

Abstract
Recent trials have shown that the prostaglandin E2 EP1 receptor is responsible for NMDA excitotoxicity in the brain after injury. Consequently, in this study, we investigated the use of SC-51089, a selective prostaglandin E2 EP1 receptor antagonist, as a pre-treatment modality to decrease cell death, reduce brain edema, and improve neurobehavioral function after surgically induced brain injury (SBI) in mice. Eleven-week-old C57 black mice (n = 82) were randomly assigned to four groups: sham (n = 31), SBI (n = 27), SBI treated with SC51089 at 10 μg/kg (n = 7), and SBI treated with SC51089 at 100 μg/kg (n = 17). Treated groups received a single dose of SC51089 intrapertioneally at 12 and 1 h pre-surgery. SBI was performed by resecting the right frontal lobe using a frontal craniotomy. Postoperative assessment occurred at 24 and 72 h, and included neurobehavioral testing and measurement of brain water content and cell death. Results indicated that neither low- nor high-dose EP1 receptor inhibition protected against the SBI-related effects on brain edema formation or cell death. There was however a significant improvement in neurobehavioral function 24 h post-SBI with both dosing regimens. Further studies will be needed to assess the potential therapeutic role of EP1 receptor targeting in SBI.
Keywords: SC-51089, Prostaglandin E2, EP1 Receptor, Surgically induced brain injury (SBI), Neurosurgery
Introduction
Neurosurgical procedures can damage viable brain tissue unintentionally by a wide range of methods. These surgically induced brain injuries (SBI) can be a result of direct surgical trauma, intraoperative bleeding, thermal injury from electrocautery, or stretch damage from tissue retraction [1, 2]. The concern with these injuries is the heightened inflammatory response that is mounted in an attempt to combat the effects of brain tissue damage. Propagation of this local response may result in direct cell death, enhanced disruption of the blood-brain-barrier (BBB) causing an increase in brain edema formation, and subsequent deterioration in neurobehavioral function [3]. Unfortunately, current therapies, such as steroids and diuretics, are relatively non-specific and focus mostly on reducing the postoperative edema that has already transpired [4]. Therefore, recent studies have looked into targeting specific anti-inflammatory mediators, which may be more effective at reducing cell death and brain edema while improving neurobehavior.
Cyclooxygenase-2 (COX-2) is a rate-limiting enzyme responsible for catalyzing the synthesis of prostaglandins at sites of injury. In the brain, COX-2 acts as a key mediator of inflammation, orchestrating a wide spectrum of brain injuries, including excitotoxic brain injury, cerebral ischemia, traumatic brain injury, and neurodegenerative disorders [5]. A key downstream effector of COX-2 neurotoxicity is prostaglandin E2 (PGE2), an endogenous signaling molecule produced by the enzymatic transformation of arachidonic acid by COX-2 6, 7]. PGE2 has been linked to BBB disruption during CNS inflammation [8–10], and through its interaction with the G-protein coupled receptor, EP1, it can enhance peripheral microvessel endothelial cell permeability and thereby cause a substantial amount of brain edema accumulation. Aside from its role in BBB disruption and edema formation, the EP1 receptor may also be involved in excitotoxic cell death [7].
Recently, selective COX-2 enzyme inhibitors have come under scrutiny because of an increased occurrence of cardiovascular events [11, 12], suggesting that downstream signaling pathways in the inflammatory cascade may be a safer target. Consequently, in this study, we investigated the role of the PGE2 EP1 receptor on brain edema formation, neuronal cell death, and neurobehavioral function after SBI. We hypothesized that EP1 receptor inhibition will result in marked improvement in these three parameters in our mouse population.
Materials and Methods
Animal Groups
All procedures for this study were approved by the Animal Care and Use Committee at Loma Linda University and complied with the NIH Guide for the Care and Use of Laboratory Animals. Eighty-two male Sprague-Dawley mice weighing between 35 and 45 g were randomly divided into the following four groups: sham (n = 31), SBI (n = 27), SBI treated with SC51089 at 10 μg/kg (n = 7), and SBI treated with SC51089 at 100 μg/kg (n = 17).
Operative Procedure
The SBI model was adapted as previously described in mice [4]. Briefly, mice were anesthetized with a ketamine (100 mg/kg)/xylazine (10 mg/kg) combination intraperitoneal injection and positioned prone in a stereotaxic head frame (Stoelting, Wood Dale, IL). A 3 mm by 3 mm cranial window was created 1 mm anterior and 2 mm lateral to the coronal and sagittal sutures, respectively. Using a flat blade (6 mm × 1.5 mm), two incisions were made along the sagittal and coronal planes, leading away from the bregma and extending to the edge of the craniotomy window. The brain sections were weighed and were not significantly different between animals. Electrocautery was utilized for 2 s along the medial coronal and posterior sagittal borders at a power level consistent with the coagulation setting used in the operating room. Sham surgery included only a craniotomy window and replacement of the bone flap without any dural incisions.
Treatment Method
SC51089 (Enzo Life Sciences, Plymouth Meeting, PA) was dissolved in 0.5% DMSO and administered intraperitoneally approximately 12 h and 1 h before SBI induction. Treated mice were divided into two groups, depending on the concentration of drug received – either a low-dose concentration (10 μg/kg) or a high-dose concentration (100 μg/kg).
Assessment of Neurobehavioral Deficits
Neurological outcomes were assessed by a blind observer at 24 h post-SBI using the Modified Garcia Score [13], beam balance test, and modified wire hanging test [3].
The Modified Garcia Score is a 21-point sensorimotor assessment system consisting of seven tests with scores of 0–3 for each test (maximum score = 21). These seven tests included: (1) spontaneous activity, (2) side stroking, (3) vibris touch, (4) limb symmetry, (5) climbing, (6) lateral turning, and (7) forelimb walking.
Additionally, beam balance and wire hanging tests were performed. Both the beam (590 cm in length by 51 cm in width) and wire (550 cm in length by 51 mm in width) were constructed and held in place by two platforms on each side. Mice were observed for both their time and behavior until they reached one platform, and were scored according to six grades. The test was repeated three times, and an average score was taken [minimum score 0; maximum score (healthy rat) 5].
Brain Water Content
Brain water content was measured as previously described [14]. Briefly, mice were killed at 24 and 72 h post SBI, and brains were immediately removed and divided into three parts: ipsilateral frontal, contralateral frontal, and cerebellum. The cerebellum was used as an internal control for brain water content. Tissue samples were then weighed on an electronic analytical balance (APX-60, Denver Instrument; Arvada, CO) to the nearest 0.1 mg to obtain the wet weight (WW). The tissue was then dried at 105°C for 48 h to determine the dry weight (DW). The percent brain water content was calculated as [(WW − DW)/WW] × 100.
Assessing Cell Death
The Cell Death Detection ELISA kit (Roche Applied Science) was used to quantify cell death in the ipsilateral frontal cortex 24 h after SBI. For quantification of DNA fragmentation, which indicates apoptotic cell death, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals).
Statistical Analysis
Quantitative data were expressed as the mean ± SEM. One way ANOVA and Tukey tests were used to determine significance in differences between the means. Neurological scores were evaluated using the Dunn method. A p-value < 0.05 was considered statistically significant.
Results
PGE2 EP1 Receptor Inhibition Failed to Reduce Brain Edema After SBI
Brain water content was measured at 24 and 72 h post-SBI (Fig. 1). The results showed that vehicle mice presented with significantly worse brain edema compared to sham mice. After treatment with low-dose (10 μg/kg) or high-dose (100 μg/kg) SC51089, brain edema failed to reduce significantly in the ipsilateral and contralateral frontal cortex compared to vehicle groups.
Fig. 1.
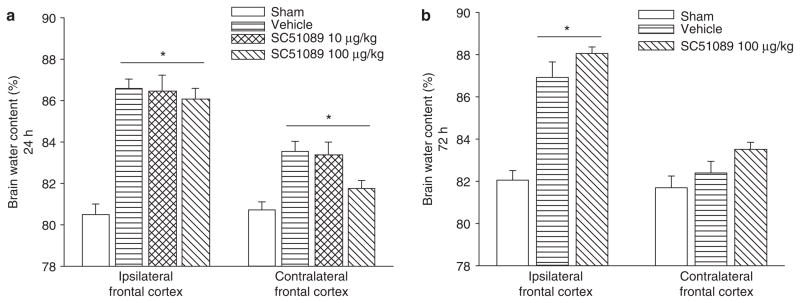
Brain water content. Brain water content increased significantly in the ipsilateral basal ganglia at 24 and 72 h post-SBI injury. This increase was not affected by SC51089 treatment. *Significant difference vs. sham (p < 0.05); (a) 24 h: sham = 14; (vehicle) = 14; (ICH + SC51089 10 μg/kg) = 7; (ICH + SC51089 100 μg/kg) = 6. (b) 72 h: sham = 11; (vehicle) = 7; (ICH + SC51089 100 μg/kg) = 5
PGE2 EP1 Receptor Inhibition Transiently Improves Neurobehavioral Scores at 24 h
To evaluate the sensorimotor deficits after SBI, the modified Garcia test, wire hanging test, and beam balance test were conducted at 24 and 72 h post-SBI. The results showed that vehicle mice presented with severe neurobehavioral deficits compared to sham mice. After treatment with low-dose or high-dose SC51098, a significant improvement in neurobehavioral function was seen with the modified Garcia test at 24 h (Fig. 2a); however, with the wire hanging and beam balance tests, there was no improvement in neurobehavioral function noted compared to the vehicle group (Fig. 2b, c). At 72 h, there was no improvement seen with either treatment dose compared to vehicle.
Fig. 2.

Neurobehavoiral function. Neurobehavioral deficits were present in all vehicle compared to sham animals. With SC51089 treatment, there was a significant improvement in Garcia behavioral testing scores at 24 h post SBI. *Significant difference vs. sham (p < 0.05); #significant difference vs. vehicle (p < 0.05); (a) modified Garcia scoring; (b) wire hanging score; (c) beam balance score
Cell Death Fails to Reduce with EP1 Receptor Inhibition
Cell death was also measured via an absorbance assay at 24 h post-SBI (Fig. 3). The results showed that vehicle mice presented with a significant increase in the number of cell deaths compared to sham mice. After treatment with high-dose (100 μg/kg) SC51089, the number of cell deaths failed to reduce significantly.
Fig. 3.

Cell death measurement 24 h post SBI. There were significant differences between the sham and vehicles groups. However, no improvement was seen with SC51089 treatment *Significant difference vs. sham (p < 0.05); #significant difference vs. vehicle (p < 0.05); (A) 24 h: sham = 6; (vehicle) = 6; (ICH + SC51089 100 μg/kg) = 6
Discussion
In this study, we used a mouse SBI model to investigate the role by which the COX-2 downstream receptor, EP1, contributed to brain injury outcomes. Using SC51089, a selective EP1 receptor inhibitor, we showed for the first time that receptor inhibition failed to attenuate cell death and reduce brain edema after SBI. Additionally, at 24 h post injury, we observed a marked improvement in neurobehavioral deficits; however, those improvements reversed by 72 h. Consequently, this study suggests that EP1 receptor inhibition has limited neuroprotective effects in SBI mice.
The downstream COX-2 effecter, PGE2, has been implicated in causing neuronal cell death in the brain [15]. By binding to the G-protein coupled receptor, EP1, PGE2 can activate the IP3 signaling pathway causing an influx of calcium into the cell and in doing so disrupt normal calcium homeostasis. As a result of this calcium influx, NMDA receptor activation also occurs, causing significant excitotoxic damage and neuronal cell death [16]. This being true, blocking of the EP1 receptor should prevent damage and reduce neuronal cell death; however, this was not the case in our study. Instead, we found that neuronal cell death did not improve with EP1 receptor inhibition at 24 h post-injury. There was neither a reduction nor an increase in the number of cell deaths, implying that SC51089 had no effect on our outcome. This begs the question of whether COX-2-mediated inflammation is involved with delayed cell death rather than the acute phase of injury after SBI. Therefore, further studies will need to investigate the effects of COX-2 inhibition on neuronal cell death at later time points.
Aside from neuronal cell death, brain edema can be an additionally encountered postoperative complication following neurosurgical procedures. Its accumulation leads to significant brain swelling [11, 12], resulting in a dramatic rise in intracranial pressure. In SBI, disruption of the BBB is responsible for brain edema formation. In our study we found that brain edema formation was not affected by SC51089 treatment.
Although neuronal cell death and brain edema did not improve, we did find a reduction in neurobehavioral deficits in our mouse population. At 24 h post-injury, there was an improvement in neurobehavioral deficits with EP1 receptor inhibition. Testing was conducted using the modified Garcia test and both the wire hanging and beam balance tests. Improvements with treatment were only seen with the modified Garcia test, which is more sensitive to acute neurological deficits than the other tests [13]. However, the improvement in neurobehavioral function was transient, quickly deteriorating by 72 h after injury. This is most likely a result of delayed cell death in brain regions associated with neurobehavioral function [17, 18]. In the central region of injury, blood flow is compromised, which leads to energy failure and subsequent cell death; however, in the surrounding peri-injury sites, neurons remain viable for prolonged periods [19, 20]. Hence, the area of the brain responsible for neurobehavioral function may not have been affected until later in the course of the injury.
There is one main limitation that needs to be acknowledged and addressed regarding the present study. The limitation concerns the timing of treatment. In this study, we chose to give SC51089 treatment at 12 and 1 h pre-surgery as a means of preventing and/or decreasing potential damage from neurological surgery. However, we failed to take into account the half-life of the drug. We recognize the need for studies to investigate the pharmacodynamics of SC51089 in order to best determine when to give treatment.
Conclusion
We conclude that EP1 receptor inhibition fails to improve the injuries of an SBI in adult male mice. Further studies will be needed to confirm this relationship and determine the future clinical direction.
Acknowledgments
This study is partially supported by NIH NS053407 to J.H. Zhang and NS060936 to J. Tang.
Footnotes
Conflict of interest statement We declare that we have no conflict of interest.
Contributor Information
Nikan H. Khatibi, Department of Anesthesiology, Loma Linda Medical Center, Loma Linda, CA, USA
Vikram Jadhav, Department of Physiology, Loma Linda University, School of Medicine, Loma Linda, CA, USA.
Brenden Matus, Department of Physiology, Loma Linda University, School of Medicine, Loma Linda, CA, USA.
Nancy Fathali, Department of Physiology, Loma Linda University, School of Medicine, Loma Linda, CA, USA.
Robert Martin, Department of Anesthesiology, Loma Linda Medical Center, Loma Linda, CA, USA.
Richard Applegate, Department of Anesthesiology, Loma Linda Medical Center, Loma Linda, CA, USA.
Jiping Tang, Email: jtang@llu.edu, Department of Physiology, Loma Linda University, School of Medicine, Loma Linda, CA, USA, Department of Physiology and Pharmacology, Loma Linda University, School of Medicine, Loma Linda, CA 92354, USA.
John H. Zhang, Department of Anesthesiology, Loma Linda Medical Center, Loma Linda, CA, USA, Department of Physiology, Loma Linda University, School of Medicine, Loma Linda, CA, USA, Department of Neurosurgery, Loma Linda Medical Center, Loma Linda, CA, USA
References
- 1.Andrews RJ, Muto RP. Retraction brain ischaemia: cerebral blood flow, evoked potentials, hypotension and hyperventilation in a new animal model. Neurol Res. 1992;14(1):12–18. doi: 10.1080/01616412.1992.11740004. [DOI] [PubMed] [Google Scholar]
- 2.Hernesniemi J, Leivo S. Management outcome in third ventricular colloid cysts in a defined population: a series of 40 patients treated mainly by transcallosal microsurgery. Surg Neurol. 1996;45(1):2–14. doi: 10.1016/0090-3019(95)00379-7. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi M, et al. Matrix metalloproteinase inhibition attenuates brain edema in an in vivo model of surgically-induced brain injury. Neurosurgery. 2007;61(5):1067–1075. doi: 10.1227/01.neu.0000303203.07866.18. discussion 1075–1076. [DOI] [PubMed] [Google Scholar]
- 4.Jadhav V, et al. Neuroprotection against surgically induced brain injury. Surg Neurol. 2007;67(1):15–20. doi: 10.1016/j.surneu.2006.07.014. discussion 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63(9):901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- 6.Carlson NG. Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res. 2003;71(1):79–88. doi: 10.1002/jnr.10465. [DOI] [PubMed] [Google Scholar]
- 7.Kawano T, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12(2):225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 8.Jaworowicz DJ, Jr, et al. Nitric oxide and prostaglandin E2 formation parallels blood-brain barrier disruption in an experimental rat model of bacterial meningitis. Brain Res Bull. 1998;46(6):541–546. doi: 10.1016/s0361-9230(98)00052-5. [DOI] [PubMed] [Google Scholar]
- 9.Mark KS, Trickler WJ, Miller DW. Tumor necrosis factor-alpha induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J Pharmacol Exp Ther. 2001;297(3):1051–1058. [PubMed] [Google Scholar]
- 10.Payne DK, Fuseler JW, Owens MW. Modulation of endothelial cell permeability by lung carcinoma cells: a potential mechanism of malignant pleural effusion formation. Inflammation. 1994;18(4):407–417. doi: 10.1007/BF01534438. [DOI] [PubMed] [Google Scholar]
- 11.Marmarou A, et al. Contribution of edema and cerebral blood volume to traumatic brain swelling in head-injured patients. J Neurosurg. 2000;93(2):183–193. doi: 10.3171/jns.2000.93.2.0183. [DOI] [PubMed] [Google Scholar]
- 12.Marmarou A, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104(5):720–730. doi: 10.3171/jns.2006.104.5.720. [DOI] [PubMed] [Google Scholar]
- 13.Garcia JH, et al. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 14.Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 15.Andreasson K. Emerging roles of PGE(2) receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010;91(3–4):104–112. doi: 10.1016/j.prostaglandins.2009.04.003. Epub 2009 Apr 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou P, et al. Neuroprotection by PGE2 receptor EP1 inhibition involves the PTEN/AKT pathway. Neurobiol Dis. 2008;29(3):543–551. doi: 10.1016/j.nbd.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2, but not lipoxygenase, in regions synaptically distant from spreading depression. J Comp Neurol. 1996;376(3):447–462. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewett SJ, et al. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293(2):417–425. [PubMed] [Google Scholar]
- 19.Dereski MO, et al. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 1993;85(3):327–333. doi: 10.1007/BF00227730. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JH, et al. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142(2):623–635. [PMC free article] [PubMed] [Google Scholar]