Nafion alone is unable to exclude all electroactive interfering species, in particular neutral and cationic molecules, other strategies have been used to eliminate interference from these species. Mitchell and Michaelis employed both additional membranes and an enzyme system to improve the selectivity of Ni-porphyrin/Nafion sensors.37 To eliminate interference from ascorbic acid (present in high concentrations in the brain), a Ni-porphyrin/Nafion-coated carbon fiber electrode was further modified with an additional membrane containing ascorbic acid oxidase. As such, the sensor membrane converted the ascorbic acid into dehydroascorbate, a non-electroactive molecule. When compared to the unmodified and Nafion-coated porphyrin electrodes, the selectivity of the ascorbic acid oxidase/Nafion-modified electrodes for NO over ascorbic acid was increased by 3900- and 56-fold, respectively. A polylysine or polypyridinium membrane was also added to create a slight positive charge on the electrode surface, thus providing selectivity over dopamine. Although both membranes improved selectivity over a range of cationic interferences, the polypyridinium provided the greatest enhancement. The authors reported little response to 1 mM solutions of CO with this multi-membrane sensor, but without rationale or speculation.
Calibration Methods
Proper calibration of electrochemical sensors is imperative for the accuracy of analytical measurements, especially when measuring an analyte at concentrations as low as that for NO. During calibration, efforts must be made to mimic the measurement environment including pH, ionic strength, temperature, shear stress, etc. Calibration of NO sensors for use in vivo is especially challenging as endogenous species like hemoglobin and oxygen quickly scavenge NO, often circumventing in vivo calibration.35
Controlled amounts of NO for sensor calibration are most often obtained by reaction of iodide in acidified nitrite, NO release via NO donor compounds such as S-nitroso-acetyl-DL-penicilamine (SNAP), or saturating aqueous solutions with gaseous NO.51 A review by Davies and Zhang should be consulted for details pertaining to the preparation of NO standard solutions using these approaches.36 Below we briefly summarize each approach with key advantages and disadvantages that should be considered when choosing a sensor calibration method.
Calibration using the chemical generation of NO is performed by reacting potassium nitrite with acidified iodide, resulting in the liberation of NO:
| (5) |
Iodine generation of NO is useful because the chemicals are simple and inexpensive, and the amount of NO produced is stoichiometrically equivalent to the initial concentration of KNO2. However, the harsh acidic conditions required may damage sensor membranes, presents a safety hazard, and requires careful disposal.
Another widely accepted method of calibrating electrochemical sensors utilizes the Cu(I)-catalyzed decomposition of SNAP, a small molecule NO-donor, in aqueous solutions to form one molar equivalent of NO.51 Briefly, the sensor is placed into a measured amount of a deoxygenated cuprous chloride saturated solution. A carefully determined volume of a deoxygenated buffer (pH 9.0) containing a known amount of SNAP and ethylenediaminetetraacetic acid (EDTA) is then added to the CuCl solution, catalyzing the release of NO. The primary advantages for this method include the commercial availability of SNAP, the ease of preparation of standard solutions, and the relatively mild conditions needed. However, this method is heavily dependent on both the purity of SNAP and proper storage of the light- and heat-sensitive CuCl and SNAP solutions.36 Furthermore, the pH of the aqueous solution used in this method lacks physiological relevance.
The most common method for calibrating NO sensors is based on sparging aqueous solutions with NO gas. A water or buffer solution is first deoxygenated with an inert gas (nitrogen or argon) to prevent reaction of NO with O2. Purified NO gas is then bubbled into the deoxygenated solution for several minutes to NO saturation ~2 mM at room temperature).36 Standards are prepared by mixing carefully measured aliquots of the saturated NO solution with fresh deoxygenated solutions. Alternatively, less concentrated NO solutions are prepared by sparging solutions with calibrated NO gas mixtures (i.e., NO and N2). The resulting concentration of NO, CNO, in solution is then determined using Henry’s law:
| (6) |
where PNO is the pressure of NO (in mmHg) and SNO is the solubility of NO in the aqueous solution.40 A typical calibration curve utilizing NO solutions is shown in Figure 5, where aliquots of a standard solution prepared from a calibrated NO gas mixture were injected into degassed buffer. The NO levels were measured using a solid permselective electrode.40 The advantages of gas sparging include simplicity, easy application to any aqueous solution, and freedom from using chemicals (e.g., NO donors or acid/iodide). Extreme caution (i.e., fume hood, proper ventilation, etc.) must be exercised when using NO gas due to safety and toxicity concerns related to NO gas.
Figure 5.
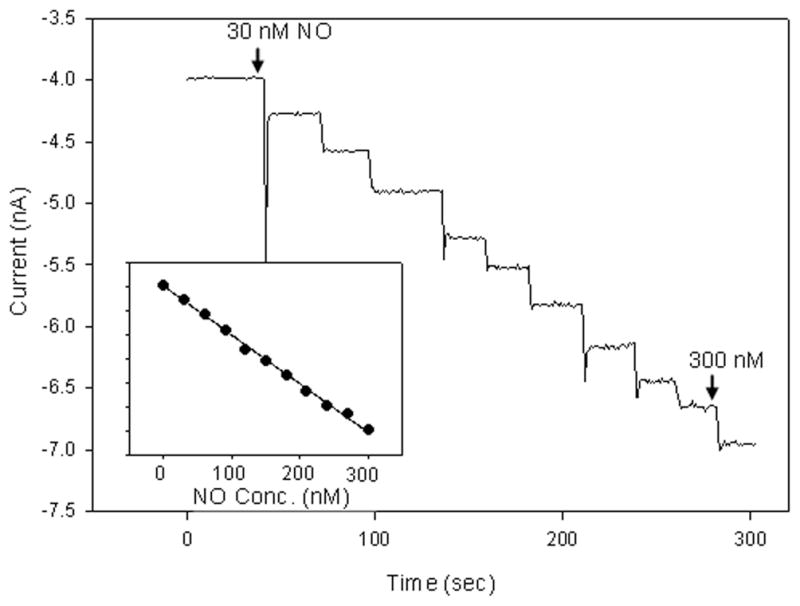
Response of fluorosilane-coated platinum microelectrode to successive additions of NO solution. Inset shows resulting calibration curve. Reprinted with permission from reference 40. Copyright 2008 American Chemical Society.
Nitric Oxide Measurements in Biological Systems
A review of the NO sensor literature reveals numerous examples of electrochemical NO sensors applied to biological systems. Previous reviews have examined the various biological applications of NO sensors.30, 35, 36, 52 Below we detail a small selection of NO sensors applied to biological measurements as examples of the diversity of problems studied by these devices. Only sensors operating via electrooxidation (direct or catalytic) mode are described due to the interference from O2 encountered for electroreductive NO sensors.
Measurement of NO in blood is of particular interest due to its role in regulating vascular tone.13 Furthermore, the bioavailability of endothelium-derived NO is an important indicator of cardiovascular risk, and has been found to be decreased in patients with conditions such as hypertension and diabetes mellitus.53 Traditionally, evaluations of NO bioavailability has been performed indirectly by observing the extent of vasodilation upon stimulation of NO release by bradykinin or acetylcholine (ACh).53 With the development of NO sensors, induced NO response is more accurately determined by directly measuring the resulting NO release. However, blood is a challenging environment for NO measurement due to the presence of both dissolved oxygen and hemoglobin, scavengers of NO that lower circulating NO concentrations. Sensors applied to in vivo NO measurement in blood must have excellent sensitivity to NO and selectivity for NO over the NO oxidation byproducts nitrate and nitrite. In an early example of blood-based electrochemical NO measurements in human vasculature, a Nafion-coated metalloporphyrinic NO sensor was inserted into the hand vein to monitor stimulated NO release via infusions bradykinin and acetylcholine.54 The resulting NO measurement revealed a dose-dependent relationship between the concentrations of the stimulating species and NO released. Injection of N-monomethyl-L-arginine (L-NAME), an inhibitor of NOS, attenuated the release of NO, indicating that the source of the observed signal was NO. The testing of selectivity during calibration indicated that the sensor was unresponsive to the chemical stimulants and inhibitor used during the course of the study. In addition, no signal was detected in response to nitrite or nitrate.
In contrast, measurements of NO released in biological tissues requires rugged sensors capable of penetrating dense structures without sustaining damage or diminished performance. While small sensors are preferred for such measurements to decrease the extent of tissue damage during implantation, the fragility associated with small devices precludes their practical use. Malinski’s group addressed this problem by sheathing a shortened version of their Nafion-coated porphyrinic NO sensor (0.5 – 8 μm diameter) in a needle to protect the sensor tip during NO measurements in rat tissue.55 As shown in Figure 6 the NO sensor was capable of measuring NO in rat lung tissue following the injection of lipopolysaccharide (LPS), a component of bacterial cells that induces an immune response. The resulting stimulated NO production exceeded 100 nM and remained elevated for 20 min. The sensor was also implanted into rat heart tissue, where a significant increase in NO was detected (910 nM) upon stimulation of the endocardium via injection of a calcium ionophore.
Figure 6.
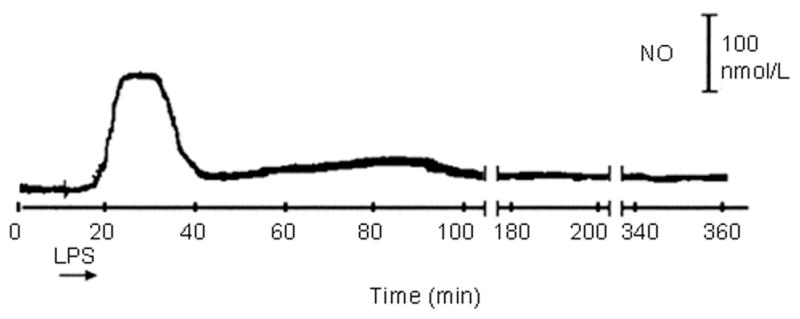
Measurement of LPS-induced NO in rat lung tissue at a ruggedized Nafion-coated porphyrinic microsensor. Adapted from reference 55 with permission from Elsevier.
The brain represents perhaps the most challenging environment for measuring NO, primarily due to low NO concentration, the need for ultrasmall sensors, and the presence of multiple interfering species at high concentrations including ascorbic acid, uric acid, and dopamine. Neurosensors must thus offer superior NO selectivity over these interfering species. Brown et al. reported the fabrication of an NO sensor for real-time monitoring of NO in the striatum.56 A working electrode was fabricated by cutting an insulated Pt/iridium (Ir) (90% Pt) wire (125 m bare diameter) to form a Pt micro-disk. A Nafion membrane was then applied to the electrode, resulting in an NO-permeable membrane that excluded dopamine, nitrite, ascorbic acid, and uric acid. The resulting sensor had a detection limit of 5 nM. In vivo measurements were performed in the striatum of freely-moving rats, where injections of both NO solutions and L-arginine separately elicited a significant NO response at the electrode over saline.
Due to the identification of NO identified as a potent tumoricidal agent at elevated concentrations, compounds capable of generating NO at tumors are being studied as potential anti-cancer therapeutics.57 The ability to assess the efficacy and mechanism of NO-based drugs by quantifying local NO concentrations in/near tumors is clearly integral to the success of such approaches. Electrochemical NO sensors are well-suited for this role, owing to the excellent NO sensitivity and spatial resolution. Griveau and coworkers have gone as far as implanting NO sensors into Lewis lung carcinoma tumors in mice.57 The NO sensors were fabricated by coating a 125 μm diameter Pt/Ir wire with a catalytic Ni phthalocyanine and two NO selective membranes (Nafion and o-phenylenediamine). The sensors were then placed into holes drilled into the tumors. Well-defined response to NO was observed after injections of exogenous NO donors such as 3-(2-hydroxy-2-nitroso-1-propylhydrazino)-1-propanamine (PAPA-NONOate), diethylammonium(Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA-NONOate),57 or nitroglycerin.58 Furthermore, the resulting sensors were selective over species such as nitrite, ascorbic acid, and hydrogen peroxide.
When making NO measurements in biological systems, it is important to consider the distance of the sensor from the NO source. Due to its rapid diffusion (3300 μm2 s−1) and reactivity in biological milieu, the concentration of NO will decay rapidly with increasing distance from the point of generation.13 Malinski’s group observed an exponential decrease in the NO concentration measured at a porphyrinic sensor with increasing distance from the source via stimulated NO release from a single endothelial cell (Figure 7). At the cell surface, a concentration of roughly 950 nM of NO was detected while NO was not measurable at distances >50 μm from the cell.55 Indeed, maintaining a controllable distance from the NO source is critical for measuring NO release from single cells or small groups of cells with high spatial resolution and reproducibility. Pailleret and coworkers utilized scanning electrochemical microscopy to precisely control the distance between an NO electrode and endothelial cells adhered to a substrate.59 In this unique strategy, a bare Pt microelectrode was first positioned via an oxygen reduction approach curve and then modified with Ni tetrasulfonated phthalocyanine in situ, resulting in a precisely positioned NO-sensitive sensor. The sensor was then used to monitor bradykinin-stimulated NO release via catalytic electrooxidation.
Figure 7.
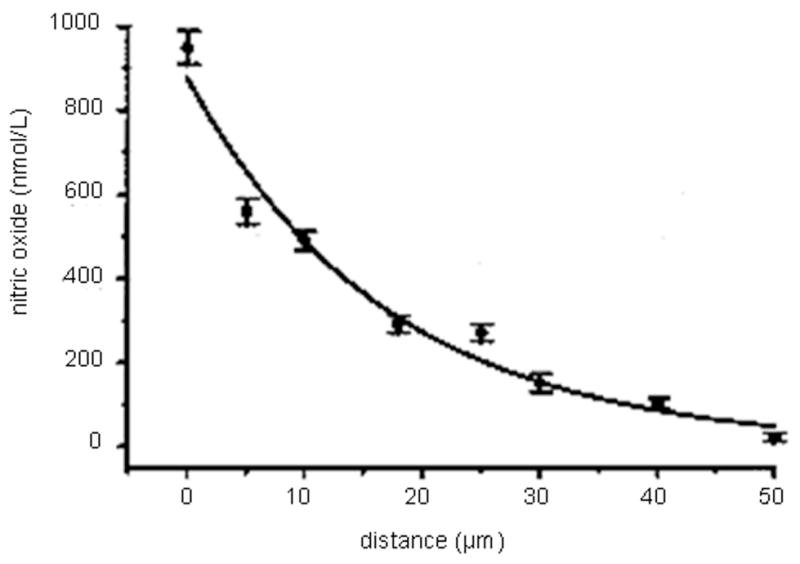
Decrease in NO concentration as distance increases between a porphyrinic NO microsensor and an endothelial cell. Adapted from reference 55 with permission from Elsevier.
Size is another important parameter for consideration when choosing a NO sensor design for a particular biological purpose. The diameter of most published sensors range from a few hundred nanometers to greater than two millimeters.35 Since the NO sensitivity of the sensor is directly proportional to the electroactive surface area, larger working electrodes typically offer greater NO sensitivity. However, the size requirement for NO sensors depends greatly on the intended measurement location. For example, a micro- or ultramicroelectrode (< 1 μm) is suitable for measuring NO in a single cell or a cluster of cells, while a larger sensor may be desirable for in vivo bulk NO measurements in blood. Ultramicroelectrode sensors offer the additional advantage of minimal perturbation of the surrounding environment upon implantation. Conversely, larger sensors are typically more robust and therefore less susceptible to failure over time.
Future Directions and Conclusions
While a continued focus on improving the analytical performance of current electrochemical NO sensors is important, future research must address and improve the ability of NO sensors to resist biofouling for more reliable use in blood (i.e., protein adsorption, platelet adhesion, and thrombus formation) and tissue (i.e., fibrous encapsulation and infection). Indeed, sensor biofouling often results in diminished analytical performance, poor reproducibility, and even failure. Previously published strategies for reducing biofouling on implantable sensors for other chemical species are reviewed elsewhere.60, 61 Briefly, strategies for improving biocompatibility include passive protection of the sensor through the use of sensor membranes that resist biofouling (e.g., polyurethanes, polyethylene glycol, Nafion, and phospholipids), and polymers that actively release antifouling agents.62 Ironically, a most promising approach for reducing biofouling of implantable sensors is based on NO-release from sensor membranes.62, 63 Clearly, such a strategy would be problematic for NO sensors.
As researchers continue to unravel the complex biological roles of NO and develop therapies based on NO, the need for sensitive, selective, and accurate NO measurement devices will increase. Specifically, the use of clinical NO measurements as diagnostic and prognostic indicators necessitates inexpensive, small, and simple point-of-care devices. Electrochemical NO sensors, more than any other type of NO measurement technique, are well suited to fill this role, and for practical purposes remain most attractive for real-time in vivo NO quantification in biological systems. In addition to their ease of fabrication and miniaturization, the instrumentation required to perform ultra sensitive measurements is both affordable and potentially portable for field use. While few analytical sensors work well for all applications, the most successful designs are characterized by both desirable analytical performance criteria and ruggedness. The emerging development of new approaches using nano-structured polymers and carbon nanotubes should further enable sensitive, selective, and accurate determinations of NO in challenging environments.
Acknowledgments
The authors (M.H.S. and J.H.S.) gratefully acknowledge the National Institutes of Health (EB000708) and Kwangwoon University for support of their research programs.
References
- 1.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan MT, Furchgott RF. Fed Proc. 1987;46:385–385. [Google Scholar]
- 3.Rapoport RM, Murad F. Circ Res. 1983;52:352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- 4.Masters BS. In: Nitric Oxide: biology and pathobiology. Ignarro LJ, editor. Academic Press; San Diego: 2000. pp. 3–19. [Google Scholar]
- 5.Stamler JS. Coronary Artery Dis. 1999;10:273–276. [Google Scholar]
- 6.Guix FX, Uribesalgo I, Coma M, Munoz FJ. Prog Neurobiol. 2005;76:126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Bogdan C. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 8.MacMicking J, Xie QW, Nathan C. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 9.Luo JD, Chen AF. Acta Pharmacol Sin. 2005;26:259–264. doi: 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- 10.Hetrick EM, Shin JH, Stasko NA, Johnson CB, Wespe DA, Holmuhamedov E, Schoenfisch MH. ACS Nano. 2008;2:235–246. doi: 10.1021/nn700191f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494–4502. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wink DA, Mitchell JB. Free Radical Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 13.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. Free Radical Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lancaster JR. Nitric Oxide-Biol Chem. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 15.Hetrick EM, Schoenfisch MH. Annu Rev Anal Chem. 2009;2:409–433. doi: 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang KJ, Zhang M, Xie WZ, Zhang HS, Feng YQ, Wang H. Anal Bioanal Chem. 2007;388:939–946. doi: 10.1007/s00216-007-1283-2. [DOI] [PubMed] [Google Scholar]
- 17.Ichimori K, Ishida H, Fukahori M, Nakazawa H, Murakami E. Rev Sci Instrum. 1994;65:2714–2718. [Google Scholar]
- 18.Malinski T, Taha Z. Nature. 1992;358:676–678. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- 19.Casero E, Pariente F, Lorenzo E, Beyer L, Losada J. Electroanalysis. 2001;13:1411–1416. [Google Scholar]
- 20.Bedioui F, Trevin S, Devynck J. J Electroanal Chem. 1994;377:295–298. [Google Scholar]
- 21.Lee Y, Oh BK, Meyerhoff ME. Anal Chem. 2004;76:536–544. doi: 10.1021/ac035064h. [DOI] [PubMed] [Google Scholar]
- 22.Burtis CA, Ashwood ER, Tietz NW. Tietz Textbook of Clinical Chemistry. 3. W.B. Saunders Co; Philadelphia, PA: 1999. [Google Scholar]
- 23.Goldstein DS, Stull R, Zimlichman R, Levinson PD, Smith H, Keiser HR. Clin Chem. 1984;30:815–816. [PubMed] [Google Scholar]
- 24.Lee Y, Kim J. Anal Chem. 2007;79:7669–7675. doi: 10.1021/ac070814z. [DOI] [PubMed] [Google Scholar]
- 25.Pelletier MM, Kleinbongard P, Ringwood L, Hito R, Hunter CJ, Schechter AN, Gladwin MT, Dejain A. Free Radical Biol Med. 2006;41:541–548. doi: 10.1016/j.freeradbiomed.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Wu LY, Wang R. Pharmacol Rev. 2005;57:585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 27.Hartsfield CL. Antioxid Redox Sign. 2002;4:301–307. doi: 10.1089/152308602753666352. [DOI] [PubMed] [Google Scholar]
- 28.Clark LC. T Am Soc Art Int Org. 1956;2:41. [Google Scholar]
- 29.Shibuki K. Neurosci Res. 1990;9:69–76. doi: 10.1016/0168-0102(90)90048-j. [DOI] [PubMed] [Google Scholar]
- 30.Ciszewski A, Milczarek G. Talanta. 2003;61:11–26. doi: 10.1016/S0039-9140(03)00355-2. [DOI] [PubMed] [Google Scholar]
- 31.Liu XJ, Shang LB, Pang JT, Li GX. Biotechnol Appl Biochem. 2003;38:119–122. doi: 10.1042/BA20030056. [DOI] [PubMed] [Google Scholar]
- 32.Maskus M, Pariente F, Wu Q, Toffanin A, Shapleigh JP, Abruna HD. Anal Chem. 1996;68:3128–3134. doi: 10.1021/ac951063g. [DOI] [PubMed] [Google Scholar]
- 33.Meulemans A. Neurosci Lett. 1993;157:7–12. doi: 10.1016/0304-3940(93)90630-4. [DOI] [PubMed] [Google Scholar]
- 34.Zen JM, Kumar AS, Wang HF. Analyst. 2000;125:2169–2172. doi: 10.1039/b008176k. [DOI] [PubMed] [Google Scholar]
- 35.Bedioui F, Villeneuve N. Electroanalysis. 2003;15:5–18. [Google Scholar]
- 36.Davies IR, Zhang XJ. Methods in Enzymol. 2008;436:63–95. doi: 10.1016/S0076-6879(08)36005-4. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell KM, Michaelis EK. Electroanalysis. 1998;10:81–88. [Google Scholar]
- 38.Prakash R, Srivastava RC, Seth PK. Polym Bull. 2001;46:487–490. [Google Scholar]
- 39.Kitamura Y, Uzawa T, Oka K, Komai Y, Ogawa H, Takizawa N, Kobayashi H, Tanishita K. Anal Chem. 2000;72:2957–2962. doi: 10.1021/ac000165q. [DOI] [PubMed] [Google Scholar]
- 40.Shin JH, Privett BJ, Kita JM, Wightman RM, Schoenfisch MH. Anal Chem. 2008;80:6850–6859. doi: 10.1021/ac800185x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedemann MN, Robinson SW, Gerhardt GA. Anal Chem. 1996;68:2621–2628. doi: 10.1021/ac960093w. [DOI] [PubMed] [Google Scholar]
- 42.Hayon J, Ozer D, Rishpon J, Bettelheim A. J Chem Soc, Chem Commun. 1994:619–620. [Google Scholar]
- 43.Bedioui F, Trevin S, Albin V, Villegas MGG, Devynck J. Anal Chim Acta. 1997;341:177–185. [Google Scholar]
- 44.Diab N, Schuhmann W. Electrochim Acta. 2001;47:265–273. [Google Scholar]
- 45.Nyokong T, Vilakazi S. Talanta. 2003;61:27–35. doi: 10.1016/S0039-9140(03)00356-4. [DOI] [PubMed] [Google Scholar]
- 46.Vilakazi SL, Nyokong T. J Electroanal Chem. 2001;512:56–63. [Google Scholar]
- 47.Silva JF, Griveau S, Richard C, Zagal JH, Bedioui F. Electrochem Commun. 2007;9:1629–1634. [Google Scholar]
- 48.Pereira-Rodrigues N, Albin V, Koudelka-Hep M, Auger V, Pailleret A, Bedioui F. Electrochem Commun. 2002;4:922–927. [Google Scholar]
- 49.Miserere S, Ledru S, Ruille N, Griveau S, Boujtita M, Bedioui F. Electrochem Commun. 2006;8:238–244. [Google Scholar]
- 50.Mao LQ, Yamamoto K, Zhou WL, Jin LT. Electroanalysis. 2000;12:72–77. [Google Scholar]
- 51.Zhang XJ, Cardosa L, Broderick M, Fein H, Davies IR. Electroanalysis. 2000;12:425–428. [Google Scholar]
- 52.Bedioui F, Trevin S, Devynck J. Electroanalysis. 1996;8:1085–1091. [Google Scholar]
- 53.Neishi Y, Mochizuki S, Miyasaka T, Kawamoto T, Kume T, Sukmawan R, Tsukiji M, Ogasawara Y, Kajiya F, Akasaka T, Yoshida K, Goto M. Proc Natl Acad Sci U S A. 2005;102:11456–11461. doi: 10.1073/pnas.0501392102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallance P, Patton S, Bhagat K, Macallister R, Radomski M, Moncada S, Malinski T. Lancet. 1995;346:153–154. doi: 10.1016/s0140-6736(95)91211-8. [DOI] [PubMed] [Google Scholar]
- 55.Brovkovych V, Stolarczyk E, Oman J, Tomboulian P, Malinski T. J Pharm Biomed Anal. 1999;19:135–143. doi: 10.1016/s0731-7085(98)00090-9. [DOI] [PubMed] [Google Scholar]
- 56.Brown FO, Finnerty NJ, Lowry JP. Analyst. 2009;134:2012–2020. doi: 10.1039/b909005c. [DOI] [PubMed] [Google Scholar]
- 57.Griveau S, Dumezy C, Seguin J, Chabot GG, Scherman D, Bedioui F. Anal Chem. 2007;79:1030–1033. doi: 10.1021/ac061634c. [DOI] [PubMed] [Google Scholar]
- 58.Griveau S, Seguin J, Scherman D, Chabot GG, Bedioui F. Electroanalysis. 2009;21:631–634. [Google Scholar]
- 59.Pailleret A, Oni J, Reiter S, Isik S, Etienne M, Bedioui F, Schuhmann W. Electrochem Commun. 2003;5:847–852. [Google Scholar]
- 60.Frost M, Meyerhoff ME. Anal Chem. 2006;78:7370–7377. doi: 10.1021/ac069475k. [DOI] [PubMed] [Google Scholar]
- 61.Li CM, Dong H, Cao XD, Luong JHT, Zhang XJ. Curr Med Chem. 2007;14:937–951. [PubMed] [Google Scholar]
- 62.Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 63.Hetrick EM, Prichard HL, Klitzman B, Schoenfisch MH. Biomaterials. 2007;28:4571–4580. doi: 10.1016/j.biomaterials.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]