

Abstract
Clinical refractoriness to nucleoside analogs (e.g., gemcitabine, capecitabine) is a major scientific problem and is one of the main reasons underlying the extremely poor prognostic state of pancreatic cancer. The drugs’ effects are suboptimal partly due to cellular mechanisms limiting their transport, activation, and overall efficacy. Nonetheless, novel therapeutic approaches are presently under study to circumvent nucleoside analog resistance in pancreatic cancer. With these new approaches come additional challenges to be addressed. This review describes the determinants of chemoresistance in the gemcitabine cytotoxicity pathways, provides an overview of investigational approaches for overcoming chemoresistance, and discusses new challenges presented. Understanding the future directions of the field may assist in the successful development of novel treatment strategies for enhancing chemotherapeutic efficacy in pancreatic cancer.
Keywords: Pancreatic cancer, Gemcitabine, Chemoresistance, Therapeutic, Nucleoside analog
1. Introduction
Pancreatic cancer continues to be a devastating malignancy with a 5-year patient survival rate remaining less than 6% [1]. Since the approval of the chemotherapeutic agent gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) in 1996 [2,3], the advancement in the treatment of pancreatic cancer has progressed slowly. Therefore, gemcitabine monotherapy currently remains the standard of care, although it only extends survival by a mere 5 weeks [4]. In addition, the innate and acquired chemoresistance of patients to the drug has emerged as a major concern as observed by a very low patient response rate [5]. The chemoresistance of gemcitabine in pancreatic cancer can be attributed to several anatomical (e.g., dense desmoplastic stroma), pathophysiological (e.g., amplification of growth, cell survival, and anti-apoptotic pathways), and pharmacological (e.g., requirement of phosphorylation for drug activation) barriers. Innumerable clinical trials have ensued over the past decade investigating the efficacy of gemcitabine in chemotherapeutic combinations [6]. Unfortunately, this strategy has proven largely unsuccessful, leading to further evaluation of the molecular aspects of the disease biology as well as approaches for overcoming chemoresistance. As the field shifts towards targeted therapy, several new approaches have recently developed. This review highlights the cellular and molecular mechanisms that contribute directly to gemcitabine chemoresistance as well as the contemporary approaches under investigation for overcoming chemoresistance and the therapeutic challenges they pose. Understanding the future directions of the pancreatic cancer treatment may lead to the successful development of new therapeutic strategies for enhancing chemotherapeutic efficacy.
2. Determinants of chemoresistance in the gemcitabine pathway
The dismal circumstance of pancreatic cancer is largely due to late diagnoses (i.e., locally-advanced or metastatic disease) at which chemoresistance in patients is a critical issue. The driving force for metastasis has been a long-standing question in the field, with precise answers not yet available. However, with the advent of tumor genomic sequencing projects, genetic mutations in both the primary carcinoma and subsequent metastatic tumors have been compared and contrasted. These studies have concluded that although genomic instability leading to genetic mutations contributes to metastasis, non-genetic factors are involved as well [7,8]. Particularly, there is evolutionary selection that drives clones with particular genetic mutations or epigenetic changes to form metastatic tumors.
Among the factors for the evolutionary drive includes cellular signaling that favor expansion of cellular populations with specific mutations. Several cell signaling pathways vital for both intrinsic and acquired chemoresistance in pancreatic cancer have been identified [4,9]. Although not necessarily directly related to the gemcitabine cytotoxicity mechanisms, these pathways involved in both the tumor and stromal microenvironment play critical roles in determining drug resistance. Specifically, signaling pathways involved in growth, proliferation, differentiation, apoptosis, invasion, and angiogenesis, such as the notable Akt, EGFR, SHH, Notch, MAPK, and NFκB pathways, appear to either directly or indirectly influence chemosensitivity. Furthermore, with respect to differentiation, epithelial-to-mesenchymal transition (EMT) and cancer stem cells are two emerging aspects of interest that drive chemoresistance and clonal selection in pancreatic cancer.
Recently, Yachida et al. showed that the progression of pancreatic cancer takes place over approximately 17 years: over a decade is needed to progress from the initiating mutation to a primary carcinoma, about five years is needed for the tumor to obtain metastatic ability, and patients die approximately two years after [6]. Although this confirms that current intervention is indeed at the terminal phase of pancreatic cancer, it is apparent that a large window of opportunity exists for early detection and diagnosis of the disease. However, since early diagnostic methods are not yet available, it is also important to focus on treatment aspects at the end stage of pancreatic cancer, that is, when diagnosis occurs. Undoubtedly, there is a large spectrum of genetic and molecular alterations that contributes to chemoresistance in pancreatic cancer, many of which have been extensively reviewed elsewhere [4,9-11]. Therefore, this review will focus primarily on chemoresistance due to alterations in the gemcitabine pathway.
2.1. Transporters and metabolic enzymes
Due to its hydrophilicity (logP of – 1.33) [12], gemcitabine relies on numerous transporters to pass through the cellular lipid bilayer and exert its cytotoxicity. Earlier studies by Mackey et al. identified key transporters involved in the uptake of gemcitabine and demonstrated the need for their activity in order to confer gemcitabine sensitivity [13,14]. While they found that the human equilibrative transporters 1 and 2 (hENT1 and hENT2) were able to mediate gemcitabine transport (Km of 160 and 740, respectively), the human concentrative nucleoside transporter 1 (hCNT1) had the greatest intrinsic transport activity (Vmax:Km of 0.24) [14]. Since then, both hENT1 and hCNT1 have been highly implicated in gemcitabine chemoresistance as well as patient outcome. Although the clinical correlation between hENT1 expression, both transcriptionally and immunohistochemically, and disease-free as well as overall survival of pancreatic cancer patients has been well-established [15-19], the correlation between hENT1 expression and innate and acquired chemoresistance in cultured pancreatic cancer cells remains somewhat incongruous and context-dependent [13,20-23]. This may be due to the bidirectional nature of the transporter resulting in drug efflux at higher concentrations, the relative role of the transporter among the presence of other nucleoside transporters, and the expression characteristics of the transporter in normal versus tumor cells [21] including variations in cell cycle characteristics [24]. In pancreatic cells, mislocalization of hENT2, rather than changes in expression levels, proposes an alternative mechanism for gemcitabine chemoresistance [25]. Similarly, the loss of hCNT1 cell surface expression and activity frequently observed in pancreatic cancer cells was found to correlate directly with high gemcitabine chemoresistance [21,24]. Structure-activity characterization of hCNT3 revealed electrostatic interaction by the 3′-hydroxyl position to play a major role in the transport of substrates, including gemcitabine [26]. While the exact role of hCNT3 in gemcitabine chemoresistance has yet to be evaluated, patients with elevated immunocytochemical expression of the transporter were observed to experience a lower risk of disease recurrence and longer overall survival [18].
In addition to influx transporters, a few efflux transporters have also been implicated in gemcitabine resistance of pancreatic cancer including the ATP-binding cassette (ABC) family of multidrug resistance- associated proteins (MRPs). Increased expression of MRP2 mRNA and protein in pancreatic cancer tissues has been associated with both intrinsic and acquired resistance to a regimen of gemcitabine plus cisplatin [27]. Likewise, MRP7, expressed in both normal pancreas [28] and most established pancreatic carcinoma cell lines [29], has been demonstrated to be able to transport gemcitabine, although direct correlations with cytotoxicity have not yet been demonstrated [30]. This may be in part due to the ability of MRPs to efflux the active metabolite of gemcitabine, allowing for reduction of drug concentrations inside the cell [31]. Although the overall significance of transporters in determining gemcitabine chemosensitivity has been well demonstrated, the precise mechanisms and alterations that occur in pancreatic tumors resulting in chemoresistance remain unclear.
Once inside the cell, gemcitabine is phosphorylated by deoxycytidine kinase (dCK) before further phosphorylation into its active diphosphate (dFdC-DP) and triphosphate (dFdC-TP) forms. Activated gemcitabine, in the form of dFdC-TP, induces masked chain termination during which the nucleotide is incorporated into DNA, followed by another deoxynucleotide, prohibiting further DNA polymerase action [32]. Since involved in the rate-limiting step for gemcitabine cellular activation, a deficiency in dCK expression and activity has been shown to be associated with gemcitabine resistance in pancreatic cancer. Loss of dCK mRNA in highly-resistant pancreatic cancer cells has been observed [20,33,34], and a clear correlation between dCK activity and gemcitabine sensitivity in murine and human pancreatic tumor xenografts has been demonstrated [35]. Overexpression of dCK was found to significantly enhance gemcitabine sensitivity in two of three pancreatic cancer cell lines [36]. Furthermore, simultaneous expression of dCK and p8 in a gemcitabine-resistant pancreatic cancer cell line, PANC-1, significantly decreased the cytotoxic IC50 of gemcitabine and enhanced apoptosis and caspase-3 activity; tumor growth inhibition was also noticeably improved in nude mice [37]. Clinically, low immunohistochemical expression of dCK was correlated with both decreased overall survival as well as older age of patients, suggesting a role of age-related methylation in patients [38]. Low expression of the RNA-binding protein HuR has also been shown to correlate with a 7-fold increase in the risk of mortality for pancreatic cancer patients due to its ability to regulate dCK protein levels and confer gemcitabine chemoresistance [39,40].
Gemcitabine is rendered inactive by cytidine deaminase (CDA), which removes the NH2 group from the pyrimidine [41], allowing the uracil metabolite to be exported from the cell. Interestingly, gemcitabine-induced inhibition of cytidine deaminase activity leads to a decrease in dFdC-TP catabolism, hence propelling the self-potentiation of gemcitabine activity [42]. Although the focus of most studies involving CDA and gemcitabine in pancreatic cancer has been on generalized adverse effects (i.e., high-grade neutropenia) due to genetic polymorphisms rather than cancer cell cytotoxicity [43-47], a few studies have identified a dramatic increase in chemosensitivity, up to 54-fold, of cell lines with the inhibition of CDA [36,48]. Although further studies are necessary, alterations in CDA expression levels in tumors could be a promising mechanism for improving gemcitabine sensitivity.
2.2. Other molecular targets
Additional candidates of interest for the manipulation of gemcitabine cytotoxicity include effectors of DNA synthesis and repair. In particular, ribonucleotide reductase subunits 1 and 2 (RRM1 and RRM2) are inhibited by dFdC-DP and dFdC-TP and hindered from repairing flawed DNA. In addition to impeding DNA repair, inhibition of RR, the rate-limiting enzyme in deoxyribonucleoside triphosphate (dNTP) synthesis, reduces the endogenous dNTP pool, lessening competition and indirectly facilitating dFdC-TP incorporation into DNA [49]. The transcriptional upregulation of the larger subunit, RRM1, has been consistently observed as pancreatic cancer cell lines acquire gemcitabine resistance [20,50]. Consistently, low expression of RRM1 in tumors is correlated with enhanced response to gemcitabine specifically in recurrent cases [50,51]. The subunit appears to have no correlation with disease-free or progression-free survival, and its correlation with overall survival is variable [51,52]. Therefore, the expression of RRM1 seems to be more relevant to acquired rather than innate gemcitabine resistance in both pancreatic cancer cell lines and patients.
For RRM2, some studies have shown increased transcript and protein expressions in pancreatic cancer cell lines [20,53], while others have additionally identified the significance of the alteration in determining gemcitabine sensitivity in pancreatic cancer. Specifically, RNA interference of RRM2 attenuated chemoresistance and invasiveness in cells [33,54], while it suppressed tumor growth, enhanced apoptosis, and inhibited metastasis in xenograft models [55]. Moreover, Ohhashi et al. found a reduction in cellular proliferation with the inhibition of RRM1 and RRM2 even in the absence of gemcitabine treatment [33]. Clinically, low RRM2 mRNA expression levels correlated with significantly enhanced disease-free, median, and overall survival as well as overall response rate in gemcitabine-treated patients [56,57]. Reduction of the dNTP pool by gemcitabine inhibition of RRM2 is clear in its effects on nuclear DNA (i.e., facilitating the incorporation of dFdC-TP into replicating DNA). However, this mechanism may also hold true for mitochondrial DNA (mtDNA) although evidence has not yet been provided. Nevertheless, gemcitabine has been shown to directly affect mtDNA by inhibiting its γ-polymerase [58].
3. Approaches to enhancing gemcitabine delivery
3.1. Prodrugs by chemical modification
An apparent approach for enhancing gemcitabine delivery to cells is to bypass its dependence on transporters for entering the cell. This can be achieved by chemically modifying the drug and creating various prodrugs, such as acyl derivatives, with increased lipophilicity. For example, lipophilic prodrugs of troxacitabine, a nucleoside analog drug with an unnatural L-configuration, were created with the addition of linear aliphatic chains to the amino group; sensitivity of pancreatic cancer cells to the modified drugs was greater than 100-fold compared with troxacitabine [59]. Likewise, lipophilic prodrugs of gemcitabine were synthesized by linking the 4-amino group with acyl derivatives (i.e., valeroyl, heptanoyl, lauroyl, and stearoyl) [60,61]. In addition, by masking the N-terminus, this modification also protects gemcitabine from rapid deamination and improves its half-life. In particular, 4-(N)-stearoyl-gemcitabine was found to decrease the cytotoxic IC50 of a cervix, breast, colorectal, and nasopharyngeal cell line by 1.5-5-fold [62,63]. Furthermore, a novel gemcitabine-cardiolipin conjugate was shown to induce cytotoxicity in several gemcitabine-resistant cell lines independent of nucleoside transporter activity. In vivo, the conjugate demonstrated less adverse effects compared with gemcitabine as measured by body weight and white blood cell count, while inhibiting tumor growth by 20% greater than gemcitabine. A high dose of the treatment increased the median survival of tumor-bearing mice by 73%, while the same dose and schedule of gemcitabine led to toxic death of all mice [64]. Another lipophilic prodrug of gemcitabine, CP-4126, was created by esterifying an elaidic fatty acid at the 5’ position. The compound was found to be as effective as gemcitabine in chemoresistant cell lines, but both were ineffective in cells devoid of dCK activity. However, CP-4126 maintained its efficacy in nucleoside transporter-inhibited cells, while the IC50 for gemcitabine increased 200-fold. In various xenograft models, both gemcitabine and CP-4126 were equally effective, but the prodrug was able to be administered orally [65]. The potential of this approach has extended to a phase I study with a novel oral gemcitabine prodrug, LY2334737, evaluated in patients with advanced or metastatic solid tumors. This prodrug protects the amine group of gemcitabine with a covalent bond to valproic acid, preventing extensive pre-systemic deamination. As the first human clinical trial for LY2334737, the study demonstrated safe administration of the drug, despite the caveat of blood-brain barrier effects, with minor adverse effects and observations of antitumor activity [66].
An alternative approach for enhancing efficacy includes the creation of a phosphoramidate prodrug. By creating a variant of the monophosphate form (i.e., dFdC-MP), the rate-limiting phosphorylation step is bypassed. Wu et al. demonstrated that a phosphoramidate prodrug of gemcitabine was approximately 4-fold more effective than gemcitabine in dCK-deficient variants of cancerous cell lines. Furthermore, inhibition of transporter activity did not diminish the prodrug’s activity in the dCK variants, although the same did not hold true for the parental cell lines [67]. Similarly, another phosphoramidate prodrug, GemMP [10], was found to reduce thyroid cancer cell proliferation by arresting cells in S phase at concentrations 5-10-fold lower than gemcitabine [68]. In addition to increasing lipophilicity, reducing deamination, and bypassing the rate-limiting phosphorylation step, chemical modifications of gemcitabine can also be conducted in order to enhance its interaction with delivery vesicles such as liposomes [60,61], which will be further discussed below.
3.2. Nanoparticle drug delivery
Nanoparticle drug delivery has been shown to be a promising approach for overcoming several anatomical, pathophysiological, and pharmacological barriers [69] and attenuating chemoresistance in pancreatic cancer. By encapsulating or adsorbing gemcitabine in nanoparticles, reduced pre-systemic metabolism, lower dosages, and sustained release are possible. For example, encapsulation of gemcitabine into chitosan and albumin nanoparticles produced sustained release profiles as well as improved antitumor activity in vitro compared with the administration of free drug [70]. Additional therapeutic advantages of nanoparticle drug delivery include altered pharmacokinetic parameters (e.g., decreased drug clearance), increased drug concentration in tumor tissues via the enhanced permeability and retention (EPR) effect (i.e., nanoparticles tend to greater accumulate in tumor rather than normal tissues) [71], increased local plasma T3 levels, and the potential for targeted delivery.
Furthermore, combination therapies can also be utilized with nanotechnology. One study reported that a combination of gemcitabine and curcumin in nanoparticles enhanced the inhibition of tumor growth, abolished systemic metastases, and reduced the activation of nuclear factor κB in a pancreatic cancer xenograft model as compared with either agent alone [72]. Similarly, gemcitabine and paclitaxel preconjugated with a hydrolysable linker and subsequently loaded into a drug carrier were found to significantly improve the chemotherapeutic activity as compared with their free form [73].
As previously noted, decreasing the hydrophilicity of gemcitabine aids in the incorporation of the drug into liposomal formulations. In particular, stearoyl gemcitabine nanoparticles better managed tumor growth as compared with free gemcitabine, and results were further enhanced by PEGylation which increases drug half-life [74]. Such nanoparticles were also shown to overcome gemcitabine resistance related to the overexpression of RRM1 in both cell culture and mice. Furthermore, the enhanced cytotoxicity of these nanoparticles was additionally observed in dCK-deficient tumor cells with the induction of apoptosis through caspase activation [75]. Similarly, conjugation of gemcitabine with squalene, a precursor to cholesterol synthesis, resulted in amphiphilic molecules that were shown to have enhanced anticancer activity due to protection of the drug from rapid deamination [76]. Such nanoparticles were demonstrated to possess greater ability to induce S phase arrest and apoptosis of cancer cells as compared with free gemcitabine [77]. Furthermore, it has been reported that albumin enhanced the passive diffusion of squalenoyl gemcitabine nanoparticles rather than relying on membrane transporters [78]. This was confirmed by higher activity of the nanoparticles than free gemcitabine in transporter-deficient, gemcitabine-resistant tumor cells [79]. Furthermore, the penetration of the gemcitabine-squalene molecules was increased due to the presence of cholesterol in the monolayer, which may contribute to the increased anticancer activity since cancer cell membranes are rich in cholesterol [80]. Squalenoyl-based gemcitabine nanoparticles were found to overcome gemcitabine resistance with increased cytotoxicity in gemcitabine- resistant cell lines deficient in both dCK and hENT1 [77].
Nanoparticles can also be modified for targeted delivery of gemcitabine to pancreatic cancer cells. The addition of monoclonal antibodies has been utilized as a targeting moiety for pancreatic tumors. A recent study demonstrated that Herceptin (HER2)-conjugated chitosan nanoparticles loaded with gemcitabine led to an increase in antiproliferative activity in vitro along with enhanced S-phase arrest as compared with free gemcitabine. In addition, the targeted nanoparticles were efficiently taken up by the cells and prolonged intracellular retention was obtained. Sustained in vitro release of the drug from the nanoparticulate system indicated proper diffusion of the drug from the polymeric matrix [80]. Likewise, gold nanoparticles with anti-epidermal growth receptor antibodies demonstrated that a lower dose of gemcitabine was able to inhibit the proliferation of pancreatic cancer cells as well as orthotopic tumor growth [81].
An additional method for targeting includes the use of magnets which are directed to the desired site upon application of an external magnetic field. The drug can then be released with the help of various triggering factors like ultrasound or changes in physiological conditions (e.g., pH, temperature) or by simple diffusion [82,83]. Such systems prepared by the inclusion of magnetic nanocrystals into squalenoyl gemcitabine bioconjugates have been reported. Upon injection into a mouse tumor model, magneticallyguided nanoparticles showed enhanced anticancer activity [84].
Nanogel formulations with tumor-specific molecules were also efficient in overcoming resistance by enhancing tumor growth inhibition with stable release of the drug over several days in nucleoside transporter-deficient and dCK-deficient lymphogenic cancer cells [85]. Such nanogel formulations containing the active form of the nucleoside analog (i.e., dFdC-TP) demonstrated greater cytotoxicity and reduced resistance as compared with nucleoside analog prodrugs. The drug delivery systems aid in the protection of the active drugs and enhance intracellular retention [86,87].
3.3. Dosage and schedule modifications
The mechanism of action of gemcitabine is atypical since the drug depends on the cell cycle and only a few key proteins for efficacy. The importance of these key players for gemcitabine is unique and evident by the significant influence on active metabolite concentration and drug efficacy with the modification of only a single target. Therefore, the efficacy of gemcitabine is dependent on a balance of both dosage and schedule.
Gemcitabine pharmacokinetics can be described by a linear, 2-compartment model with a half-life of 42–94 min (short infusion of <70 min) depending on age and gender. While the rate of clearance varies with age and gender, 92–98% of a typical gemcitabine dose (i.e., 1000 mg/m2/30 min infusion) is excreted, predominantly in the urine. The volume of distribution of gemcitabine is highly affected by infusion duration and gender, while effects of plasma protein binding are negligible [2].
Currently, the standard regimen consists of 1000 mg/m2 gemcitabine given weekly as a 30 min infusion for 3 weeks followed by one week of rest. While that dosage is believed to saturate dCK activity, pharmacokinetic studies have determined that a 1000 mg/m2/h infusion rate was optimum for intracellular phosphorylation of gemcitabine [88]. Nonetheless, several studies have shown 1500 mg/m2 as the maximum tolerated dose (MTD), with high-grade neutropenia, granulocytopenia, and thrombocytopenia being dose-limiting factors [89,90]. Trials comparing a fixed dose rate (FDR) of gemcitabine (1500 mg/m2 at a rate of 10 mg/m2/min weekly for 3 weeks out of a 4-week cycle) with standard gemcitabine treatment indicated increased efficacy of the FDR but with much greater hematological toxicities (i.e., neutropenia, anemia, and thrombocytopenia) in patients [91-93]. Furthermore, although one study established the MTD of gemcitabine at 6500 mg/m2 with hematopoietic progenitor support, its efficacy was not superior than that reported with lower-dosage FDR schedules [94]. A high dose of gemcitabine was also investigated with 2200 mg/m2 administered as an infusion for 30 min on days 1 and 15 for 6 months. The regimen was deemed safe, tolerable, and effective for palliative treatment of advanced pancreatic cancer patients [95]. Collectively, these data indicate that altering the exposure level of gemcitabine in patients is vital for obtaining specific desired effects.
When comparing schedules, 1000 mg/m2 gemcitabine as a 30 min infusion given weekly for either 3 consecutive weeks every 4 weeks or 2 consecutive weeks every 3 weeks indicated comparable efficacy although the 3-week schedule produced lower toxicity in patients [96]. Another schedule of 1000 mg/m2 gemcitabine given for 7 consecutive weeks followed by a week of rest and then weekly for 3 weeks out of 4 identified increased toxicities compared with the conventional regimen [97]. Metronomic dosing, or continuous and timed administration of low dosage, of gemcitabine (1 mg/kg/d for a month) was found to exert equal cytotoxicity in orthotopic models of human pancreatic carcinoma in nude mice compared with the conventional schedule of 100 mg/kg/d 0, 3, 6, and 9 post-implantation. However, an anti-angiogenic effect was additionally observed with the metronomic regimen [98].
Infusion duration has also been identified as a key factor that influences gemcitabine efficacy. One study demonstrated that the chemotherapeutic retained its antitumor activity at doses as low as 300 mg/m2 when infusion time was prolonged [99]. Other trials increased the infusion rate of 100 mg/m2 to 24 h weekly for 3 weeks in a 28-day cycle and found an improvement in the quality of life of patients but also only marginal antitumor activity [100]. Furthermore, an investigational method of hypoxic abdominal stop-flow perfusion demonstrated increased efficacy of gemcitabine in patients compared with the standard treatment at doses up to 1125 mg/m2. However, this complicated modality remains in its infancy, and further studies are needed for its complete assessment [101].
4. Approaches to directly modifying determinants of gemcitabine cytotoxicity
4.1. Gene therapy
Even though gene therapy has not yet been well-developed for the application of cancer (as compared with other disorders such as cystic fibrosis), the approach has become appealing for altering specific targets with its potential for producing a bystander effect within heterogeneous tumors. A fusion gene of dCK and uridine monophosphate kinase (UMK) (dCK∷UMK) was found to sensitize pancreatic cancer cells to gemcitabine by markedly decreasing viability. Tumor volume was also reduced as an antitumoral bystander effect was observed due to apoptosis of untransduced cells. Additionally, the use of a synthetic carrier (i.e., polyethyleneimine (PEI)) further induced tumor regression [102]. A combination of dCK∷UMK with siRNA against RRM2 and thymidylate synthase (TS), whose inhibition activates hENT1, and gemcitabine promoted chemosensitivity even more with a 40-fold decrease in cytotoxic IC50 in PANC-1 cells. Tumor volume was reduced dramatically and mouse survival prolonged significantly due to an increase in apoptosis and decrease in cellular proliferation [103]. Recently, it has also been found that the overexpression of dCK and knockdown of p8 with recombinant adenoviral vectors also significantly decreased gemcitabine resistance in PANC-1 cells and inhibited tumor growth with enhanced apoptosis and caspase-3 activity [37].
The herpes simplex virus 1 thymidine kinase (HSV-TK)/ganciclovir (GCV) strategy has thus far been successful up to the clinical level, and the same technique may be adapted to target increased RR in cancer cells. RR has been a target of interest with gene therapy since its overexpression is an attribute of mitotic cancerous cells. Therefore, vectors lacking the RR gene are attractive options due to their ability to complement with overexpressed mammalian RR and selectively target rapidly dividing cells. For example, a herpes simplex virus (HSV) with an RRM1 deletion mutant (i.e., ICP6Δ) was found to enhance the expression of adeno-associated viruses along with their kinase genes both in pancreatic cancer cells and xenografts, demonstrating potential for the combination therapy [104]. Similarly, an HSV vector lacking the RR gene (i.e., hrR3) was found to extend survival in 70% of mice receiving both the vector and ganciclovir, 40% of mice receiving the vector alone, and 0% of untreated mice [105].
Overexpression of hENT1 has also been conducted with a recombinant adenovirus (i.e., Ad-hENT1) in human pancreatic cancer cells. Although expression of the transporter is known to vary with cell cycle and cell type, treatment with the vector improved response to gemcitabine [106]. Other targets of gene therapy in pancreatic cancer include p53 and p16 [107] and Bax [108], although they are not directly related to gemcitabine metabolism.
4.2. Epigenetic approaches
The field of epigenetics has been rapidly emerging since the identification of its influence on the initiation, progression, and resistance of various cancers. The ability to reverse aberrant epigenetic alterations renders this targeted approach highly attractive. Among the various mechanisms, DNA methylation has been the most widely studied. Abnormal promoter hypermethylation can silence tumor suppressor genes and have been implicated in the clinicopathological features and promotion of tumorigenic properties of pancreatic cancer. One study identified that low expression of dCK in pancreatic cancer patients correlated with overall survival as well as age, suggesting a role of age-related methylation of the dCK gene [38]. It was also found that the CDA gene was methylated in an entire cohort of colorectal cancer patient samples, although no correlation was observed between methylation status and clinicopathological parameters [109]. By utilizing microarrays, countless other aberrantly methylated genes have been identified in pancreatic cancer, particularly those related to growth, differentiation, angiogenesis, and apoptotic signaling [110-113]. However, further information about the methylation status of the targets of the gemcitabine pathway is limited.
FDA-approved inhibitors of DNAmethylation are currently available with several more under investigation. For example, the nucleoside analogs 5-azacytidine (5-aza-C) and 5-aza-2′-deoxycytidine (5-aza-dC) have been approved for the treatment of high risk myelodysplastic syndromes [114] and are currently under study for acute and chronic myeloid leukemias and ovarian cancer [115]. Both compounds have been found to be substrates for hCNT1 [116] and rely on dCK for their phosphorylation [117]. Combination of 5-aza-dC with gemcitabine was shown to inhibit pancreatic cancer cell growth to a greater extent than with gemcitabine alone [118]. The DNA methylation inhibitor cladribine, also a nucleoside analog, has been studied in conjunction with rituximab for the treatment of mantle cell lymphoma; high levels of activity with minimal toxicity were found [119].
In addition to DNA, epigenetic modifications, namely methylation and acetylation, can also occur at the histone level. Unlike DNA methylation, histone methylation is a mechanism that has yet to be thoroughly studied in the context of cancer. Histone acetylation, on the other hand, is a mechanism well-studied in the development and progression of cancer since aberrant activity promotes the expression of various oncogenes. This process is reversibly catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Currently, FDA-approved HDAC inhibitors include vorinostat and romidepsin for the treatment of T cell lymphoma, although many others remain under study in clinical trials [115]. Romidepsin has also been demonstrated to have antiproliferative activity infive human pancreatic cancer cell lines (IC50: 1–500 nM) via cell cycle arrest at both G1 and G2/M phases followed by apoptosis [120]. Additionally, the HDAC inhibitor MS-27-275 was shown to decrease cells in S phase cells while increasing G1-phase cells. Antitumor efficacy was observed in both Capan-1 cells andmouse xenograft [121]. Trichostatin A [122], another HDAC inhibitor, has been widely studied and shown to induce antitumor effects in pancreatic cancer cell lines at submicromolar concentrations [123,124]. Furthermore, a novel compound derived from both MS-27-275 and TSA was shown to cause cell cycle arrest and subsequent apoptosis in pancreatic cancer cell lines [125]. Several studies have shown the increased efficacy of combining an HDAC inhibitor with gemcitabine in pancreatic cancer cell lines including the synergistic apoptotic effects of TSA with gemcitabine in pancreatic ductal carcinoma cells [126-130]. With 50% reduction of pancreatic cancer xenografts and no observed toxicity, Donadelli et al. further identified that TSA did not affect the gene expressions of targets in the gemcitabine pathway [127]. Nonetheless, a phase II trial of CI994 (n-acetyl dinaline), an HDAC inhibitor, with gemcitabine demonstrated no additional advantage in survival rate compared with gemcitabine alone [131], warranting further studies for the better understanding of this therapeutic approach.
The rapidly expanding field of microRNAs (miRNAs), short (~22 nt) RNA sequences that post-transcriptionally regulate gene expression by binding to complementary mRNA, is also appealing for its potential to influence cancer epigenetic therapy. Their ability to regulate expression of canonical oncogenes and tumor suppressor genes as well as the recent demonstration of their direct involvement in oncogenic processes emphasizes their potential to intervene in dysfunctional cancer pathways. There is also increasing evidence demonstrating that miRNAs are critical regulators of drug resistance in pancreatic cancer. For example, re-expression of the miR-200 family or downregulation of miR-21 in gemcitabine-resistant cells led to sensitization of the cells to gemcitabine [4]. Since miRNA expression patterns are unique to each tumor type, aberrant expression profiles (e.g., the downregulation of the let-7 family) have been shown to contribute to cancer pathogenesis and tumor development and progression [4,132]. In addition to being epigenetic targets, miRNA activity can also be modulated by inhibition with antisense oligonucleotides or overexpression with synthetic miRNAs, as reviewed recently [4,132]. The development of curcumin as a potential therapeutic for pancreatic cancer patients has been under investigation due to its interactions with DNA methyltransferase, HDACs, and HATs and ability to alter miRNA expression, including upregulation of miR-200 and downregulation of miR-21 [133]. In this regard, several recent reviews identify key growth, proliferation, and differentiation (e.g., PI3K/Akt, MAPK, K-ras, STAT-3, Notch-1, Notch-2), invasion (e.g., ABCG2, cadherin, ZEB1, vimentin), and apoptosis (e.g., CDK6, p53, caspase 3) proteins altered by miRNAs [4,132,134]. Therefore, targeting miRNAs offers great promise for tackling pancreatic cancer chemoresistance and providing diagnostic and therapeutic values.
Studies from our laboratory show miRNAs influencing the expression of direct targets of the gemcitabine pathway. We have identified that miR-214, miR-339-3p, and miR-650 overexpression significantly reduced CNT1, but not ENT1, protein levels. Furthermore, several miRNAs were found to markedly reduce both CNT1 transport activity as well as gemcitabine cytotoxicity in a pancreatic cancer cell line [24]. Likewise, our recent study showed that the differential processing of the let-7 family of miRNAs altered gemcitabine chemosensitivity in pancreatic cancer cells through its activity on RR (unpublished data). Both studies identify the potential for modulating miRNA expression to enhance the efficacy of gemcitabine, although the ability of pancreatic cancer cells to process miRNA precursors should be carefully considered while choosing precursor miRNA-based therapies. In addition to overcoming chemoresistance, epigenetic and miRNA alterations also have the potential to act as prognostic and predictive biomarkers for the guidance of therapy.
4.3. Molecular therapeutic agents
Several small and large molecule inhibitors demonstrate potential as therapeutic agents. For example, proteasomal inhibitors, such as bortezomib, have been shown to increase cytotoxicity in combination with gemcitabine [24,135]. Similarly, tetrahydrouridine (ThU), a competitive inhibitor of CDA, has been shown to sensitize pancreatic cancer cells to gemcitabine up to 54-fold [36]. Although pharmacokinetic studies have been performed on the compound, its therapeutic value as well as safety and toxicity profiles remain under study in humans [136]. RNA interference has also been commonly used for the study of specific targets in pancreatic cancer chemosensitivity. While Ohhashi et al. noticed no change in gemcitabine sensitivity or cellular proliferation with hENT1-targeted siRNA [33], others found that knockout of TS with siRNA decreased chemoresistance in pancreatic cancer cell lines [137]. Furthermore, shRNA against hENT1 combined with 5-fluorouracil (5-FU) treatment attenuated cell viability and gemcitabine cytotoxic IC50 values. Given the dual treatment, cells were arrested in G0/G1 phase, while mouse xenografts had diminished tumor volumes [26]. Furthermore, modulation of transporter activity and, consequently, gemcitabine uptake was recently conducted with siRNA against hCNT1 [24]. Similarly, silencing of dCK with siRNA in gemcitabine-resistant cells further reduced gemcitabine sensitivity without affecting cellular proliferation [33]. Treatment of gemcitabine-resistant PANC-1 cells with hydroxyurea led to a 4-fold increase in chemosensitivity [138]. RRM1- and RRM2-targeted siRNA in pancreatic carcinoma cell lines increased gemcitabine sensitivity, apoptosis, and caspase-3 activity while lessened cell proliferation and invasiveness [33,54,55,139]. In an orthotopic xenograft model, synergism was observed between RRM2 silencing and gemcitabine, leading to reduced tumor growth, enhanced tumor apoptosis, and inhibition of metastasis [55].
5. Novel therapeutic combinations with gemcitabine
In addition to radiosensitization, potentiating chemosensitization of gemcitabine by other pharmaceutical agents has been a longstanding area of interest. Although combination therapies with gemcitabine have been studied extensively both in vitro and in clinical trials, only a few have consistently resulted with significant improvements in patient survival. Since several mechanisms of drug resistance exist, combination therapy can simultaneously undertake multiple mechanisms and pathways, allowing the drugs to act in concert. This rationale is well-illustrated by the current approach towards breast cancer treatment. Nonetheless, combinations of gemcitabine with other chemotherapeutics such as other nucleoside analogs (i.e., 5-FU, capecitabine), platins (i.e., cisplatin, oxaliplatin), and taxoids (i.e., docetaxel [122,140], paclitaxel [141]) have shown variable results with no significant enhancement of patient survival [6].
With the interests geared towards targeted therapy, numerous novel therapeutics have been studied with gemcitabine: antibodies (i.e., bevacizumab, cetuximab), growth factor inhibitors (i.e., erlotinib, aflibercept, axitinib [142], saracatinib [143]), topoisomerase I inhibitors (i.e., irinotecan, exatecan, rubitecan), MMP inhibitors (i.e., marimastat, BAY 12-9566), tyrosine kinase inhibitors (i.e., masitinib, sorafenib, sunitinib [144], ARQ 197 [145]), and other inhibitors (i.e., pemetrexed, tipifarnib, celecoxib, imitinib [146], PX-12 (a novel inhibitor of thioredoxin) [141] and enzastaurin [147]) [6]. Among the sizeable list, several agents show promise. However, erlotinib has been the only agent thus far to have consistently demonstrated equal or significantly enhanced median overall survival compared with gemcitabine (1-year survival rates of 23% and 17%, respectively) [6]. Therefore, the EGFR inhibitor is currently FDA-approved for use in combination with gemcitabine for the treatment of pancreatic cancer. Other agents of interest include curcumin (a natural product) [6], leucovorin (an adjuvant agent) [6], tegafur (a prodrug of 5-FU) [148,149], and MGCD0103 and valproic acid (HDAC inhibitors) [150]. In particular, notable combinations under study include GTX (gemcitabine, docetaxel, and capecitabine) [151,152], GemOx (gemcitabine, oxaliplatin) [153], GemOxCet (gemcitabine, oxaliplatin, cetuximab), and Gemoxel (gemcitabine, oxaliplatin, capecitabine) [154]. Nonetheless, novel combinations not including gemcitabine such as Xeliri (capecitabine, irinotecan) [155], Folfiri (leucovorin, 5-FU, irinotecan) [155], and Folfirinox (leucovorin, 5-FU, irinotecan, oxaliplatin) [156] have also shown promise in combating pancreatic cancer, although more studies are needed to determine toxicity and efficacy profiles.
An alternative approach studied is the use of agents for modulating the tumor microenvironment. Several clinical trials have been conducted with prophylactic anticoagulants (i.e., nadroparin, enoxaparin, dalteparin) with gemcitabine, although minimal significant results for the advancement of pancreatic cancer therapy were obtained [6]. Similarly, the use of pomalidomide, an investigational immunomodulating drug that both inhibits angiogenesis as well as exerts antitumoral effects, was found to be feasible and safe in most patients [157]. When co-administered with gemcitabine, a drug depleting tumor-associated stromal tissue via inhibition of the Hedgehog cellular signaling pathway, IPI-926, was found to facilitate gemcitabine intratumoral delivery and extend survival of a spontaneous mouse pancreatic tumor model [158]. Futhermore, a novel ceramide analog, AL6, has been shown to increase the dCK/CDA gene expression ratio, leading to increased apoptosis in two pancreatic ductal adenocarcinoma cell lines [159]. A study of gemcitabine in combination with imexon, a pro-oxidant small molecule, was found to significantly increase chemotherapeutic efficacy in PANC-1 xenograft tumors. This is thought to be due to the inhibition of RR by imexon, with inhibition effects even greater with the addition of gemcitabine [160]. Overall, combinations of a targeting agent with gemcitabine have shown to be well-tolerated with promise for enhanced efficacy. Further studies as well as meta-analysis of data from clinical trials are warranted for consistent efficacy results.
6. Therapeutic challenges in overcoming chemoresistance
Despite the abundance of common and novel chemotherapies, gemcitabine remains the superior drug of choice for the treatment of pancreatic cancer due to its ability to extend survival, although only by a mere few weeks, and improve the quality of life with better tolerance. As one of the many enduring nucleoside analog drugs, gemcitabine has been predicted to remain as the drug of choice for pancreatic cancer for the near future even though its effects are truly suboptimal. In spite of considerable efforts to improve the drug of choice, several other approaches (e.g., targeting stromal pathways in tumors) for overcoming chemoresistance in pancreatic cancer have been underway, bringing with them the new challenges they pose.
It is inarguable the abundance of consistent genetic and molecular alterations observed in panIN, primary, and metastatic pancreatic neoplasms. Numerous large-scale genomic sequencings and proteomic screenings have been conducted with novel discoveries identified from the experiments. However, the direct applicability of these potential biomarkers in patient populations remains unknown. For example, perhaps the most recognized alteration noted in pancreatic cancer is the mutation of Kras. Although targeting this player has thus far been unsuccessful due to the redundancy of cellular signaling pathways and perhaps also the lessened dependency of cells on this pathway as the disease progresses, a novel small molecule inhibitor of the Ras oncoprotein may develop with promise [161]. While the utilization and targeting of such genetic and molecular modifications remain a challenge in overcoming chemoresistance, the identification of precise biomarkers is vital since they may be used for targeting tumor cells and for guiding therapy. Identifying cell surface markers could allow for the targeting of tumor cells alone, leaving surrounding healthy cells in the heterogeneous tumor microenvironment to thrive. Also, utilizing these markers could stratify patient groups for individualized and tailored treatments. In addition to earlier diagnosis, biomarkers also have the potential for therapeutic monitoring of patients, especially since pancreatic cancer is known to undergo genetic mutations at a high frequency and rapid changes in disease biology. Currently, challenges arise in both identifying appropriate biomarkers for use in patient populations as well as in the practicality of screening a patient in the narrow time period between initial diagnosis and start of treatment. Clinical trial inconsistencies pose another challenge as the patient populations are often low in number and heterogeneous (e.g., with some patients already under treatment and some chemo-naïve). Well-annotated, patient-derived tumor samples remain a poorly available resource for further research.
In addition to identifying genetic and molecular alterations, it is crucial to understand when the changes occur during the progression of the disease. Pancreatic cancer, like any other solid tumor, consists of multiple defined stages, yet all patients are treated with the same drug (mainly due to diagnoses occurring at advance-stage disease). With better comprehension of the changes that occur throughout the cancer, including variations between the primary and secondary tumors and the primary and recurrent tumors, tailored drug therapies can be developed. Furthermore, management of genetic and molecular changes and disease monitoring are vital with disease progression. Identification of genetic and molecular signatures that can segregate pancreatic tumors into categories, such as those from Collison et al. (i.e., classical, quasimesenchymal, and exocrine-like) [162], could lead to separate and appropriate treatment and management strategies. Therefore, it is imperative to understand how the chemotherapeutic drugs exert their efficacy as this information may be particularly useful when considering the heterogeneity of pancreatic tumors.
Pancreatic tumors are well-known for their dense desmoplastic stroma and sparse, collapsed vasculature, all characteristics impeding effective chemotherapy. Although the tumors consist of both stromal and epithelial tissues, most earlier studies have focused mainly on epithelial components. However, currently, an increased number of studies are investigating the stroma of pancreatic tumors, including methods to target eminent stromal pathways such as Hedgehog, TGF-β, and Wnt/PI3K as well as deplete stromal tissues in order to stimulate angiogenesis and increase drug delivery. The latter approach is currently under investigation in numerous Phase I-III clinical trials. Enhanced knowledge of the heterogeneity of pancreatic tumors may lead to improved methods for overcoming chemoresistance due to tumor diversity. Furthermore, accurate methods for diagnosis as well as disease and therapeutic monitoring can be developed.
Significant advances have been made in the study of pancreatic cancer, including the development of in vitro cell lines and in vivo mouse models. Although a plethora of pancreatic cancer cell lines exist for study, their ability to mimic the wide range of pancreatic tumor characteristics is lacking. For example, Collison et al. identified cells lines that represented the classical and quasimesenchymal subtypes of pancreatic cancer but not the exocrine-like subtype [162]. Although the use of cell lines for cancer study remains valid, it is important to note the potential flaws of the model systems. Furthermore, although novel mouse models such as the orthotopic and K-rasLSL-G12D/+; Trp53LSL-R172H/+;Cre (KPC) have been developed, there remains the issue of interspecies differences in the development of chemoresistance. It is not yet fully determined how well these murine models represent human pancreatic tumors in both development and treatment outcomes. For example, the orthotopic model does not generate enough stroma to mimic human tumors, and the KPC model does not contain the entire spectrum of genetic mutations observed in human tumors. Although developmental issues are evident, the relative significance of these differences on influencing therapeutic outcome remains unknown. Ultimately, although great strides have been made with the development and use of novel mouse models, there remains a lack of model that has the ability to completely recapitulate human disease.
In addition to identifying new and far more improved drugs, an integrated approach is needed to overcome chemoresistance. For example, the rational combinations of the reviewed therapies (Fig. 1), such as epigenetic agents, gene therapy, antibodies, and signaling inhibitors, along with the use of chemotherapeutics still need further explication. The direct applicability of these therapies in combination (e.g., precise biomarker combinations to target cancer cells of all stages) in guiding treatment remains unknown. Furthermore, disconnect between benchtop, clinical, and regulatory science propels an additional, complex challenge in overcoming pancreatic cancer. Currently, numerous clinical trials remain at early investigational stages and are difficult to escalate to the level of approved patient-use. Although divisions between specialized fields still exist, the process of streamlining translational research is gaining momentum, and its persistence may ultimately lead to overcoming the therapeutic challenges of pancreatic cancer.
Fig. 1.
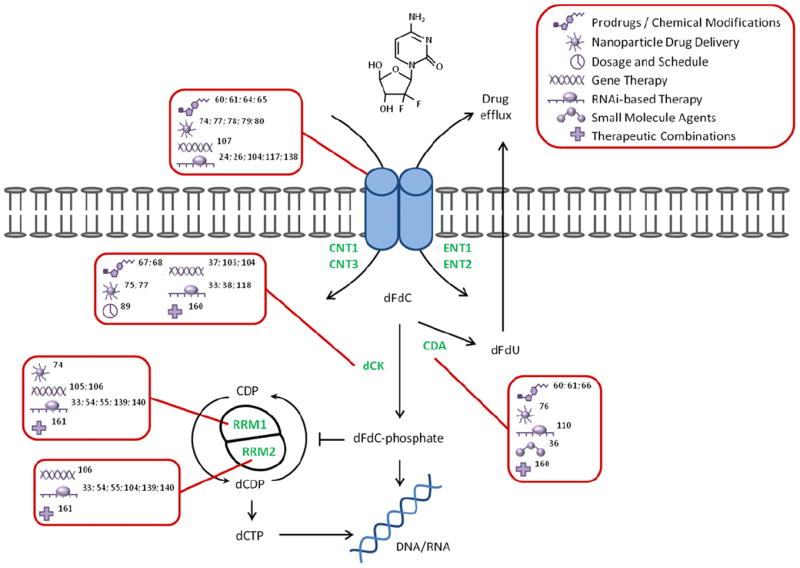
Novel therapeutic approaches for overcoming limitations in the gemcitabine pathway. Gemcitabine enters the cell via nucleoside transporters (i.e., CNT1, CNT3, ENT1, ENT2). The drug is either phosphorylated into its active form (i.e., dFdC-phosphate) by dCK or deaminated into dFdU by CDA and eliminated from the cell. Activated gemcitabine can then terminate the cell by directly targeting DNA or inhibiting RRM1 and RRM2 to deplete the dNTP pool necessary for DNA replication. Descriptions in boxes (red) indicate known methods for targeting a particular determinant (green) of gemcitabine chemosensitivity in pancreatic cancer. Abbreviations: CNT, concentrative nucleoside transporter; ENT, equilibrative nucleoside transporter; dCK, deoxycytidine kinase; CDA, cytidine deaminase; RR, ribonucleotide reductase; dFdC, 2′,2′-difluorodeoxycytidine; dFdU, 2′,2′-difluorodeoxyuridine; CDP, cytidine diphosphate; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate.
7. Conclusions
As the current standard of care, gemcitabine is predicted to remain as the drug of choice for the treatment of pancreatic cancer for the near future. Determinants of chemoresistance in the gemcitabine pathway are well-known, yet many challenges remain in pursuing the targets of interest to improve efficacy. Furthermore, other than the gemcitabine pathway, numerous other cellular and tumoral determinants pose as obstacles in overcoming chemoresistance. Nonetheless, novel therapeutic approaches are currently underway to circumvent limitations in gemcitabine transport, phosphorylation, and overall efficacy. Although therapeutic challenges are clear and present, countless studies, both at the benchtop and clinical levels, continue to provide clues for maximizing chemotherapeutic efficacy in patients.
Acknowledgments
This work was supported by NCI 1R03CA161832-01 (RG), NCI 5 P50 CA128613 SPORE (RG), the Georgia Cancer Coalition (RG), and the Department of Pharmaceutical and Biomedical Sciences, University of Georgia, Athens, GA. We thank Dr. Robert Arnold and Dr. Mandi Murph for critically reviewing this article.
References
- 1.Cancer Facts and Figures 2011. American Cancer Society. Atlanta, GA: 2011. [Google Scholar]
- 2.Gemzar Patient Therapy Guide: Pancreatic Cancer. Eli Lilly and Company; Indianapolis, IN: 2011. Beginning the journey: locally advanced or metastatic pancreatic cancer; pp. 13–14. [Google Scholar]
- 3.Hertel L, Kroin J, Misner J, Tustin J. Synthesis of 2-deoxy-2, 2-difluoro-D-ribose and 2-deoxy-2,2′-difluoro-D-ribofuranosyl nucleosides. The Journal of Organic Chemistry. 1988;53:2406–2409. [Google Scholar]
- 4.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH. Pancreatic cancer: understanding and overcoming chemoresistance. Nature Reviews Gastroenterology and Hepatology. 2010;8:27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 5.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of Clinical Oncology. 1997;15:2403. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 6.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nature Reviews Clinical Oncology. 2010;7:163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 7.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 10.Vaccaro V, Melisi D, Bria E, Cuppone F, Ciuffreda L, Pino MS, Gelibter A, Tortora G, Cognetti F, Milella M. Emerging pathways and future targets for the molecular therapy of pancreatic cancer. Expert Opinion on Therapeutic Targets. 2011:1–14. doi: 10.1517/14728222.2011.607438. [DOI] [PubMed] [Google Scholar]
- 11.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nature Reviews Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 12.National Institute of Allergy and Infectious Diseases N.I.H. Anti-HIV/OI Chemical Compound Database [Google Scholar]
- 13.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Research. 1998;58:4349. [PubMed] [Google Scholar]
- 14.Mackey JR, Yao SYM, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. Journal of the National Cancer Institute. 1999;91:1876. doi: 10.1093/jnci/91.21.1876. [DOI] [PubMed] [Google Scholar]
- 15.Giovannetti E, Del Tacca M, Mey V, Funel N, Nannizzi S, Ricci S, Orlandini C, Boggi U, Campani D, Del Chiaro M. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Research. 2006;66:3928. doi: 10.1158/0008-5472.CAN-05-4203. [DOI] [PubMed] [Google Scholar]
- 16.Farrell J, Garcia M, Lai R, Ammar A, Regine W, Abrams R, Bowen Benson A, Macdonald J, Cass C, Elsaleh H. Human Ent1 is predictive of response in patients with pancreatic cancer treated with gemcitabine: results from the Rtog 9704 prospective randomized trial. Pancreas. 2007;35:401. [Google Scholar]
- 17.Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet C, Cass C, Lai R, Mackey JR. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clinical Cancer Research. 2004;10:6956. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 18.Maréchal R, Mackey JR, Lai R, Demetter P, Peeters M, Polus M, Cass CE, Young J, Salmon I, Devière J. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clinical Cancer Research. 2009;15:2913. doi: 10.1158/1078-0432.CCR-08-2080. [DOI] [PubMed] [Google Scholar]
- 19.Robins MJ, Peng Y, Damaraju VL, Mowles D, Barron G, Tackaberry T, Young JD, Cass CE. Improved syntheses of 5′-S-(2-aminoethyl)-6-N-(4- nitrobenzyl)-5′-thioadenosine (SAENTA), analogues, and fluorescent probe conjugates: analysis of Cell-surface human equilibrative nucleoside Transporter 1 (hENT1) levels for prediction of the antitumor efficacy of gemcitabine. Journal of medicinal chemistry. 2010 doi: 10.1021/jm100432w. [DOI] [PubMed] [Google Scholar]
- 20.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T, Kohgo Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. British Journal of Cancer. 2007;96:457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Manteiga J, Molina-Arcas M, Casado FJ, Mazo A, Pastor-Anglada M. Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2′ 2′-difluorodeoxycytidine-induced cytotoxicity. Clinical Cancer Research. 2003;9:5000–5008. [PubMed] [Google Scholar]
- 22.Rauchwerger DR, Firby PS, Hedley DW, Moore MJ. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Research. 2000;60:6075. [PubMed] [Google Scholar]
- 23.Murata Y, Hamada T, Kishiwada M, Ohsawa I, Mizuno S, Usui M, Sakurai H, Tabata M, Ii N, Inoue H. Human equilibrative nucleoside transporter 1 expression is a strong independent prognostic factor in UICC T3–T4 pancreatic cancer patients treated with preoperative gemcitabine-based chemoradiotherapy. Journal of Hepato-Biliary-Pancreatic Sciences. 2011:1–13. doi: 10.1007/s00534-011-0440-3. [DOI] [PubMed] [Google Scholar]
- 24.Bhutia YD, Hung SW, Patel B, Lovin D, Govindarajan R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Research. 2011;71:1825. doi: 10.1158/0008-5472.CAN-10-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishio R, Tsuchiya H, Yasui T, Matsuura S, Kanki K, Kurimasa A, Hisatome I, Shiota G. Disrupted plasma membrane localization of equilibrative nucleoside transporter 2 in the chemoresistance to gemcitabine (dFdCyd) of human pancreatic cancer cells. Cancer Science. 2011;102:622–629. doi: 10.1111/j.1349-7006.2010.01837.x. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, Chen W, Xu J. Downregulation of human equilibrative nucleoside transporter 1 by RNAi Enhances 5–fluorouracil response in pancreatic cancer. Hepato-Gastroenterology. 2010;57:1567–1572. [PubMed] [Google Scholar]
- 27.Noma B, Sasaki T, Fujimoto Y, Serikawa M, Kobayashi K, Inoue M, Itsuki H, Kamigaki M, Minami T, Chayama K. Expression of multidrug resistance-associated protein 2 is inved in chemotherapy resistance in human pancreatic cancer. International Journal of Oncology. 2008;33:1187. [PubMed] [Google Scholar]
- 28.Kruh GD, Guo Y, Hopper-Borge E, Belinsky MG, Chen ZS. Chen ABCC10, ABCC11, and ABCC12. Pflügers Archiv European Journal of Physiology. 2007;453:675–684. doi: 10.1007/s00424-006-0114-1. [DOI] [PubMed] [Google Scholar]
- 29.Hagmann W, Jesnowski R, Faissner R, Guo C, Löhr JM. ATP-binding cassette C transporters in human pancreatic carcinoma cell lines. Pancreatology. 2008;9:136–144. doi: 10.1159/000178884. [DOI] [PubMed] [Google Scholar]
- 30.Hopper-Borge E, Xu X, Shen T, Shi Z, Chen ZS, Kruh GD. Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Research. 2009;69:178. doi: 10.1158/0008-5472.CAN-08-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudin D, Li L, Niu N, Kalari K, Gilbert J, Ames M, Wang L. Gemcitabine cytotoxicity: interaction of efflux and deamination. Journal of Drug Metabolism and Toxicology. 2011;2:1. doi: 10.4172/2157-7609.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang P, Plunkett W. Induction of apoptosis by gemcitabine. 1995:19. [PubMed] [Google Scholar]
- 33.Ohhashi S, Ohuchida K, MIzumoto K, Fujita H, Egami T, Yu J, Toma H, Sadatomi S, Nagai E, Tanaka M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Research. 2008;28:2205. [PubMed] [Google Scholar]
- 34.Yao J, Feng F, Lin C, Zhang X, Fu M, Liang X, Yang Y. The mechanism of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine) in a pancreatic cancer cell line. Chinese Journal of Oncology. 2005;27:721. [PubMed] [Google Scholar]
- 35.Kroep JR, Loves WJP, van der Wilt CL, Alvarez E, Talianidis I, Boven E, Braakhuis BJM, van Groeningen CJ, Pinedo HM, Peters GJ. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Molecular Cancer Therapeutics. 2002;1:371. [PubMed] [Google Scholar]
- 36.Funamizu N, Okamoto A, Kamata Y, Misawa T, Uwagawa T, Gocho T, Yanaga K, Manome Y. Is the resistance of gemcitabine for pancreatic cancer settled only by overexpression of deoxycytidine kinase? Oncology Reports. 2010;23:471–475. [PubMed] [Google Scholar]
- 37.Tang K, Zhang Z, Bai Z, Ma X, Guo W, Wang Y. Enhancement of gemcitabine sensitivity in pancreatic cancer by co-regulation of dCK and p8 expression. Oncology Reports. 2011;25:963–970. doi: 10.3892/or.2011.1139. [DOI] [PubMed] [Google Scholar]
- 38.Sebastiani V, Ricci F, Rubio-Viquiera B, Kulesza P, Yeo CJ, Hidalgo M, Klein A, Laheru D, Iacobuzio-Donahue CA. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: relationship to molecular mechanisms of gemcitabine resistance and survival. Clinical Cancer Research. 2006;12:2492. doi: 10.1158/1078-0432.CCR-05-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams TK, Costantino CL, Bildzukewicz NA, Richards NG, Rittenhouse DW, Einstein L, Cozzitorto JA, Keen JC, Dasgupta A, Gorospe M. pp 32 (ANP32A) expression inhibits pancreatic cancer cell growth and induces gemcitabine resistance by disrupting HuR binding to mRNAs. PloS One. 2010;5:e15455. doi: 10.1371/journal.pone.0015455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A, Keen JC, Yeo CJ, Gorospe M, Brody JR. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Research. 2009;69:4567. doi: 10.1158/0008-5472.CAN-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metabolism and Disposition. 1992;20:849–855. [PubMed] [Google Scholar]
- 42.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of self-potentiation. Cancer Research. 1992;52:533. [PubMed] [Google Scholar]
- 43.Mercier C, Raynal C, Dahan L, Ortiz A, Evrard A, Dupuis C, Blesius A, Duluc M, Franceschini F, Giacometti S. Toxic death case in a patient undergoing gemcitabine-based chemotherapy in relation with cytidine deaminase downregulation. Pharmacogenetics and Genomics. 2007;17:841. doi: 10.1097/FPC.0b013e32825ea6e3. [DOI] [PubMed] [Google Scholar]
- 44.Farrell J, Bae K, Wong J, Guha C, Dicker A, Elsaleh H. Cytidine deaminase single-nucleotide polymorphism is predictive of toxicity from gemcitabine in patients with pancreatic cancer: RTOG 9704. The Pharmacogenomics Journal. 2011 doi: 10.1038/tpj.2011.22. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka M, Javle M, Dong X, Eng C, Abbruzzese JL, Li D. Gemcitabine metabolic and transporter gene polymorphisms are associated with drug toxicity and efficacy in patients with locally advanced pancreatic cancer. Cancer. 2010;116:5325–5335. doi: 10.1002/cncr.25282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugiyama E, Kaniwa N, Kim SR, Hasegawa R, Saito Y, Ueno H, Okusaka T, Ikeda M, Morizane C, Kondo S. Population pharmacokinetics of gemcitabine and its metabolite in Japanese cancer patients: impact of genetic polymorphisms. Clinical Pharmacokinetics. 2010;49:549–558. doi: 10.2165/11532970-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 47.Ueno H, Kaniwa N, Okusaka T, Ikeda M, Morizane C, Kondo S, Sugiyama E, Kim S, Hasegawa R, Saito Y. Homozygous CDA*3 is a major cause of life-threatening toxicities in gemcitabine-treated Japanese cancer patients. British Journal of Cancer. 2009;100:870–873. doi: 10.1038/sj.bjc.6604971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eda H, Ura M. The antiproliferative activity of DMDC is modulated by inhibition of cytidine deaminase. Cancer Research. 1998;58:1165. [PubMed] [Google Scholar]
- 49.Pereira S, Fernandes PA, Ramos MJ. Mechanism for ribonucleotide reductase inactivation by the anticancer drug gemcitabine. Journal of Computational Chemistry. 2004;25:1286–1294. doi: 10.1002/jcc.20054. [DOI] [PubMed] [Google Scholar]
- 50.Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, Yamasaki M, Marubashi S, Takemasa I, Miyamoto A. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. International Journal of Cancer. 2007;120:1355–1363. doi: 10.1002/ijc.22390. [DOI] [PubMed] [Google Scholar]
- 51.Akita H, Zheng Z, Takeda Y, Kim C, Kittaka N, Kobayashi S, Marubashi S, Takemasa I, Nagano H, Dono K. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. 2009;28:2903–2909. doi: 10.1038/onc.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim R, Tan A, Lai KK, Jiang J, Wang Y, Rybicki LA, Liu X. Prognostic roles of human equilibrative transporter 1 (hENT 1) and ribonucleoside reductase subunit M1 (RRM1) in resected pancreatic cancer. Cancer. 2011 doi: 10.1002/cncr.25883. [DOI] [PubMed] [Google Scholar]
- 53.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clinical Cancer Research. 2004;10:2307. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- 54.Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. Retrovirally mediated RNA interference targeting the M2 subunit of ribonucleotide reductase: a novel therapeutic strategy in pancreatic cancer. Surgery. 2004;136:261–269. doi: 10.1016/j.surg.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 55.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 2004;23:1539–1548. doi: 10.1038/sj.onc.1207272. [DOI] [PubMed] [Google Scholar]
- 56.Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, Yu J, Kayashima T, Souzaki R, Tajiri T. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia (New York, NY) 2010;12:807. doi: 10.1593/neo.10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itoi T, Sofuni A, Fukushima N, Itokawa F, Tsuchiya T, Kurihara T, Moriyasu F, Tsuchida A, Kasuya K. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. Journal of Gastroenterology. 2007;42:389–394. doi: 10.1007/s00535-007-2017-0. [DOI] [PubMed] [Google Scholar]
- 58.Fowler JD, Brown JA, Johnson KA, Suo Z. Kinetic investigation of the inhibitory effect of gemcitabine on DNA polymerization catalyzed by human mitochondrial DNA polymerase. Journal of Biological Chemistry. 2008;283:15339. doi: 10.1074/jbc.M800310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adema A, Radi M, Daft J, Narayanasamy J, Hoebe E, Alexander L, Chu C, Peters G. Troxacitabine prodrugs for pancreatic cancer. Nucleosides, Nucleotides &# 38; Nucleic Acids. 2007;26(8):1073–1077. doi: 10.1080/15257770701515591. [DOI] [PubMed] [Google Scholar]
- 60.Castelli F, Sarpietro MG, Ceruti M, Rocco F, Cattel L. Characterization of lipophilic gemcitabine prodrug-liposomal membrane interaction by differential scanning calorimetry. Molecular Pharmaceutics. 2006;3:737–744. doi: 10.1021/mp060059y. [DOI] [PubMed] [Google Scholar]
- 61.Brusa P, Immordino ML, Rocco F, Cattel L. Antitumor activity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Anticancer Research. 2007;27:195. [PubMed] [Google Scholar]
- 62.Stella B, Arpicco S, Rocco F, Marsaud V, Renoir JM, Cattel L, Couvreur P. Encapsulation of gemcitabine lipophilic derivatives into polycyanoacrylate nanospheres and nanocapsules. International Journal of Pharmaceutics. 2007;344:71–77. doi: 10.1016/j.ijpharm.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Journal of Controlled Release. 2004;100:331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Chen P, Chien PY, Khan AR, Sheikh S, Ali SM, Ahmad MU, Ahmad I. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anti-Cancer Drugs. 2006;17:53. doi: 10.1097/01.cad.0000185182.80227.48. [DOI] [PubMed] [Google Scholar]
- 65.Bergman AM, Adema AD, Balzarini J, Bruheim S, Fichtner I, Noordhuis P, Fodstad Ø, Myhren F, Sandvold ML, Hendriks HR. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Investigational New Drugs. 2011:1–11. doi: 10.1007/s10637-009-9377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koolen S, Witteveen P, Jansen R, Langenberg M, Kronemeijer R, Nol A, Garcia-Ribas I, Callies S, Benhadji K, Slapak C. Phase I study of oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors intravenous-to-oral switch in anticancer chemotherapy focus on taxanes and gemcitabine. 2011:135. doi: 10.1158/1078-0432.CCR-11-0353. [DOI] [PubMed] [Google Scholar]
- 67.Wu W, Sigmond J, Peters GJ, Borch RF. Synthesis and biological activity of a gemcitabine phosphoramidate prodrug. Journal of Medicinal Chemistry. 2007;50:3743–3746. doi: 10.1021/jm070269u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kotchetkov R, Groschel B, Gmeiner WH, Krivtchik AA, Trump E, Bitoova M, Cinatl J, Kornhuber B. Antineoplastic activity of a novel multimeric gemcitabine-monophosphate prodrug against thyroid cancer cells in vitro. Anticancer Research. 2000;20:2915–2922. [PubMed] [Google Scholar]
- 69.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature Reviews Clinical Oncology. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arias JL, Reddy LH, Couvreur P. Superior preclinical efficacy of gemcitabine developed as chitosan nanoparticulate system. Biomacromolecules. 2011;12:97–104. doi: 10.1021/bm101044h. [DOI] [PubMed] [Google Scholar]
- 71.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjugate Chemistry. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 72.Bisht S, Mizuma M, Feldmann G, Ottenhof NA, Hong SM, Pramanik D, Chenna V, Karikari C, Sharma R, Goggins MG, Rudek MA, Ravi R, Maitra A. Systemic administration of polymeric nanoparticle-encapsulated curcumin (NanoCurc) blocks tumor growth and metastases in preclinical models of pancreatic cancer. Molecular Cancer Therapeutics. 2010;9:2255–2264. doi: 10.1158/1535-7163.MCT-10-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aryal S, Hu CM, Zhang L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small. 2010;6:1442–1448. doi: 10.1002/smll.201000631. [DOI] [PubMed] [Google Scholar]
- 74.Sloat BR, Sandoval MA, Li D, Chung WG, Lansakara PD, Proteau PJ, Kiguchi K, DiGiovanni J, Cui Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. International Journal of Pharmaceutics. 2011;409:278–288. doi: 10.1016/j.ijpharm.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chung G, Sandoval MA, Sloat BR, Lansakara PD, Cui Z. Stearoyl gemcitabine nanoparticles overcome resistance related to the overexpression of ribonucleotide reductase subunit M1. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2011 doi: 10.1016/j.jconrel.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine: Nanotechnology, Biology, and Medicine. 2011 doi: 10.1016/j.nano.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 77.Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 78.Reddy LH, Khoury H, Paci A, Deroussent A, Ferreira H, Dubernet C, Decleves X, Besnard M, Chacun H, Lepetre-Mouelhi S, Desmaele D, Rousseau B, Laugier C, Cintrat JC, Vassal G, Couvreur P. Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice. Drug Metabolism and Disposition: The Biological Fate of Chemicals. 2008;36:1570–1577. doi: 10.1124/dmd.108.020735. [DOI] [PubMed] [Google Scholar]
- 79.Bildstein L, Dubernet C, Marsaud V, Chacun H, Nicolas V, Gueutin C, Sarasin A, Benech H, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Transmembrane diffusion of gemcitabine by a nanoparticulate squalenoyl prodrug: an original drug delivery pathway. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2010;147:163–170. doi: 10.1016/j.jconrel.2010.07.120. [DOI] [PubMed] [Google Scholar]
- 80.Arya G, Vandana M, Acharya S, Sahoo SK. Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine: Nanotechnology, Biology, and Medicine. 2011 doi: 10.1016/j.nano.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 81.Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Yaszemski MJ, Reid JM, Ames MM, Mukherjee P, Mukhopadhyay D. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer research. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 82.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Advanced Drug Delivery Reviews. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 83.Arias JL, Reddy LH, Couvreur P. Magnetoresponsive squalenoyl gemcitabine composite nanoparticles for cancer active targeting. Langmuir: The ACS Journal of Surfaces and Colloids. 2008;24:7512–7519. doi: 10.1021/la800547s. [DOI] [PubMed] [Google Scholar]
- 84.Arias JL, Reddy LH, Othman M, Gillet B, Desmaele D, Zouhiri F, Dosio F, Gref R, Couvreur P. Couvreur Squalene based nanocomposites: a new platform for the design of multifunctional pharmaceutical theragnostics. ACS Nano. 2011;5:1513–1521. doi: 10.1021/nn1034197. [DOI] [PubMed] [Google Scholar]
- 85.Galmarini CM, Warren G, Senanayake MT, Vinogradov SV. Efficient overcoming of drug resistance to anticancer nucleoside analogs by nanodelivery of active phosphorylated drugs. International Journal of Pharmaceutics. 2010;395:281–289. doi: 10.1016/j.ijpharm.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hillaireau H, Le Doan T, Appel M, Couvreur P. Hybrid polymer nanocapsules enhance in vitro delivery of azidothymidine-triphosphate to macrophages. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2006;116:346–352. doi: 10.1016/j.jconrel.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 87.Saiyed ZM, Gandhi NH, Nair MP. AZT 5′-triphosphate nanoformulation suppresses human immunodeficiency virus type 1 replication in peripheral blood mononuclear cells. Journal of Neurovirology. 2009;15:343–347. doi: 10.1080/13550280903062813. [DOI] [PubMed] [Google Scholar]
- 88.Rivera F, López-Tarruella S, Vega-Villegas ME, Salcedo M. Treatment of advanced pancreatic cancer: from gemcitabine single agent to combinations and targeted therapy. Cancer Treatment Reviews. 2009;35:335–339. doi: 10.1016/j.ctrv.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 89.Furuse J, Ishii H, Okusaka T, Nagase M, Nakachi K, Ueno H, Ikeda M, Morizane C, Yoshino M. Phase I study of fixed dose rate infusion of gemcitabine in patients with unresectable pancreatic cancer. Japanese Journal of Clinical Oncology. 2005;35:733. doi: 10.1093/jjco/hyi190. [DOI] [PubMed] [Google Scholar]
- 90.Brand R, Capadano M, Tempero M. A phase I trial of weekly gemcitabine administered as a prolonged infusion in patients with pancreatic cancer and other solid tumors. Investigational New Drugs. 1997;15:331–341. doi: 10.1023/a:1005981317532. [DOI] [PubMed] [Google Scholar]
- 91.Tempero M, Plunkett W, Ruiz van Haperen V, Hainsworth J, Hochster H, Lenzi R, Abbruzzese J. Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. Journal of Clinical Oncology. 2003;21:3402. doi: 10.1200/JCO.2003.09.140. [DOI] [PubMed] [Google Scholar]
- 92.Poplin E, Levy D, Berlin J, Rothenberg M, Cella D, Mitchell E, Alberts S, Benson A., III Phase III trial of gemcitabine (30-minute infusion) versus gemcitabine (fixed-dose-rate infusion [FDR]) versus gemcitabine+ oxaliplatin (GEMOX) in patients with advanced pancreatic cancer (E6201) Journal of Clinical Oncology. 2006;24:4004. doi: 10.1200/JCO.2008.20.9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mané JM, Sancho A, Muñoz A, Rubio I, Fernández R, Carrera S, Fuente N, Ballesteros D, Casas R, Marrodán I. Fixed-dose-rate gemcitabine infusion in patients with advanced pancreatic or biliary tree adenocarcinoma. Tumori. 2010;96:405–410. doi: 10.1177/030089161009600305. [DOI] [PubMed] [Google Scholar]
- 94.Bengala C, Guarneri V, Giovannetti E, Lencioni M, Fontana E, Mey V, Fontana A, Boggi U, Del Chiaro M, Danesi R. Prolonged fixed dose rate infusion of gemcitabine with autologous haemopoietic support in advanced pancreatic adenocarcinoma. British Journal of Cancer. 2005;93:35–40. doi: 10.1038/sj.bjc.6602673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ulrich Pur H, Kornek GV, Raderer M, Haider K, Kwasny W, Depisch D, Greul R, Schneeweiss B, Krauss G, Funovics J. A phase II trial of biweekly high dose gemcitabine for patients with metastatic pancreatic adenocarcinoma. Cancer. 2000;88:2505–2511. doi: 10.1002/1097-0142(20000601)88:11<2505::aid-cncr11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 96.Hirao K, Kawamoto H, Sakakihara I, Noma Y, Yamamoto N, Harada R, Tsutsumi K, Fujii M, Kato H, Kurihara N. A 4-week versus a 3-week schedule of gemcitabine monotherapy for advanced pancreatic cancer: a randomized phase II study to evaluate toxicity and dose intensity. International Journal of Clinical Oncology. 2011:1–9. doi: 10.1007/s10147-011-0237-z. [DOI] [PubMed] [Google Scholar]
- 97.Okada S, Ueno H, Okusaka T, Ikeda M, Furuse J, Maru Y. Phase I trial of gemcitabine in patients with advanced pancreatic cancer. Japanese Journal of Clinical Oncology. 2001;31:7. doi: 10.1093/jjco/hye003. [DOI] [PubMed] [Google Scholar]
- 98.Laquente B, Lacasa C, Ginestà MM, Casanovas O, Figueras A, Galán M, Ribas IG, Germà JR, Capellà G, Viñals F. Antiangiogenic effect of gemcitabine following metronomic administration in a pancreas cancer model. Molecular Cancer Therapeutics. 2008;7:638. doi: 10.1158/1535-7163.MCT-07-2122. [DOI] [PubMed] [Google Scholar]
- 99.Pollera CF, Ceribelli A, Crecco M, Oliva C. Prolonged infusion gemcitabine: a clinical phase I study at low-(300 mg/m 2) and high-dose (875 mg/m 2) levels. Investigational New Drugs. 1997;15:115–121. doi: 10.1023/a:1005817024382. [DOI] [PubMed] [Google Scholar]
- 100.Eckel F, Schmelz R, Erdmann J, Mayr M, Lersch C. Phase II trial of a 24-hour infusion of gemcitabine in previously untreated patients with advanced pancreatic adenocarcinoma. Cancer Investigation. 2003;21:690–694. doi: 10.1081/cnv-120023767. [DOI] [PubMed] [Google Scholar]
- 101.Kuemmerle A, Decosterd LA, Buclin T, Liénard D, Stupp R, Chassot PG, Mosimann F, Lejeune F. A phase I pharmacokinetic study of hypoxic abdominal stop-flow perfusion with gemcitabine in patients with advanced pancreatic cancer and refractory malignant ascites. Cancer Chemotherapy and Pharmacology. 2009;63:331–341. doi: 10.1007/s00280-008-0743-5. [DOI] [PubMed] [Google Scholar]
- 102.Vernejoul F, Ghénassia L, Souque A, Lulka H, Drocourt D, Cordelier P, Pradayrol L, Pyronnet S, Buscail L, Tiraby G. Gene therapy based on gemcitabine chemosensitization suppresses pancreatic tumor growth. Molecular Therapy. 2004;14:758–767. doi: 10.1016/j.ymthe.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 103.Réjiba S, Bigand C, Parmentier C, Hajri A. Gemcitabine-based chemogene therapy for pancreatic cancer using Ad-dCK: UMK GDEPT and TS/RR siRNA strategies. Neoplasia (New York, NY) 2009;11:637. doi: 10.1593/neo.81686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kasuya H, Mizuno M, Yoshida J, Nishiyama Y, Nomoto S, Nakao A. Combined effects of adeno associated virus vector and a herpes simplex virus mutant as neoplastic therapy. Journal of Surgical Oncology. 2000;74:214–218. doi: 10.1002/1096-9098(200007)74:3<214::aid-jso12>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 105.Kasuya H, Nishiyama Y, Nomoto S, Hosono J, Takeda S, Nakao A. Intraperitoneal delivery of hrR3 and ganciclovir prolongs survival in mice with disseminated pancreatic cancer. Journal of Surgical Oncology. 1999;72:136–141. doi: 10.1002/(sici)1096-9098(199911)72:3<136::aid-jso5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 106.Pérez-Torras S, Garcia-Manteiga J, Mercadé E, Casado FJ, Carbo N, Pastor-Anglada M, Mazo A. Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochemical Pharmacology. 2008;76:322–329. doi: 10.1016/j.bcp.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 107.Halloran CM, Ghaneh P, Shore S, Greenhalf W, Zumstein L, Wilson D, Neoptolemos JP, Costello E. 5 Fluorouracil or gemcitabine combined with adenoviral mediated reintroduction of p16INK4A greatly enhanced cytotoxicity in Panc 1 pancreatic adenocarcinoma cells. The Journal of Gene Medicine. 2004;6:514–525. doi: 10.1002/jgm.540. [DOI] [PubMed] [Google Scholar]
- 108.Xu ZW, Friess H, Büchler MW, Solioz M. Overexpression of Bax sensitizes human pancreatic cancer cells to apoptosis induced by chemotherapeutic agents. Cancer Chemotherapy and Pharmacology. 2002;49:504–510. doi: 10.1007/s00280-002-0435-5. [DOI] [PubMed] [Google Scholar]
- 109.Prabhu JS, Korlimarla A, Banerjee A, Wani S, PAYAL K, SAHOO R. Gene-specific methylation: potential markers for colorectal cancer. The International Journal of Biological Markers. 2009;24:57–62. doi: 10.1177/172460080902400109. [DOI] [PubMed] [Google Scholar]
- 110.Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, Van Heek NT, Rosty C, Walter K, Sato N, Parker A, Ashfaq R. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. American Journal of Pathology. 2003;162:1151. doi: 10.1016/S0002-9440(10)63911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fry LC, Mönkemüller K, Malfertheiner P. Molecular markers of pancreatic cancer: development and clinical relevance. Langenbeck’s Archives of Surgery. 2008;393:883–890. doi: 10.1007/s00423-007-0276-0. [DOI] [PubMed] [Google Scholar]
- 112.Habbe N, Bert T, Simon B. Identification of methylation-associated gene expression in neuroendocrine pancreatic tumor cells. Pancreatology. 2007;7:352–359. doi: 10.1159/000107270. [DOI] [PubMed] [Google Scholar]
- 113.Yu XJ, Long J, Fu DL, Zhang QH, Ni QX. Analysis of gene expression profiles in pancreatic carcinoma by using cDNA microarray. HBPD International. 2003;2:467–470. [PubMed] [Google Scholar]
- 114.Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, Shu J, He R, Zhang N, Chung W. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PloS One. 2011;6:e23372. doi: 10.1371/journal.pone.0023372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nature Biotechnology. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rius M, Stresemann C, Keller D, Brom M, Schirrmacher E, Keppler D, Lyko F. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Molecular Cancer Therapeutics. 2009;8:225. doi: 10.1158/1535-7163.MCT-08-0743. [DOI] [PubMed] [Google Scholar]
- 117.Geutjes EJ, Tian S, Roepman P, Bernards R. Deoxycytidine kinase is overexpressed in poor outcome breast cancer and determines responsiveness to nucleoside analogs. Breast Cancer Research and Treatment. 2012:1–10. doi: 10.1007/s10549-011-1477-3. [DOI] [PubMed] [Google Scholar]
- 118.Missiaglia E, Donadelli M, Palmieri M, Crnogorac-Jurcevic T, Scarpa A, Lemoine NR. Growth delay of human pancreatic cancer cells by methylase inhibitor 5-aza-2-deoxycytidine treatment is associated with activation of the interferon signalling pathway. Oncogene. 2005;24:199–211. doi: 10.1038/sj.onc.1208018. [DOI] [PubMed] [Google Scholar]
- 119.Yu M, Epner E. The epigenetics of mantle cell lymphoma. Current Treatment Options in Oncology. 2007;8:375–381. doi: 10.1007/s11864-007-0047-8. [DOI] [PubMed] [Google Scholar]
- 120.Sato N, Ohta T, Kitagawa H, Kayahara M, Ninomiya I, Fushida S, Fujimura T, Nishimura GI, Shimizu K, Miwa K. FR901228, a novel histone deacetylase inhibitor, induces cell cycle arrest and subsequent apoptosis in refractory human pancreatic cancer cells. International Journal of Oncology. 2004;24:679–685. [PubMed] [Google Scholar]
- 121.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proceedings of the National Academy of Sciences. 1999;96:4592. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xenidis N, Chelis L, Amarantidis K, Chamalidou E, Dimopoulos P, Courcoutsakis N, Tentes A, Chiotis A, Prassopoulos P, Kakolyris S. Docetaxel plus gemcitabine in combination with capecitabine as treatment for inoperable pancreatic cancer: a phase II study. Cancer Chemotherapy and Pharmacology. 2011:1–8. doi: 10.1007/s00280-011-1717-6. [DOI] [PubMed] [Google Scholar]
- 123.Donadelli M, Costanzo C, Faggioli L, Scupoli MT, Moore PS, Bassi C, Scarpa A, Palmieri M. Trichostatin A, an inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Molecular Carcinogenesis. 2003;38:59–69. doi: 10.1002/mc.10145. [DOI] [PubMed] [Google Scholar]
- 124.Moore PS, Barbi S, Donadelli M, Costanzo C, Bassi C, Palmieri M, Scarpa A. Gene expression profiling after treatment with the histone deacetylase inhibitor trichostatin A reveals altered expression of both pro-and anti-apoptotic genes in pancreatic adenocarcinoma cells. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2004;1693:167–176. doi: 10.1016/j.bbamcr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 125.Ryu JK, Lee WJ, Lee KH, Hwang JH, Kim YT, Yoon YB, Kim CY. SK-7041, a new histone deacetylase inhibitor, induces G2-M cell cycle arrest and apoptosis in pancreatic cancer cell lines. Cancer Letters. 2006;237:143–154. doi: 10.1016/j.canlet.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 126.Cecconi D, Donadelli M, Rinalducci S, Zolla L, Scupoli MT, Scarpa A, Palmieri M, Righetti PG. Proteomic analysis of pancreatic endocrine tumor cell lines treated with the histone deacetylase inhibitor trichostatin A. Proteomics. 2007;7:1644–1653. doi: 10.1002/pmic.200600811. [DOI] [PubMed] [Google Scholar]
- 127.Donadelli M, Costanzo C, Beghelli S, Scupoli MT, Dandrea M, Bonora A, Piacentini P, Budillon A, Caraglia M, Scarpa A. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2007;1773:1095–1106. doi: 10.1016/j.bbamcr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 128.Gahr S, Ocker M, Ganslmayer M, Zopf S, Okamoto K, Hartl A, Leitner S, Hahn EG, Herold C. The combination of the histone-deacetylase inhibitor trichostatin A and gemcitabine induces inhibition of proliferation and increased apoptosis in pancreatic carcinoma cells. International Journal of Oncology. 2007;31:567–576. [PubMed] [Google Scholar]
- 129.Piacentini P, Donadelli M, Costanzo C, Moore PS, Palmieri M, Scarpa A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Archiv. 2006;448:797–804. doi: 10.1007/s00428-006-0173-x. [DOI] [PubMed] [Google Scholar]
- 130.Arnold NB, Arkus N, Gunn J, Korc M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clinical Cancer Research. 2007;13:18. doi: 10.1158/1078-0432.CCR-06-0914. [DOI] [PubMed] [Google Scholar]
- 131.Richards D, Boehm K, Waterhouse D, Wagener D, Krishnamurthi S, Rosemurgy A, Grove W, Macdonald K, Gulyas S, Clark M. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Annals of Oncology. 2006;17:1096. doi: 10.1093/annonc/mdl081. [DOI] [PubMed] [Google Scholar]
- 132.Rachagani S, Kumar S, Batra SK. MicroRNA in pancreatic cancer: pathological, diagnostic and therapeutic implications. Cancer Letters. 2010;292:8–16. doi: 10.1016/j.canlet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116:4670–4676. doi: 10.1002/cncr.25414. [DOI] [PubMed] [Google Scholar]
- 134.Dhayat, Mardin WA, Mees ST, Haier J. Epigenetic markers for chemosensitivity and chemoresistance in pancreatic cancer—a review. International Journal of Cancer. 2011 doi: 10.1002/ijc.26078. [DOI] [PubMed] [Google Scholar]
- 135.Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Schedule-dependent molecular effects of the proteasome inhibitor bortezomib and gemcitabine in pancreatic cancer. Journal of Surgical Research. 2003;113:88–95. doi: 10.1016/s0022-4804(03)00201-4. [DOI] [PubMed] [Google Scholar]
- 136.Beumer JH, Eiseman JL, Parise RA, Florian JA, Joseph E, D’Argenio DZ, Parker RS, Kay B, Covey JM, Egorin MJ. Plasma pharmacokinetics and oral bioavailability of 3,4,5,6-tetrahydrouridine, a cytidine deaminase inhibitor, in mice. Cancer Chemotherapy and Pharmacology. 2008;62:457–464. doi: 10.1007/s00280-007-0625-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Komori S, Osada S, Mori R, Matsui S, Sanada Y, Tomita H, Tokuyama Y, Takahashi T, Yamaguchi K, Yoshida K. Contribution of thymidylate synthase to gemcitabine therapy for advanced pancreatic cancer. Pancreas. 2010;39:1284. doi: 10.1097/MPA.0b013e3181dec17d. [DOI] [PubMed] [Google Scholar]
- 138.Funamizu N, Kamata Y, Misawa T, Uwagawa T, Lacy CR, Yanaga K, Manome Y. Hydroxyurea decreases gemcitabine resistance in pancreatic carcinoma cells with highly expressed ribonucleotide reductase. Pancreas. 2011 doi: 10.1097/MPA.0b013e318224b5fb. [DOI] [PubMed] [Google Scholar]
- 139.Mitsuno M, Kitajima Y, Ohtaka K, Kai K, Hashiguchi K, Nakamura J, Hiraki M, Noshiro H, Miyazaki K. Tranilast strongly sensitizes pancreatic cancer cells to gemcitabine via decreasing protein expression of ribonucleotide reductase 1. International Journal of Oncology. 2010;36:341–349. [PubMed] [Google Scholar]
- 140.Binder D, Hübner RH, Temmesfeld-Wollbrück B, Schlattmann P. Pulmonary toxicity among cancer patients treated with a combination of docetaxel and gemcitabine: a meta-analysis of clinical trials. Cancer Chemotherapy and Pharmacology. 2011:1–9. doi: 10.1007/s00280-011-1648-2. [DOI] [PubMed] [Google Scholar]
- 141.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a Phase I/II Trial. Journal of Clinical Oncology. 2011 doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spano JP, Moore MJ, Pithavala YK, Ricart AD, Kim S, Rixe O. Phase I study of axitinib (AG-013736) in combination with gemcitabine in patients with advanced pancreatic cancer. Investigational New Drugs. 2011:1–9. doi: 10.1007/s10637-011-9697-2. [DOI] [PubMed] [Google Scholar]
- 143.Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Investigational New Drugs. 2010:1–8. doi: 10.1007/s10637-010-9611-3. [DOI] [PubMed] [Google Scholar]
- 144.Awasthi N, Schwarz MA, Schwarz RE. Antitumour activity of sunitinib in combination with gemcitabine in experimental pancreatic cancer. HPB. 2011 doi: 10.1111/j.1477-2574.2011.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Adjei AA, Schwartz B, Garmey E. Early clinical development of ARQ 197, a selective, Non–ATP-competitive inhibitor targeting MET tyrosine kinase for the treatment of advanced cancers. The Oncologist. 2011;16:788–799. doi: 10.1634/theoncologist.2010-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Starling N, Hawkes E, Chau I, Watkins D, Thomas J, Webb J, Brown G, Thomas K, Barbachano Y, Oates J. A dose escalation study of gemcitabine plus oxaliplatin in combination with imatinib for gemcitabine-refractory advanced pancreatic adenocarcinoma. Annals of Oncology. 2011 doi: 10.1093/annonc/mdr317. [DOI] [PubMed] [Google Scholar]
- 147.Richards DA, Kuefler PR, Becerra C, Wilfong LS, Gersh RH, Boehm KA, Zhan F, Asmar L, Myrand SP, Hozak RR. Gemcitabine plus enzastaurin or single-agent gemcitabine in locally advanced or metastatic pancreatic cancer: results of a phase II, randomized, noncomparative study. Investigational New Drugs. 2011;29:144–153. doi: 10.1007/s10637-009-9307-8. [DOI] [PubMed] [Google Scholar]
- 148.Nakamori S, Endo W, Ozato H, Shibata T, Takeda Y, Tohno K, Hasuike Y, Masutani S, Morimoto T, Doki Y. Multicenter phase II study of pre-administered uracil/tegafur (UFT) plus gemcitabine for unresectable/recurrent pancreatic cancer gan to kagaku ryoho. Cancer and Chemotherapy. 2011;3(8):789. [PubMed] [Google Scholar]
- 149.Pericay Pijaume C, Escudero Emperador P, Bastús Piulats R, Campos Cervera JM, Esquerdo Galiana G, Gallén Castillo M, Alfaro Gamero J, Dotor Navarro E, Pisa Gatell A, Guasch Jordán I. Open-label trial on efficacy and security of treatment with gemcitabine and oral modulation with tegafur and levofolinic acid (GEMTG) in patients with advanced pancreatic cancer. Clinical and Translational Oncology. 2011;13:61–66. doi: 10.1007/s12094-011-0618-9. [DOI] [PubMed] [Google Scholar]
- 150.Sung V, Richard N, Brady H, Maier A, Kelter G, Heise C. Histone deacetylase inhibitor MGCD0103 synergizes with gemcitabine in human pancreatic cells. Cancer Science. 2011 doi: 10.1111/j.1349-7006.2011.01921.x. [DOI] [PubMed] [Google Scholar]
- 151.De Jesus-Acosta A, Oliver GR, Blackford A, Kinsman K, Flores EI, Wilfong LS, Zheng L, Donehower RC, Cosgrove D, Laheru D. A multicenter analysis of GTX chemotherapy in patients with locally advanced and metastatic pancreatic adenocarcinoma. Cancer Chemotherapy and Pharmacology. 2011:1–10. doi: 10.1007/s00280-011-1704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hill ME, Li X, Kim S, Campbell A, Culler K, Bloomston M, Zalupski M, Hejna G, Bekaii-Saab T. A phase I study of the biomodulation of capecitabine by docetaxel and gemcitabine (mGTX) in previously untreated patients with metastatic adenocarcinoma of the pancreas. Cancer Chemotherapy and Pharmacology. 2011;67:511–517. doi: 10.1007/s00280-010-1348-3. [DOI] [PubMed] [Google Scholar]
- 153.Sahora K, Kuehrer I, Eisenhut A, Akan B, Koellblinger C, Goetzinger P, Teleky B, Jakesz R, Peck-Radosavljevic M, Ba’ssalamah A. NeoGemOx: gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery. 2011;149:311–320. doi: 10.1016/j.surg.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 154.Hess V, Pratsch S, Potthast S, Lee L, Winterhalder R, Widmer L, Cescato C, Lohri A, Jost L, Stillhart P. Combining gemcitabine, oxaliplatin and capecitabine (GEMOXEL) for patients with advanced pancreatic carcinoma (APC): a phase I/II trial. Annals of Oncology. 2010;21:2390. doi: 10.1093/annonc/mdq242. [DOI] [PubMed] [Google Scholar]
- 155.Cereda S, Reni M, Rognone A, Ghidini M, Belli C, Longoni S, Fugazza C, Brioschi M, Nicoletti R, Balzano G. XELIRI or FOLFIRI as salvage therapy in advanced pancreatic cancer. Anticancer Research. 2010;30:4785. [PubMed] [Google Scholar]
- 156.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New England Journal of Medicine. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 157.Infante JR, Jones SF, Bendell JC, Spigel DR, Yardley DA, Weekes CD, Messersmith WA, Hainsworth JD, Burris HA., III A phase I, dose-escalation study of pomalidomide (CC-4047) in combination with gemcitabine in metastatic pancreas cancer. European Journal of Cancer. 2011;47:199–205. doi: 10.1016/j.ejca.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 158.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Giovannetti E, Leon L, Bertini S, Macchia M, Minutolo F, Funel N, Alecci C, Giancola F, Danesi R, Peters G. Study of apoptosis induction and deoxycytidine kinase/cytidine deaminase modulation in the synergistic interaction of a novel ceramide analog and gemcitabine in pancreatic cancer cells. Nucleosides, Nucleotides and Nucleic Acids. 2010;29:419–426. doi: 10.1080/15257771003730193. [DOI] [PubMed] [Google Scholar]
- 160.Roman NO, Samulitis BK, Wisner L, Landowski TH, Dorr RT. Imexon enhances gemcitabine cytotoxicity by inhibition of ribonucleotide reductase. Cancer Chemotherapy and Pharmacology. 2011;67:183–192. doi: 10.1007/s00280-010-1306-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maurer T, Garrenton L, Oh A, Pitts K, Skelton N, Fauber B, Pan B, Malek S, Stokoe D, Bowman K, Wu J, Giannetti A, Starovasnik M, Mellman I, Jackson P, Rudolph J, Wang W, Fang G. Drugging the undruggable: small-molecule inhibition of the ras oncoprotein. Molecular Biology Cell. 2011;22 Abstract No. 24. [Google Scholar]
- 162.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature Medicine. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]