

Abstract
A high-throughput screen of the NIH molecular libraries sample collection and subsequent optimization of a lead dipeptide-like series of severe acute respiratory syndrome (SARS) main protease (3CLpro) inhibitors led to the identification of probe compound ML188 (16-(R), (R)-N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)furan-2-carboxamide, Pubchem CID: 46897844). Unlike the majority of reported coronavirus 3CLpro inhibitors that act via covalent modification of the enzyme, 16-(R) is a non-covalent SARS-CoV 3CLpro inhibitor with moderate MW and good enzyme and antiviral inhibitory activity. A multi-component Ugi reaction was utilized to rapidly explore structure activity relationships within S1′, S1, and S2 enzyme binding pockets. The X-ray structure of SARS-CoV 3CLpro bound with 16-(R) was instrumental in guiding subsequent rounds of chemistry optimization. 16-(R) provides an excellent starting point for the further design and refinement of 3CLpro inhibitors that act by a non-covalent mechanism of action.
INTRODUCTION
Coronaviruses (CoV) are enveloped, large plus-strand RNA viruses that cause medical disorders such as the common cold, lower respiratory tract infections and diarrhea.1 The first characterized human CoV strains, 229E and OC43, were identified and studied extensively from 1965 to the mid-1980s.2 In 2003, the novel SARS-CoV was identified3,4 as the etiological agent of the global pandemic of severe acute respiratory syndrome (SARS), an atypical pneumonia that led to progressive respiratory failure in over 8,000 individuals and 800 deaths by July of that year.5 The death rate of approximately 10% for the SARS virus and its ability to spread person-to-person via respiratory droplets make it particularly deadly among worldwide pandemic threats. With the cooperation of leading nations, a rigorous public healthcare campaign was fortunately successful in controlling the outbreak. However, a reemergence of the SARS-CoV is still considered a potential pandemic risk and potentially new strains of SARS could be more severe than that found from the 2003 outbreak. Since 2003, two additional human coronaviruses, NL63 and HKU1, have been identified in patients around the world and the viruses have been characterized and found to be significantly less lethal than SARS-CoV.6-8 Most recently, a new SARS-like virus, called HCoV-EMC, has been identified in at least two individuals, one of whom died.9 Sequence analysis of HCoV-EMC indicates that this virus is more closely related to bat coronaviruses than to SARS-CoV. Therefore, the possibility of a future SARS-like pandemic remains possible and to date, there are still no vaccines or antiviral agents available to prevent or treat SARS-like infections.
The SARS-CoV genome encodes a large polyprotein that is proteolytically processed by two cysteine proteases including the 3C-Chymotrypsin-Like protease (3CLpro) and the Papain-Like protease (PLpro). 3CLpro is essential for proteolytic processing at 11 different cleavage sites within the coronavirus polyprotein and is thus vital for viral replication.10 The 3CLpro enzyme exists primarily as a dimer in solution and the dimer has been confirmed to be the active species for the enzyme reaction.11 The cloning and expression of recombinant SARS 3CLpro,12 along with studies showing that 3CLpro is essential for the viral life cycle,13 support a role for 3CLpro as an important pathogenic component of SARS-CoV and therefore a viable target for antiviral drug development.
The SARS-CoV 3CLpro has three domains: I (residues 8–101), II (residues 102–184), and III (residues 201–301). Domains I and II, which contain the active site region, are β-barrel domains, and domain III is an α-helical domain. The active site contains a catalytic dyad consisting of a cysteine residue (Cys-145) that acts as a nucleophile and a histidine residue (His-41) that acts as the general acid-base. Optimized octapeptide-based inhibitors using mutational and CoMFA models have been reported,14 and more recently, a systematic saturation mutagenesis study was conducted at the P5 through P3′ positions of the substrate.15 These results demonstrate a strong structure-activity relationship between 3CLpro and its substrate and have provided a basis for peptidomimetic inhibitor design. X-ray structures of the SARS-CoV 3CLpro enzyme bound to hexapeptidyl chloromethyl ketone inhibitors were first reported16-18 and numerous peptidic structures now exist in the context of targeted anti-viral drug design.19-24 These first generation protease inhibitors maintain a peptidic nature, often five residues in length, and bear a reactive warhead group at the terminus which forms a covalent interaction with Cys-145 (Figure 1, 1-7). Reactive “warhead” groups for 3CLpro have included aldehydes, epoxy-ketones, halo-methyl ketones, trifluoromethyl ketones, and a number of examples of Michael acceptors.19-25 These inhibitors often first form a noncovalent interaction complex with the enzyme, positioning the warhead in close proximity to the catalytic cysteine. Attack of the thiolate anion of the catalytic cysteine onto the reactive atom of the warhead leads to formation of the covalent adduct, inactivating the enzyme. One of these compounds, TG-0205221 (5), reacts with SARS 3CLpro with a reported Ki value of ~60 nM.22
Figure 1.

Representative first generation peptidic 3CLpro inhibitors 1-7 highlighting reactive warhead groups (red) and side chain residues (P1 blue).
These first generation inhibitors achieved sub-micromolar activity and provided valuable insights into further structure-based inhibitor design, quickly leading to non-peptidic, warhead-based small molecule inhibitors (Figure 2, 8-11).19, 26-35 Efforts utilizing virtual screening approaches also proved successful leading to non-peptidic inhibitors (e.g. 12-13).32-33 One of the early compounds disclosed bearing a cinnamyl amide, cinanserin (Figure 2, 8), has a reported IC50 value of ~ 5 μM.26 Low molecular weight non-peptide inhibitors bearing reactive esters (9-10b)27-29 and ketones moieties (11a-b)30-31 have demonstrated moderate to good micromolar inhibition and in the case of pyridyl ester 10b29 achieved inhibition in a cell-based assay below 10 μM.
Figure 2.

Representative second generation non-peptidic 3CLpro inhibitors highlighting potential warhead groups (red).
The utility and development of using covalently bound inhibitors can unfortunately be limited due to their potential for off-target side effects and toxicity.36 Despite the fact that there are currently 39 commercially available therapeutics that act through covalent-modification mechanisms, none of these target cysteine proteases.36 The fact that all of the protease classes (serine, threonine, aspartyl and metallo), with the exception of cysteine, have been targeted by marketed therapeutics suggests that getting covalent cysteine-protease inhibitors to market may have unique challenges.37 Therefore, researchers are now focusing more attention on the identification of non-covalent inhibitors. Legitimate non-covalent inhibitors described to date have been limited to high molecular weight peptidomimetics (MW > 800 amu), with low ligand efficiency,38-39 and have included known protease drugs originally developed for HIV protease.40 In an effort to identify small molecule inhibitors of the SARS 3CLpro that inhibit via a non-covalent mechanism and possess low micromolar potency and good cell permeability, a screen was conducted in 2009 against the NIH molecular libraries sample collection (~293K compounds) in collaboration with the Mesecar laboratory and the Scripps Research Institute Molecular Screening Center (SRIMSC) through the molecular libraries program center network (MLPCN).41 Herein we describe the results of this screening campaign and the prosecution of an optimization effort at the Vanderbilt Specialized Chemistry Center (VSCC) around a class of 2-(N-arylamido)-2-(pyridin-3-yl) acetamide inhibitors of moderate molecular weight. In addition, from these collaborative efforts and with the aid of X-ray crystallography, we report the details of the molecular interaction of 3CLpro inhibitor 16-(R) with the active site of 3CLpro and further describe the physicochemical and ancillary properties of 16-(R).
RESULTS AND DISCUSSION
SARS-CoV 3CL protease construct used in assays and lack of inhibition by cinanserin
The 3CLpro enzyme, also known as non-structural protein 5 (nsp5), is liberated from coronavirus polyproteins by self-cleavage at its n-terminal (nsp4/5) and c-terminal (nsp5/6) cleavage sites. Although the autocatalytic mechanism is still being elucidated, current studies suggest that it involves an initial dimerization event between two unreleased nsp5s within the polyprotein followed by a series of catalytic steps that liberate a fully active, dimeric enzyme that can then proceed to processing the remaining 9 cleavage sites of the polyprotein.42 The SARS-CoV 3CLpro is highly active as a dimer and is essentially inactive as a monomer.43 The addition of affinity tags or additional amino acids to the N- and C–termini of 3CLpro is found to substantially increase the dimer dissociation constant (Kd) and dramatically decrease the enzymatic activity of the enzyme.42 Therefore, we expressed and purified SARS-CoV 3CLpro from a newly designed expression construct that produces the authentic 3CLpro dimer that would be generated in a virus infected cell. The enzyme was purified by a multi-step chromatographic procedure and was found to be highly active.
As the compound cinanserin was reported to be a highly effective, non-covalent inhibitor of SARS-CoV and HCoV-229E 3CLpros, i.e. IC50 values of 5 μM against each enzyme,26 we sought to use this compound as a control inhibitor to calculate Z’-factors during our high-throughput screening of compound libraries. We tested the ability of cinanserin to inhibit our authentic version of SARS-CoV 3CLpro at a concentration of 100 μM and surprisingly, we found no inhibition of the enzyme. Next, we tested the ability of cinanserin to inhibit the 3CLpro enzymes from the human coronavirus HKU1 and the mouse coronavirus MHV. Once again, we found no inhibition of these 3CL proteases at a cinanserin concentration of 100 μM.
Since our kinetic inhibition results stand in contrast to those of Chen and co-workers,26 we sought an explanation for this large discrepancy in potency. Upon closer examination of the reported methods used to determine the IC50 values for cinanserin, we found that the 3CLpro expression construct utilized by Chen and coworkers incorporated a (His)6-affinity tag at the N-terminus that extended it by an additional 14 residues.44 The N-terminal affinity tag was never removed from the purified 3CLpro enzymes prior to their kinetic studies. As described above, the addition of N-terminal or C-terminal residues to SARS 3CLpro is known to have a dramatic effect in increasing the dissociation constant for dimerization of the enzyme as well as decreasing the enzymatic activity of the enzyme.43 Therefore, cinanserin may be inhibiting a non-dimeric form of the enzyme. Another possible explanation is that Triton X-100 was not included in the assay buffer to remove potential promiscuous inhibition caused by non-specific aggregation of the protein by the compounds.45 We tested cinanserin for inhibition of SARS-CoV, HKU1 and MHV 3CLpro at a concentration of 100 μM cinanserin both in the presence and absence of 0.01% Triton X-100 and found no inhibition of the enzymes in either case. Ultimately, we were unable to use cinanserin as a control inhibitor since no inhibition of SARS-CoV, HKU1 or MHV 3CL proteases was observed. We therefore used compound 10a (Figure 2), a covalent modifier, as a positive control compound.
Screening Campaign and Lead Identification at the SRMISC
High-throughput screening was conducted by evaluating the inhibition of the 3CLpro-mediated peptide cleavage event using a novel FRET-based substrate (Pubchem: AID 1859). We utilized a number of key assay features in the screening campaign that have significant advantages over previous HTS campaigns against the 3CL enzyme.46 First, we utilized a SARS-CoV 3CLpro construct that consisted of the native sequence of the enzyme that represents the in vivo, post-proteolytic form of the enzyme. As discussed above, our version of the enzyme does not contain tags or excess amino acids that are known to drastically reduce catalytic activity of the enzyme.43 Second, we maintained a high concentration of fresh reducing agent in the primary assays which helped to minimize, but did not eliminate, covalent compounds as primary leads. Finally, we utilized a highly fluorescent FRET-based 3CLpro peptide substrate composed of a HiLyte fluor ™ 488 fluorescent label attached at the N-terminus and a QXL™520 quencher dye attached to the C-terminus. This substrate allows for excitation in the spectral region of 488 nm that helps to eliminate spectral overlap with the compounds in the screen. Moreover, the high fluorescence quantum yield of the fluorescent probe and sufficiently low turnover number of the enzyme yields a highly sensitive assay that is amenable to screening in 1536 plate format with low (⪡ Km) substrate concentrations.
The primary screen of the molecular libraries small molecule repository (~293K compounds at the time of screen) was conducted using a 6 μM concentration of each compound. The primary screen produced 406 hits with greater than 12.25% inhibition. An excellent Z-prime score of 0.92 and a relatively low hit-rate of 0.14% were observed. Among the 406 hits, 380 were available for re-order and were tested in a confirmation screen (singles at 10 μM). This second screen produced 136 active compounds with greater than 12.25% inhibition. A subsequent dose-response testing of 101 compounds provided 44 actives with IC50 values below 10 μM. A number of these actives were then counter-screened against the SARS-CoV papain-like protease, PLpro, which is also a potential target since it too is vital for SARS-CoV maturation.46-47 In general, the majority of IC50 values were substantially greater than 10 μM against PLpro suggesting moderate to good selectivity for 3CLpro inhibition.
Prior to initiation of the chemistry phase of the project, SRIMSC performed structural clustering analysis of the 101 confirmed actives. Several interesting scaffold clusters and a handful of singletons were identified from this analysis. A dipeptide class, represented by hit 14 (Figure 3, SID49730186), was one of two analogues with an IC50 below 10 μM. This furyl amide series was of particular interest due to its starting potency, modular nature, structural properties and absence of reactive functionality found within typical covalent modifiers.
Figure 3.

Left panel: Hit furyl amide 14 (SID49730186), Right panel: inhibition concentration curve for 3CLpro (grey squares) and PLpro (black squares) and 14.
Probe Optimization of Furyl Amide 14
Within the furyl scaffold, an initial 80 compound library array 15 was designed based upon HTS data using an Ugi MCC (multi-component condensation) strategy (Scheme 1).48-49
Scheme 1.

Ugi strategy and monomers used to prepare library 15 holding R4 as t-butyl isocyanide.
In the initial Ugi array, the isocyanide was held constant to afford a tert-butyl group at R4. The α-aryl substituent R3 was maintained either as a 3-pyridyl, as found in the original hit 14, or as a 2-thienyl which was also identified as a tolerable group in another closely related dipeptide cluster from HTS. Five monomers were selected for the R2 amine group to examine modifications of the aryl, and a cyclopropyl was included as a potential aryl replacement as a means to reduce MW and overall lipophilicity. For the R1 carboxylic acid component, eight monomers were chosen focusing exclusively on substituted aromatics and heteroaromotics. As part of the R1 group scan, a furan-3-carboxylic acid was included as a control for SAR comparisons relative to 14. All 80 compounds were successfully prepared and submitted for testing.
Disappointingly, only two Ugi library analogues displayed inhibition levels greater than 50% at 100 μM, and subsequently were determined to have IC50 values less than 50 μM. Notably, these active analogues were exclusively within the 3-pyridyl sub-series (Figure 4, 16 and 17). Within the 3-pyridyl series, replacement of the lipophilic N-aryl R2 group with either a cyclopropyl group or a 4-fluoro phenyl derivative (Figure 4, 18 and 19) were deleterious for inhibition, indicating the importance of steric bulk for optimal interaction within this putative subpocket of the enzyme. The 3-thienyl congener 20, a direct analogue of 16, resulted in a complete loss of inhibition activity.
Figure 4.

Selected SAR highlights from First Round Ugi Library.
Concurrent with synthetic plans to prepare a second generation Ugi library, SARS-CoV 3CLpro crystals suitable for X-ray diffraction and analysis were obtained with inhibitor 16 bound to 3CLpro.50 The electron density associated with inhibitor 16 is clearly resolved and allowed for accurate refinement of its position (Figure 6). The binding orientation of 16 is overall similar to known covalent peptidomimetic inhibitors, with inhibitor 16 preferentially occupying the S3 – S1′ subpockets of the 3CLpro enzyme as the R enantiomer (Figures 5-7).21 In this orientation, the tert-butyl amide occupies the S3 pocket, the tert-butylanilido group occupies a deep S2 pocket, the 3-pyridyl group occupies the S1, and the furyl amide acts as a P1′ group. For comparison, the mechanism-based covalent inhibitor 4 (PDB code 2ALV) and its occupied subpockets are represented schematically in Figure 5 relative to inhibitor 16.21 In addition to the S3 – S1′ pockets occupied by 16, 4 occupies the S4 pocket, thus relative to 4 the MW of 16 is reduced by over 100 amu and furthermore lacks a reactive warhead.
Figure 6.

SARS-CoV 3CLpro active site with bound inhibitor 16. (Left Panel) Wall-eye stereo view of the Fo-Fc electron density omit map (3 σ) surrounding inhibitor 16. (Right Panel) Solvent accessible surface view of 16-3CLpro complex (PDB code: 3V3M, PubChem SID87915542).
Figure 5.

Schematic of enzyme pockets occupied by 16 and 4.
Figure 7.

X-ray crystal structure of 16 (capped sticks in green carbon) with SARS 3CLpro in wall-eye stereo view. Hydrogen bonds between inhibitor and 3CLpro are shown as black dotted lines.
An electron density map and a solvent accessible surface depiction of 16 bound to 3CLpro are shown in Figure 6. A stereoview of the inhibitor-bound structure and associated interactions is also shown in Figure 7. Interestingly, the 3-pyridyl ring nitrogen serves as a hydrogen bond acceptor within the S1-subpocket engaging His-163 with a N-N interatomic distance of 2.8 Å. A similar hydrogen-bond interaction with His-163 engaged is retained in other 3CLpro crystal structures.21 The furan ring oxygen and the amide carbonyl oxygen have a bi-furcated interaction with the backbone NH of Gly-143. Although the distance to the amide carbonyl is 0.5 Å closer to Gly-143 and appears to be a hydrogen bond, based upon distance (C=O to NH = 2.9 Å), the geometry is not ideal. In fact, the NH of Gly-143 is nearly coplanar with both oxygen atoms. On the floor of the S1′ pocket, the catalytic Cys-145 is positioned beneath both the amide carbonyl carbon and the furan oxygen at a distance of 3.5 Å. With this rather unique furyl amide interaction within the context of the catalytic residues and backbone Gly-143 interaction, we set out to understand this interaction further by preparing a second chemical library focusing exclusively on the P1′ group holding the P1-P3 groups constant. Results from this initial campaign are summarized in Figure 8 (21-48).
Figure 8.

28-Membered P1’ Ugi library (21-48) and subsequent exploration of 5-membered azole and furan analogues (49-52). Inhibitor data indicated for compounds with 49% or greater inhibition at 100 μM test concentration.
Replacement with 5-membered pi-excessive heterocycles proved most successful with 22, 27, and 31 having IC50’s ~50 μM and below. A fully saturated tetrahydrofuran (32) and a select set of acyclic analogues were >10-fold less active with IC50’s > 50 μM. Substitution on the furan ring negatively impacted activity (21, 26) and although replacement of the furan oxygen atom with a larger sulfur atom in the related thiophene (27) was weakly tolerated (10-fold loss), the related pyrrole (37) heterocycle was completely inactive. The nearly equipotent inhibition by the imidazole 31 (SID93616598) however was encouraging, and thus a smaller subset of analogues was prepared to complete the survey of five-membered heterocycles (49 – 52). This effort identified 2-oxazolyl analogue 49 (SID99350507) with similar potency to the 4-oxazolyl isomer 22. Interestingly, in contrast to the 2-methyl furyl derivative 21, the 2-chloro substituted furyl congener 50 (SID 99350505) was nearly equipotent to 16 with an IC50 of 5.2 μM. SAR around imidazole 31 proved steep as the N-methyl C(4) substituted analogue 52 was >10-fold less active and addition of a methylene spacer (51) was deleterious for inhibition.
A subsequent survey of P1 replacements was performed in order to identify alternate hydrogen bond acceptor groups that might engage His-163, while retaining the 2-furyl amide P1’ group.
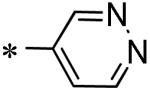
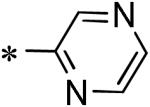
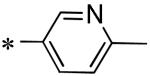
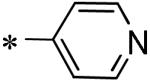
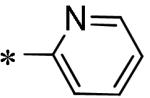
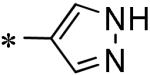
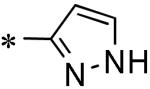
Among the various P1 replacements examined (53 – 61) pyridazine (53), and pyrazine (54) were well tolerated, although no improvement was found over lead compound 16. More dramatic modifications around the pyridyl ring were clearly limited for reasons consistent with the inhibitor bound X-ray structure as both the 4 and 2-pyridyl analogues were either weak inhibitors (56) or inactive (57) and addition of a 2-methyl in pyridyl analogue 55 was not permitted. Attempts to replace the 3-pyridyl P1 with 1,2-diazoles (58 – 59), 1,2,3-triazoles (60), or 1,3-diazoles (61) were also unsuccessful in identifying compounds with improved inhibition.
We next turned to chiral stationary phase supercritical fluid chromatography (SFC) to separate and isolate the single enantiomers of 16 (Figure 9). Highly specific inhibition was found for single stereoisomer 16-(R) with an IC50 of 1.5 ± 0.3 μM, which was identified as the first eluting peak from chiral SFC. In addition, inhibitor 16-(R) was found to specifically inhibit 3CLpro versus PLpro. Inhibitor 16-(R) was subsequently declared MLPCN probe ML188.41 Inhibitor 16-(R) was inferred as the R stereoisomer based upon the absolute configuration observed in the electron density map of the X-ray structure of the 16-3CLpro complex, and it was found to rotate plane polarized light in the positive direction. Finally, the mechanism of inhibition of SARS 3CLpro by 16-(R) was determined to be competitive based on a double-reciprocal plot of the initial rate of the SARS 3CLpro catalyzed reaction versus variable concentrations of substrate and fixed, variable concentrations of 16-(R) (see Figure 2 Supplemental Material). The resulting Ki value was determined to be 1.6 ± 0.26 μM, which is equivalent to its IC50 value and is expected since the IC50 values for the inhibitors are determined at a substrate concentration that is significantly lower than the Km value.
Figure 9.

Preparative chiral separation of 16 and identification of 16-(R).
SARS-CoV Antiviral Activity of 16-(R)
The potency of 16-(R) against SARS-CoV 3CLpro was deemed sufficient to determine the antiviral potency of the compound against SARS-CoV Urbani infected Vero E6 cells. Using our established anti-viral assay and BSL3 protocols, a dose-response curve of 16-(R) was generated against mock-infected and SARS-CoV infected cells and the results are shown in Figure 10.47
Figure 10.

Vero E6 cells were mock-infected (blue diamonds) or infected with SARS-CoV (red squares) for 1 hour prior to the addition of the antiviral compound 16-(R). At 48 hours post-infection, cell viability was determined using Cell Titer-Glo® Luminescent Cell Viability assay (Promega). The error bars represent the standard deviation of triplicate samples. Experiments were performed in duplicate on separate days.
A fit of the data presented in Figure 10 yields an antiviral EC50 value of 12.9 ± 0.7 μM. A second experiment, performed one month later, yielded an EC50 value of 13.4 ± 1.0 μM. The two independent experiments demonstrate that the resulting non-covalent inhibitor, 16-(R), can effectively inhibit SARS-CoV replication in cell culture.
We searched the literature for other non-covalent inhibitors against SARS-CoV 3CLpro that also have antiviral data associated with the compound. The compound cinanserin is reported to have antiviral activity.26 In that report, SARS-CoV infected Vero cells were treated with 50 μg/ml (134 μM) cinanserin. The authors conducted an RNA quantitation assay of the cell culture media and reported a 3 log reduction and indicate that this reduction correlates with the titer of infectious particles. The authors also did a control cytotoxicity assay and reported that the cytotoxic concentration that gives 50% reduction (CC50) is 31 μM and the CC90 is 66 μM. Given the fact that under these high cinanserin concentrations (134 μM) the majority of Vero cells (>90%) are likely killed by cinanserin, interpretation of the antiviral data in these studies is deemed ambiguous.
We next compared the EC50 of 16-(R) value to another non-covalent compound tested against SARS-CoV infected cells that targets the SARS-CoV papain-like protease (PLpro) known as GRL0617.47 Compound GRL0617 has an IC50 against the PLpro enzyme of 600 nM and an EC50 value of 14.5 μM against SARS infected Vero E6 cells.47 We next compared the EC50/IC50 ratios of the non-covalent compounds, 16-(R) and GRL0617, which are ~8.7 and ~24.1 respectively. This ratio can help guide compound design in terms of estimating what value of an IC50 needs to be achieved in order to achieve a pharmacologically relevant effect. Therefore, we compared these values to the most potent covalent 3CLpro inhibitor identified to date, TG-0205221 (5).22 This compound has an EC50/Ki value of 0.6 μM/0.052 μM or ~11.5. Thus, non-covalent and covalent inhibitors of 3CLpro can achieve similar antiviral effects.
Based upon the structural information, excellent 3CL protease inhibition activity and SARS-CoV Ubani antiviral activity, 16-(R) was elected as a first in class probe candidate from the furyl amide series. Probe 16-(R) was found to be highly selective for 3CLpro versus PLpro, and in a Ricerca lead-profiling screen51 against 68 discrete GPCRs, ion channels and transporters, no significant activity was found (<50% at 10 μM). In PBS buffer at neutral pH, probe 16-(R) was found to have excellent aqueous solubility up to 95 μg/mL or 219 μM. Extended characterization of probe inhibitor 16-(R) and analogues against other cysteine proteases and in SARS-CoV infected cells as well as in basic DMPK assays to assess metabolic stability and plasma protein binding are ongoing.
CONCLUSIONS
In summary, we have described the optimization and molecular interaction details for a potent, non-covalent inhibitor of SARS-CoV 3CLpro identified through the MLPCN initiative. A four component Ugi reaction was utilized to rapidly generate SAR and optimize potency focusing on the S1’, S1, and S2 binding pockets. X-ray analysis of the 3CLpro-16-(R) complex reveals several key interactions crucial for activity; in particular a hydrogen bonding interaction between the inhibitor 3-pyridyl ring nitrogen and the active site His-163 side-chain located within the S1-subpocket. Probe 16-(R) (ML188) is a modest molecular weight SARS-CoV 3CLpro inhibitor with demonstrated antiviral activity and a non-covalent mechanism of action, and thus provides the opportunity to facilitate further structure-based design and inhibitor refinement in the quest for potential novel anti-SARS CoV therapies. Collaborative efforts in these laboratories continue towards the identification of sub-micromolar 3CLpro inhibitors with optimal properties for anti-viral activity and potential testing in animal models. ML188 is an MLPCN probe and is freely available upon request.
EXPERIMENTAL SECTION
General
All NMR spectra were recorded on a Bruker 400 and 600 MHz instrument. 1H chemical shifts are reported in δ values in ppm downfield from TMS as the internal standard in d3-MeOH or CDCl3. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 1200 series 6130 mass spectrometer. High resolution mass spectra were recorded on a Waters Q-TOF API-US. Analytical thin layer chromatography was performed on Analtech silica gel GF 250 micron plates. Analytical HPLC was performed on an HP1100 with UV detection at 214 and 254 nm along with ELSD detection, LC-MS (J-Sphere80-C18, 3.0 × 50 mm, using either a 1.1 or 3.1 min gradient, 5%[0.05%TFA/CH3CN]:95%[0.05%TFA/H2O] to 100%[0.05%TFA/CH3CN]. Preparative RP-HPLC purification was performed on a custom HP1100 automated purification system with collection triggered by mass detection or using a Gilson Inc. preparative UV-based system using a Phenomenex Luna C18 column (50 × 30 mm I.D., 5 μm) with an acetonitrile (unmodified)-water (0.1% TFA) custom gradient. Normal-phase silica gel preparative purification was performed using an automated Combi-flash companion from ISCO. Solvents for extraction, washing and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification. All polymer-supported reagents were purchased from Argonaut Technologies and Biotage.
General Procedure for 4CC-Ugi Reaction in Library Format
To a series of 13 × 100 mm screw top glass tubes fitted with a magnetic stir bar equimolar amounts (0.08 mmol) of carboxaldehyde, amine, carboxylic acid were combined in methanol (0.2 M, 1.5 mL) and subsequently treated with tert-butylisocyanide (0.08 mmol). The mixture was stirred for 16h at ambient temperature, and then concentrated under a stream of nitrogen in a well ventilated hood. The crude mixtures were reconstituted in MeOH, treated with Argoresin MP-Trisamine (Biotage Inc.) scavenger for 2h and applied to a Celite pad using a manifold fitted with polypropylene filter tubes capable of filtering 24 samples in parallel. The filtrates were purified directly using an automated mass-guided RP-HPLC and product containing fractions were concentrated to give final products >95% purity as judged by LC-MS (215 nm and ELSD) and 1H NMR (representative library members >10%). All final library array products characterization data, 1H NMR spectra, and SARS-3CLpro % inhibition at 100 μM can be found in Supporting Information.
Analogues 21 – 48 and 49 – 61 were prepared in an analogous manner and purified by Gilson Inc. preparative UV-based RP-HPLC. Chromatographic, LC-MS data, and representative 1H and 13C NMR spectra for these remaining analogues can be found in Supporting Information. 1H NMR data for 20% of these libraries have been provided.
Preparation of (R)-N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)furan-2-carboxamide (16-(R), ML188)
To a 20 mL scintillation equimolar amounts (0.5 mmol) of pyridine-3-carboxaldehyde, 4-tert-butylaniline, furan-2-carboxylic acid were combined in methanol (0.2M, 2.5mL) and treated with tert-butylisocyanide (0.5mmol). The mixture was stirred for 16h at ambient temperature, then concentrated and applied as a DCM solution to a silica column (12g) and purified over a gradient of 0 to 100% EtOAc in Hexanes. The racemic product was obtained as a light yellow solid (198 mg, 91%): LC-MS (>98%) m/z =434 [M+H]. Separation of enantiomers was accomplished using supercritical fluid chromatography. The column was an IA (UV 250 nm, 10 × 250 mm, Chiral Technologies), eluent 6% MeOH in CO2. The desired peak as confirmed by the primary assay was identified as the first eluting peak, retention time = 2.91 min, (CID 46897844): [α]D = +38.9 (c = 1.0, CHCl3); 1H NMR (400MHz, CDCl3) δ 8.51 (1H, d, J = 2), 8.49 (1H, dd, J = 4.8, 2), 7.53 (1H, dt, J = 8, 2), 7.41 (1H, d, J = 1), 7.29 (2H, d, J =6.6), 7.23 (2H, d, J = 6.6), 7.09 (1H, dd, J = 8, 4.8), 6.18 (1H, dd, J = 3.6, 2), 6.12 (1H, s), 5.41 (1H, d, J = 3.6), 1.40 (9H, s), 1.28 (9H, s); 13C NMR (400MHz, CDCl3) δ 167.7, 159.5, 152.4, 151.3, 149.4, 146.1, 144.9, 138.1, 136.4, 130.4, 130.1, 126.0, 122.7, 117.0, 111.1, 63.6, 51.7, 34.6, 31.2, 28.6; HRMS (ESI, M+H) calcd for C26H32N3O3: 434.2440, found 434.2444.
N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-isopropylphenyl)furan-2-carboxamide, 17
1H NMR (400MHz, CDCl3) δ 8.46 (2H, m), 8.49 (1H, d, J = 7.9 Hz), 7.38 (1H, s), 7.07 (3H, m), 6.98 (2H, bs), 6.15 (2H, m), 6.10 (1H, s), 5.38 (1H, d, J = 3.6 Hz), 2.87 (1 H, p, J = 6.9 Hz), 1.37 (9H, s), 1.20 (6H, d); 13C NMR (100 MHz, CDCl3) δ 167.8, 159.5, 151.4, 150.0, 149.4, 146.1, 144.8, 138.0, 136.6, 130.5, 130.4, 127.0, 122.7, 117.0, 111.1, 63.5, 51.6, 33.6, 28.5, 23.8, 23.7; HRMS (ES+, M+H) calcd for C25H30N3O3: 420.2287, found 420.2290.
N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-cyclopropylfuran-2-carboxamide, 18
1H NMR (400MHz, CDCl3) δ 8.68 (1H, s), 8.57 (1H, d, J = 4.0 Hz), 7.97 (1H, d, J = 7.9), 7.53 (1H, s), 7.32 (1H, dd, J = 7.7, 4.8), 7.10 (1H, d, J = 3.4 Hz), 6.57 (1H, s), 6.51 (1H, dd, J = 3.4, 1.6 Hz), 5.66 (1H, s), 2.90 (1H, m), 1.36 (9H, s), 0.99 (1H, m), 0.80 (m, 1H), 0.68 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.3, 162.4, 150.5, 148.9, 147.2, 144.5, 137.1, 131.1, 122.8, 117.1, 111.2, 64.7, 51.1, 31.2, 28.2, 10.4, 9.5; HRMS (ES+, M+H) calcd for C19H24N3O3: 342.1818, found 342.1815.
N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-fluorophenyl)furan-2-carboxamide, 19
1H NMR (400MHz, CDCl3) δ 8.41 (1H, d, J = 4.0 Hz), 8.37 (1H, s), 7.43 (1H, d, J = 8.0), 7.29 (1H, s), 7.05 (2H, dd, J = 7.8, 4.8), 6.85 (2H, m), 6.29 (1H, s), 6.15 (1H, s), 6.13 (1H, dd, J = 3.5, 1.6 Hz), 5.55 (1H, d, J = 3.6 Hz), 5.24 (s, 1H), 1.30 (9H, s); 13C NMR (100 MHz, CDCl3) δ 167.8, 162.5 (d, J = 248), 159.3, 151.3, 149.5, 146.1, 144.9, 137.8, 134.9 (d, J = 3.1 Hz), 132.9 (d, J = 8.6 Hz), 130.3, 123.0, 117.2, 115.8 (d, J = 22.4 Hz), 111.1, 62.8, 51.7, 28.5; HRMS (ES+, M+H) calcd for C22H23N3O3F: 396.1723, found 396.1725.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(thiophen-3-yl)ethyl)furan-2-carboxamide, 20
1H NMR (400MHz, CDCl3) δ 7.37 (1H, s), 7.26 (2H, s), 7.24 (1H, s), 7.13 (1H, m), 6.99 (2H, bs), 6.89 (1H, d, J = 5.0 Hz), 6.13 (1H, m), 6.10 (1H, s), 6.06 (1H, s) 5.33 (1H, d, J = 3.6Hz), 1.35 (9H, s), 1.29 (9H, s); 13C NMR (100 MHz, CDCl3) δ 168.1, 159.3, 152.0, 146.4, 144.6, 137.2, 134.9, 129.6, 129.0, 126.7, 125.8, 125.1, 116.7, 111.1, 61.6, 51.4, 34.6, 31.3, 28.6; HRMS (ES+, M+H) calcd for C25H31N2O3S: 439.2055, found 439.2057.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)oxazole-5-carboxamide, 22
1H NMR (400MHz, CDCl3) δ 8.48 (2H, s), 7.84 (1H, s), 7.48 (1H, d, J = 7.9 Hz), 7.28 (2H, m), 7.08 (1H, m), 6.06 (1H, s), 5.85 (1H, bs), 5.70 (1H, s), 5.30 (1H, s) 1.37 (9H, s), 1.27 (9H, s); 13C NMR (100 MHz, CDCl3) δ 167.3, 157.7, 152.8, 152.1, 151.2, 149.3, 144.1, 137.7, 134.9, 131.1, 130.3, 129.9, 125.9, 122.7, 62.9, 51.5, 34.4, 30.9, 28.3; HRMS (ES+, M+H) calcd for C25H31N4O3: 435.2396, found 435.2397.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)thiophene-2-carboxamide, 27
1H NMR (400MHz, CDCl3) δ 8.47 (1H, s), 8.44 (1H, d, J = 4.0 Hz), 7.48 (1H, d, J = 8.0 Hz), 7.29 (1H, dd, J = 4.8, 0.9 Hz), 7.23 (2H, d, J = 8.3 Hz), 7.05 (1H, dd, J = 7.9, 4.8 Hz), 7.01 (1H, bs), 6.75 (2H, m), 6.29 (1H, bs), 6.14 (1H, s), 1.36 (9H, s), 1.26 (9H, s); 13C NMR (100 MHz, CDCl3) δ 167.9, 162.9, 152.6, 151.4, 149.4, 138.0, 137.5, 136.5, 133.1, 131.6, 130.6, 130.5, 126.7, 126.1, 122.7, 64.1, 51.7, 34.6, 31.2, 28.6; HRMS (ES+, M+H) calcd for C26H32N3O2S: 450.2215, found 450.2213.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-1H-imidazole-4-carboxamide, 31
1H NMR (600MHz, CDCl3) δ 11.66 (1H, bs), 8.47 (1H, d, J = 1.9 Hz), 8.41 (1H, dd, J = 3.2, 1.4 Hz), 7.59 (2H, s), 7.39 (1H, d, J = 5.3 Hz), 7.22 (3H, bs), 7.01 (1H, dd, J = 5.3, 4.8 Hz), 6.32 (1H, bs), 6.20 (1H, s), 5.43 (1H, bs), 1.24 (9H, s), 1.23 (9H, s). 13C NMR (150 MHz, CDCl3) δ 167.8, 161.1, 152.9, 151.4, 149.5, 137.9, 137.3, 135.6, 133.2, 130.5, 126.2, 125.0, 122.8, 63.5,51.6, 34.6, 31.2, 28.5; HRMS (ES+, M+H) calcd for C25H32N5O2: 434.2556, found 434.2555.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-2-methoxybenzamide, 33
1H NMR (400MHz, CD3OD) δ 8.72 (qH, bs), 8.60 (1H, bs), 8.24 (1H, bs), 7.73 (2H, bs), 7.30 (1H, d, J = 7.4 Hz), 7.22 (1H, t, J = 7.4 Hz), 7.11 (2H, d, J = 8.4 Hz), 7.00 (2H, d, J = 8.3 Hz), 8.87 (1H, t, J = 7.4 Hz), 6.76 (1H, d, J = 8.2 Hz), 6.25 (1H, s), 3.68 (3H, s), 1.29 (9H, s), 1.15 (9H, s); LC-MS (M+H) = 474.2
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)isonicotinamide, 35
1H NMR (400MHz, CD3OD) δ 8.57 (1H, s), 8.50 (3H, m), 7.98 (1H, m), 7.57 (2H, m), 7.51 (1H, m), 7.12 (4H, m), 6.28 (1H, s), 1.34 (9H, s), 1.14 (9H, s), LC-MS (M+H) = 445.2.
N-(tert-butyl)-2-(N-(4-(tert-butyl)phenyl)-2-hydroxyacetamido)-2-(pyridin-3-yl)acetamide, 40
1H NMR (400MHz, CD3OD) δ 8.50 (2H, m), 7.99 (1H, d, J = 6.5 Hz), 7.55 (qH, m), 7.33 (2H, d, J = 9.1 Hz), 7.q5 (2H, bs), 6.12 (1H, s), 3.83 (1H, d, J = 3.0 Hz), 1.29 (9H, s), 1.26 (9H, s); LC-MS (M+H) = 398.2
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-3-hydroxybenzamide, 44
1H NMR (400MHz, CD3OD) θ 8.63 (qH, bs), 8.56 (1H, bs), 8.16 (1H, bs), 7.66 (1H, m), 7.14 (2H, d, J = 8.8 Hz), 7.02 (2H, d, J = 8.1 Hz), 6.97 (1H, t, J = 7.9 Hz), 6.78 (1H, s), 6.74 (1H, d, J = 7.7 Hz), 6.66 (1H, dd, J = 8.2, 1.5 Hz), 6.26 (1H, s), 1.29 (9H, s), 1.17 (9H, s); LC-MS (M+H) = 460.2.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)pyrimidine-2-carboxamide, 45
1H NMR (400MHz, CD3OD) δ 8.65 (1H, d, J = 11.8 Hz), 8.54 (1H, d, J = 5.2 Hz), 8.43 (1H, s), 8.38 (1H, s), 8.10 (1H, d, J = 6.8 Hz), 7.60 (1H, m), 7.13 (1H, d, J = 8.6 Hz), 7.06 (1H, d, J = 7.9 Hz), 6.32 (1H, s), 1.31 (9H, s), 1.15 (9H, s); LC-MS (M+H) = 446.3.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-4-hydroxybenzamide, 47
1H NMR (400MHz, CD3OD) δ 8.61 (1H, s), 8.54 (1H, d, J = 5.0 Hz), 8.12 (1H, d, J = 6.4 Hz), 7.63 (1H, m), 7.19 (4H, m), 7.02 (2H, d, J = 8.2 Hz), 6.54 (2H, d, J = 8.7 Hz), 6.24 (1H, s), 1.29 (9H, s), 1.20 (9H, s); LC-MS (M+H) = 460.2.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-1-methyl-1H-imidazole-4-carboxamide, 52
1H NMR (400MHz, CD3OD) δ 8.81 (1H, s), 8.46 (1H, s), 8.43 (1H, d, J = 4.76 Hz), 7.78 (1H, d, J = 8.0 Hz), 7.37 (5H, m), 6.23 (1H, s), 5.44 (1H, s), 3.63 (3H, s), 1.33 (9H, s), 1.29 (9H, s); LC-MS (M+H) = 448.2.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-1-(6-methylpyridin-3-yl)-2-oxoethyl)furan-2-carboxamide, 55
1H NMR (400MHz, CD3OD) δ 8.56 (1H, s), 8.24 (1H, d, J = 8.3 Hz), 7.71 (1H, d, J = 8.3 Hz), 7.52 (1H, s), 7.41 (2H, d, J = 8.8 Hz), 7.22 (2H, m), 6.28 (1H, dd, J = 3.6, 3.3 Hz), 6.23 (1H, s), 5.62 (1H, d, J = 3.5 Hz), 2.69 (3H, s), 1.31 (9H, s), 1.27 (9H, s); LC-MS (M+H) = 448.3.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(1H-1,2,3-triazol-4-yl)ethyl)furan-2-carboxamide, 60
1H NMR (400MHz, CD3OD) δ 7.52 (1H, d, J = 1.2 Hz), 7.44 (1H, s), 7.34 (2H, d, J = 8.6 Hz), 7.15 (2H, bs), 6.30 (1H, s), 6.25 (1H, dd, J = 3.6, 1.6 Hz), 5.46 (1H, d, J = 3.5 Hz), 4.58 (1H, s), 1.34 (9H, s), 1.29 (9H, s); LC-MS (M+H) = 424.2.
Expression and Purification of the SARS-CoV 3CLpro enzyme
An expression construct for the SARS-CoV 3CLpro enzyme was designed to produce the exact coding region of the enzyme released in virus-infected cells. The expression construct contained a hexa-histidine affinity tag and a TEV protease site that is also recognized and cleaved by SARS-CoV 3CLpro. The coding region of SARS-CoV 3CLpro was codon-optimized and synthesized by BioBasic (Canada) and was inserted into a pET expression vector. The enzyme was expressed in E. coli BL21(DE3) and was purified via multi-step purification protocol that employed a cobalt-charged or nickel-charged metal-chelate HiTrap affinity column (GE Health sciences), DEAE anion-exchange chromatography and size-exclusion chromatography. Purified SARS-CoV 3CLpro was then stored at −80°C or used immediately for crystallization.
HTS Campaign Assays
3CLpro HTS assay protocol
The primary assay began with the addition of 4 μL of 3CLpro enzyme (150 nM final concentration) in assay buffer (50 mM HEPES, 0.1 mg/mL BSA, 0.01% Triton-X 100, 2 mM DTT) at pH 7.5 into each well of a 1536 microtiter plate. Next, 30 nL of test compound in DMSO, 3CLpro inhibitor (300 μM final concentration) in DMSO, or DMSO alone (0.6% final concentration) was added to the appropriate wells. The plates were then incubated for 10 minutes at room temperature. After incubation, 1 μL of 3Clpro peptide substrate (2 μM final concentration) in 50 mM HEPES at pH 7.5 to each well. After 30 minutes of incubation at room temperature, 1 uL of 500 mM acetic acid was added to each well to terminate the assay and well fluorescence was read on a PerkinElmer Viewlux using fluorescein filters: excitation wavelength of 480 nm (with 20 nm bandwidth) and emission wavelength of 540 nm (with 20 nm bandwidth). All data was normalized to that of the positive control (3CLPro inhibitor) and wells containing DMSO only (negative control). The same protocol was conducted for single point primary (PubChem AID 1706), triplicate point secondary (PubChem AID 1879) and confirmatory dose response assays (PubChem AID 1890).
PLpro counterscreen assay protocol
Prior to assay, PLpro peptide substrate and luciferase detection reagent were mixed in assay buffer (50 mM HEPES, 0.1 mg/mL BSA, 5 mM DTT, 0.5 mM EDTA and 1 mM Magnesium sulfate) at pH 7.5 and incubated for 60 min. The assay was begun by dispensing 2.5 μL of PLpro enzyme (7.5 nM final concentration) in assay buffer or assay buffer alone into each well of a 1536 microtiter plate. Next, 30 nL of test compound in DMSO, or DMSO alone (0.6% final concentration) was added to the appropriate wells. The plates were then incubated for 10 minutes at room temperature. Next, the enzyme reaction was initiated by dispensing 2.5 μL of the preincubated mixture containing PLpro peptide substrate and luciferase detection reagent (1 uM final substrate concentration). Finally, well luminescence was read on a PerkinElmer Viewlux after 60 minutes of incubation at room temperature. All data was normalized to that of the positive control (no enzyme) and wells containing DMSO only (negative control). This protocol was used for confirmatory dose response assays (PubChem AID 1944). Detailed assay protocols and data generated for HTS and probe development are found at PubChem AID 1859: pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1859&loc=ea
Crystallization and X-ray structure determination of SARS-CoV 3CLpro in complex with inhibitor 16-(R)
Purified SARS-CoV 3CLpro was concentrated to 16 mg/ml in a buffer composed of 20 mM HEPES pH 7.5 and 5 mM 2-mercaptoethanol. The enzyme was incubated on ice with a final concentration of 1 mM inhibitor. A 1:1 enzyme to crystallization solution ration was used and the crystallization solution consisted of 14.5% PEG 20,000, 50 mM MES pH 6.0, 50 mM potassium chloride and 1% MPD. Crystallization trials were set up at room temperature using the method of hanging drop vapor diffusion. Significantly better crystal formation was observed in the absence of sodium chloride, and crystals were sensitive to small fluctuations in pH. Single, well-formed crystals were used for soaking in mother-liquor supplemented with 2 mM of inhibitor at room temperature for 3 hours. 15% MPD was used as a cryoprotectant for freezing the crystals in liquid nitrogen where they were stored until synchrotron time was available. Crystals were transferred from liquid nitrogen into a stream of dry nitrogen gas at 100° K for X-ray data collection.
X-ray data were collected on a Rayonix-300 CCD detector at the Advanced Photon Source, Argonne National Laboratory, on beamline 21 ID-F at the Life Sciences-Collaborative Access Team (LS-CAT). X-ray data were processed and scaled using HKL2000.52 The SARS-CoV 3CLpro-ML188 complex crystallized as a single monomer in the asymmetric unit and in space group C2 with unit cell dimensions of a=106.73Å, b=82.67Å, c=53.12Å, β=106.0°. The crystal diffracted to a resolution 1.95 Å. The X-ray intensity data had a final Rmerge of 6.4% and the data were 97.8% complete overall. Additional data processing statistics are provided in Table 4 in SI.
X-ray intensities were converted to structure-factor amplitudes by the method of French and Wilson using the program TRUNCATE in the CCP4 program suite.53 The initial phases for the model were determined by the method of molecular replacement using the program MOLREP54 in the CCP4 program suite. The search model used for molecular replacement consisted of a monomer of SARS-CoV 3CLpro from PDB Code 2ALV with all side chains intact and all waters and ligands removed. The final and optimal molecular replacement solution contained a single monomer in the asymmetric unit.
An initial round of combined positional and B-factor refinement was performed with the program REFMAC55 in the CCP4 program suite using a maximum-likelihood target function and no sigma cutoff on structure factor amplitudes. Initial difference Fourier maps were calculated and visualized using the program COOT.56 The initial Fo-Fc difference maps revealed strong (+4σ), residual electron density peaks in the active site for the inhibitor 16-(R). A molecular model for the inhibitor was built using the Monomer Library Sketcher program in the CCP4 program suite, and a monomer library description was created for refinement. Iterative rounds of refinement were performed, and water molecules were added manually into strong (+4σ) difference density peaks in the initial refinement stages, and into peaks of (+ 3 σ) in the final stages of refinement. During these iterative refinements, residual electron density consistent with two DMSO molecules from the buffer solution were identified, and these molecule were built into residual density and included in all subsequent refinements.
Iterative refinement using REFMAC was continued until the Rwork and Rfree values plateaued at their lowest values which were 21.5% and 27.1%, respectively. At this point, the coordinates for the resulting model were submitted to the TLS (translation/libration/screw) server to generate a multi-group TLS model.57 The resulting TLS groups were visualized using the molecular viewer on the TLS website, and 20 TLS groups were chosen.58 Two rounds of TLS and restrained refinement59 were performed in REFMAC with the weighting term set at 0.1. The resulting and final Rwork and Rfree values were 18.7% and 23.5%, respectively, justifying the inclusion of TLS groups in the standard refinement protocol.59 The final model coordinates have been deposited in the PDB under accession code PDB:3V3M.60 A summary of the final X-ray data refinement statistics are given in Table 4 in Supporting Information.
SARS-CoVAntiviral Activity Assays
The SARS-CoV Urbani strain was used in these experiments and was provided by the Centers for Disease Control and Prevention. Maintenance of the Vero E6 cells used in these studies was achieved using Dulbecco’s Minimal Essential Media (DMEM) (Gibco) supplemented with 100 units/ml penicillin, 100 g/ml streptomycin (Gibco), and 10% FCS (Altanta Biologicals). All anti-viral experiments on SARS-CoV Urbani were carried out at Purdue University using a Biosafety Level 3 suite and approved biosafety protocols established in collaboration with the Purdue University Institutional Biosafety Committee.
Growth of Vero E6 cells was performed by seeding cells onto flat-bottom, 96-well plates at a density of approximately 9 × 103 cells per well. Vero E6 cells were either mock-infected with serum-free DMEM or infected with 100-fold the median tissue culture infective dose of SARS-CoV Urbani per well in 100 μL of serum-free MEM and incubated for 1 h at 37°C with 5% CO2. The viral inoculum was removed after the 1-h incubation period and then 100 μL of MEM, supplemented with 2% FCS and the 16-(R) inhibitor, at concentrations ranging from 30 to 0.1 M, was added. Cells were then incubated for 48 h at 37°C with 5% CO2. All controls and each inhibitor concentration was set up in triplicate, and the antiviral assays were performed independently on at least 2 separate occasions. Cell viability was determined approximately 48 h after infection using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Supplementary Material
Table 1.
Focused Pi group heterocyclic library (53-61).
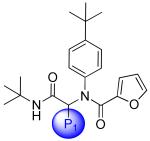
|
|||
|---|---|---|---|
|
| |||
| Compound | SID |
|
3CLpro IC50μM |
| 53 | 117686897 |
|
10 |
| 54 | 117686898 |
|
5.5 |
| 55 | 117686909 |
|
Inactive |
| 56 | 117686901 |
|
45 |
| 57 | 117686900 |
|
Inactive |
| 58 | 117686899 |
|
16% (100 μM) |
| 59 | 117686904 |
|
Inactive |
| 60 | 117686905 |
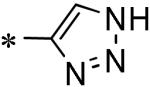
|
23% (100 μM) |
| 61 | 117686906 |
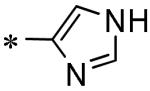
|
23% (100 μM) |
IC50values are average of two independent runs using triplicate concentrations, ‘Inactive’ defined as %inhibition <15% at 100 μM, CV <0.3
ACKNOWLEDGEMENTS
The authors thank the MLPCN (1U54 MH084659 and MH084512) and NIH via P01 AI060915 (ADM), 1R01AI085089 (ADM), 2R01AI026603 (ADM) and 1R03MH84162 (VLT) for support. Nathan Kett and Chris Denicola for preparative chiral SFC (VSCC). Julie Engers and Lauren Melancon for project team management (VSCC). Pierre Baillargeon and Lina DeLuca (Scripps) for compound management assistance. Katharine Emery, Jill Ferguson, and Becky Mercer (Scripps) for administrative assistance in reporting data and project details to the MLPCN. We gratefully acknowledge the synchrotron beamline (LS-CAT) personnel at the Advanced Photon Source at Argonne National Lab. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Abbreviations Used
- SARS
severe acute respiratory syndrome
- CoV
coronavirus
- 3CL
chymotrypsin-like protease
- PLpro
papain-like protease
- MCC
multi-component condensation
- 4CC
four-component condensation reaction
- BSL3
biosafety level 3 suite
- MLPCN
molecular libraries probe production centers network
- SID
substance identifier
- AID
assay identifier
Footnotes
Supporting Information Available:
Additional experimental procedures, analytical data for library compounds, in vitro assays, Lineweaver-Burk plot analysis, X-ray crystallography statistics and parameters. This material is free of charge via the Internet at http://pubs.acs.org.
PDB Accession Codes: 3V3M
REFERENCES
- 1.Myint SH. The Coronaviridae. In: Siddell SG, editor. Human coronavirus infections. Plenum Press; 1995. pp. 389–401. [Google Scholar]
- 2.McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci U S A. 1967;57:933–940. doi: 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol. 2004;7:412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyrc K, Berkhout B, van der Hoek L. The novel human coronaviruses NL63 and HKU1. J Virol. 2007;81:3051–3057. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fielding BC. Human coronavirus NL63: a clinically important virus? Future Microbiol. 2011;6:153–159. doi: 10.2217/fmb.10.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui L-J, Zhang C, Zhang T, Lu R-J, Xie Z-D, Zhang L-L, Liu C-Y, Zhou W-M, Ma X-J, Tan W-J. Human coronaviruses HCoV-NL63 and HCoV-HKU1 in hospitalized children with acute respiratory infections in Beijing, China. Advances in Virology. 2011:6. doi: 10.1155/2011/129134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012 doi: 10.1056/NEJMoa1211721. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Bartlam M, Rao Z. Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr Pharm Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 11.Lai L, Han X, Chen H, Wei P, Huang C, Liu S, Fan K, Zhou L, Liu Z, Pei J, Liu Y. Quaternary structure, substrate selectivity and inhibitor design for SARS 3C-like proteinase. Curr Pharm Des. 2006;12:4555–4564. doi: 10.2174/138161206779010396. [DOI] [PubMed] [Google Scholar]
- 12.Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, Lai B, Pei J, Liu Y, Chen J, Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J Biol Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel V, Herold J, Schelle B, Siddell SG. Viral replicase gene products suffice for coronavirus discontinuous transcription. J Virol. 2001;75:6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan K, Ma L, Han X, Liang H, Wei P, Liu Y, Lai L. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem Biophys Res Commun. 2005;329:934–940. doi: 10.1016/j.bbrc.2005.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuck CP, Chong LT, Chen C, Chow HF, Wan DC, Wong KB. Profiling of substrate specificity of SARS-CoV 3CL. PLoS One. 2010;5:e13197. doi: 10.1371/journal.pone.0013197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartlam M, Yang H, Rao Z. Structural insights into SARS coronavirus proteins. Curr Opin Struct Biol. 2005;15:664–672. doi: 10.1016/j.sbi.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh AK, Xi D, Johnson ME, Baker SC, Mesecar AD. Progress in Anti-SARS Coronavirus Chemistry, Biology and Chemotherapy. Ann. Rep. Med. Chem. 2006;41:183–196. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain RP, Pettersson HI, Zhang J, Aull KD, Fortin PD, Huitema C, Eltis LD, Parrish JC, James MN, Wishart DS, Vederas JC. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AK, Xi K, Ratia K, Santarsiero BD, Fu W, Harcourt BH, Rota PA, Baker SC, Johnson ME, Mesecar AD. Design and Synthesis of Peptidomimetic Severe Acute Respiratory Syndrome Chymotrypsin-like Protease Inhibitors. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 22.Yang S, Chen SJ, Hsu MF, Wu JD, Tseng CT, Liu YF, Chen HC, Kuo CW, Wu CS, Chang LW, Chen WC, Liao SY, Chang TY, Hung HH, Shr HL, Liu CY, Huang YA, Chang LY, Hsu JC, Peters CJ, Wang AH, Hsu MC. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Pettersson HI, Huitema C, Niu C, Yin J, James MNG, Eltis LD, Vederas JC. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-Like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 24.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akaji K, Konno H, Mitsui H, Teruya K, Shimamoto Y, Hattori Y, Ozaki T, Kusunoki M, Sanjoh A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Gui C, Luo X, Yang Q, Günther S, Scandella E, Drosten C, Bai D, He X, Ludewig B, Chen J, Luo H, Yang Y, Yang Y, Zou J, Thiel V, Chen K, Shen J, Shen X, Jiang H. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu CY, King KY, Kuo CJ, Fang JM, Wu YT, Ho MY, Liao CL, Shie JJ, Liang PH, Wong CH. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD, Brown ED. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh AK, Gong G, Grum-Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME, Mesecar AD. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008;18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen LR, Wang YC, Lin YW, Chou SY, Chen SF, Liu LT, Wu YT, Kuo CJ, Chen TS, Juang SH. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15:3058–3062. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Huitema C, Niu C, Yin J, James MNG, Eltis LD, Vederas JC. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C-like proteinase. Bioorg. Chem. 2008;36:229–240. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee P, Desai P, Ross L, White EL, Avery MA. Structure-based virtual screening against SARS-3CL(pro) to identify novel non-peptidic hits. Bioorg. Med. Chem. 2008;7:4138–4149. doi: 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TT, Ryu HJ, Lee SH, Hwang S, Breton V, Rhee JH, Kim D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. 2011;21:3088–3091. doi: 10.1016/j.bmcl.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeung KS, Meanwell NA. Recent developments in the virology and antiviral research of severe acute respiratory syndrome coronavirus. Infect Disord Drug Targets. 2007;7:29–41. doi: 10.2174/187152607780090739. [DOI] [PubMed] [Google Scholar]
- 35.Regnier T, Sarma D, Hidaka K, Bacha U, Freire E, Hayashi Y, Yoshiaki K. New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2009;19:2722–2727. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guterman L. Covalent drugs form long-lived ties. Chemical & Engineering News. 2011;89(36):19–26. [Google Scholar]
- 37.Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 38.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 39.Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10:464–469. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 40.Wu CY, Jan JT, Ma SH, Kuo CJ, Juan HF, Cheng YS, Hsu HH, Huang HC, Wu D, Brik A, Liang FS, Liu RS, Fang JM, Chen ST, Liang PH, Wong CH. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.For information on the MLPCN and information on how to request probe compounds, such ML188. see: http://mli.nih.gov/mli/mlpcn/
- 42.Chen S, Jonas F, Shen C, Hilgenfeld R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell. 2010;1:59–74. doi: 10.1007/s13238-010-0011-4. erratum Protein Cell. 2010, 1,307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grum-Tokars V, Ratia K, Begaye A, Baker SC, Mesecar AD. Evaluating the 3C-like protease activity of SARS-Coronavirus: Recommendations for standardized assays for drug discovery. Virus Res. 2008;133:63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun H, Luo H, Yu C, Sun T, Chen J, Peng S, Qin J, Shen J, Yang Y, Xie Y, Chen K, Wang Y, Shen X, Jiang H. Molecular cloning, expression, purification, and mass spectrometric characterization of 3C-like protease of SARS coronavirus. Protein Expr Purif. 2003;32:302–308. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 46.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ugi I, Meyr R, Fetzer U, Steinbrückner C. Versuche mit Isonitrilen. Angew. Chem. 1959;71:386. [Google Scholar]
- 49.Dömling A, Ugi II. Multicomponent Reactions with Isocyanides. Angew Chem Int Ed Engl. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 50.The protein-ligand X-ray structure of ML188-bound SARS-3CLpro has been deposited in PDB. PDB code is 3V3M.
- 51.For information on MLPCN’s probe compound ancillary screen see Ricerca LeadProfilingScreen®: https://pharmacology.ricerca.com.
- 52.Otwinowski Z, Minor W, Carter JCW, M SR. Methods in Enzymology. Academic Press; New York: 1997. Processing of X-ray Diffraction Data Collected in Oscillation Mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 53.CCP4, C.C.P.N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 54.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. Journal of Applied Crystallography. 2006;39:109–111. [Google Scholar]
- 58.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 59.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 1):122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 60.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.