

Abstract
Trisomy 22 is a common trisomy in spontaneous abortions. In contrast, live-born trisomy 22 is rarely seen due to severe organ malformations associated with this condition. Here, we report on a male infant with complete, non-mosaic trisomy 22 born at 35 + 5 weeks via caesarean section. Peripheral blood lymphocytes and fibroblasts showed an additional chromosome 22 in all metaphases analyzed (47,XY,+22). In addition, array CGH confirmed complete trisomy 22. The patient's clinical features included dolichocephalus, hypertelorism, flattened nasal bridge, dysplastic ears with preauricular sinuses and tags, medial cleft palate, anal atresia, and coronary hypospadias with scrotum bipartitum. Essential treatment was implemented in close coordination with the parents. The child died 29 days after birth due to respiratory insufficiency and deterioration of renal function. Our patient's history complements other reports illustrating that children with complete trisomy 22 may survive until birth and beyond.
Key Words: Chromosomal abnormality, Live-born, Non-mosaic, Trisomy 22
Chromosomal abnormalities represent a major cause of spontaneous abortions [Hassold et al., 1980; Warburton et al., 1991]. Trisomy 22 has been identified as the third most common trisomy in spontaneous abortions, representing 11–16% of cases [Ford et al., 1996; Menasha et al., 2005]. Due to severe organ malformations (microcephaly/cranial abnormalities, congenital heart disease, renal malformations, intrauterine growth retardation (IUGR)), term or near-term pregnancies and postnatal survival of trisomy 22 children are very rare events. Among 23 children born with non-mosaic trisomy 22, Tinkle et al. [2003] found a median survival of only 4 days. We report a live-born infant with trisomy 22 surviving for 29 days.
Clinical Report
The male patient was born at 35 + 5 weeks by caesarean section as second child of a 44-year-old Caucasian female (gravida 3, para 2) and a 40-year-old Caucasian male. The parents were healthy and unrelated. They had a 5-year-old healthy daughter. During pregnancy, sonography revealed IUGR (fig. 1), dolichocephalus, single umbilical artery, absent right kidney, left renal hypoplasia, and hypospadias (fig. 2). Due to the parents’ beliefs they did not opt for additional diagnostic procedures and chose to continue the pregnancy.
Fig. 1.

Intrauterine growth of the 47,XY,+22 patient as determined by weekly sonographic biometry assessment.
Fig. 2.

Fetal sonography of the 47,XY,+22 patient at week 34 + 5 showing dolichocephaly (a), left renal hypoplasia (b) and hypospadias (c, arrow).
Birth weight was 1,630 g (<3rd percentile) and length was 41 cm (<3rd percentile). Apgar score was 5/6*/8* (* under CPAP (continuous positive airway pressure) ventilation). The head was dolichocephalic with pale facial skin, hypertelorism, flattened nasal bridge, and dysplastic ears with preauricular sinuses and tags (fig. 3). There was a medial cleft palate. Upper arms were rotated inwards. Anal atresia was present without fistula. External genitalia showed coronary hypospadias with scrotum bipartitum (fig. 3).
Fig. 3.

Phenotypic features of the 47,XY,+22 patient. a, b Dolichocephalus; b midface hypoplasia; b, c dysplastic ears with preauricular sinuses and tags; d external genitalia showing coronary hypospadias with scrotum bipartitum; anal atresia.
Chest X-ray revealed increased perihilar streaking. Echocardiography demonstrated persistent foramen ovale and patent ductus arteriosus (Botalli duct) with systolic-diastolic low flow shunt, enlarged right ventricle and pulmonary artery and aortic stenosis.
Magnetic resonance imaging (MRI) showed heart, liver and spleen location and size within normal limits. The right kidney was missing, the left kidney was hypoplastic (2 cm), the left ureter was markedly dilated. Cranial MRI confirmed medial cleft palate and revealed hypoplasia of the corpus callosum. Sonography confirmed absence of the right and hypoplasia of the left kidney, ureteral dilatation, and pronounced dilatation of the rectum owing to anal atresia. Newborn screening for inborn errors of metabolism was unremarkable.
Initially, due to moderate dyspnoea, a CPAP-device was implemented, but increasing CO2-retention required intubation. Anal atresia was treated via installation of a descendostoma. Postnatal cardiac function was not impaired. Due to renal agenesis and hypoplasia, renal function deteriorated soon after birth, leading to increased creatinine (max. 3.3 mg/dl) and urea (max. 135 mg/dl) within the first days of life. Furosemide treatment was started at day 5 but could not compensate for further deterioration of renal function, leading to anuria and formation of anasarca. Concomitantly, pleural effusions and respiratory insufficiency required repeated paracentesis. The aggravating clinical situation and possible interventions were discussed with the family who decided against further intensification of therapy. The child died 29 days after birth. According to the parents’ wishes, autopsy was not performed. His parents felt grateful for the time they were allowed to share with him.
Cytogenetic and Molecular Cytogenetic Analysis
Postnatally, chromosome analyses were performed on phytohaemagglutinin-stimulated peripheral blood lymphocytes as well as on fibroblasts from skin biopsy using conventional techniques. GTG- and RHG-banded chromosomes (resolution: 500 bands) showed an additional chromosome 22 in all 60 metaphases examined (47,XY,+ 22; fig. 4). FISH analysis using the probes MD DGCR (specific for 22q11.2) and SHANK3 (specific for 22q13) (Kreatech Diagnostics, Poseidon DNA Probes) confirmed trisomy 22 (fig. 5). To rule out possible additional genomic rearrangements, array CGH analysis was performed after obtaining consent from the parents. Genomic DNA was isolated from pieces of umbilical cord following the standard high salt-based rapid DNA isolation method [Aljanabi and Martinez, 1997]. Array CGH was performed using a CGX-12 cytogenetics array (Roche NimbleGen, Inc., Madison, Wisc., USA). This array platform contains 135K oligonucleotide probes covering the whole genome at an average resolution of 35 kb, as well as clinically significant regions at 10 kb. The labeling of both test and reference DNA and hybridization were performed according to the manufacturer's instructions. The array was scanned using the NimbleGen MS 200 scanner. The array data analyses were performed using Genoglyphix Software (Signature Genomics Laboratories, Spokane, Wash., USA) and probe sequence annotation was based on NCBI Build 36.1 (hg18) of the human genome.
Fig. 4.

RHG- (left) and GTG-banded (right) karyotype of the patient from lymphocyte (left) and fibroblast (right) culture.
Fig. 5.
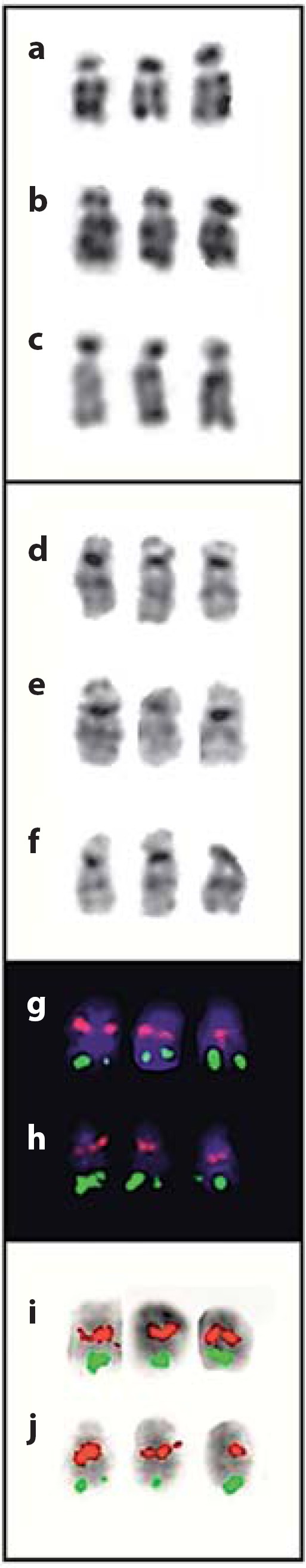
Selected examples of chromosomes 22 showing RHG-bands (a–c), GTG-bands (d–f), FISH with probes MD DGCR (red) and SHANK3 (green) (g–j). Chromosomes in g and h are stained with DAPI. Chromosomes in i and j are inverted DAPI images. Chromosomes were prepared from lymphocyte (a–c, g–j) and fibroblast (d–f) cultures.
The array CGH in the patient showed an elevation in the signal intensity of probes (logR ∼0.5) contiguously present along the long arm of chromosome 22 (22q11.21–22q13.33), indicating a copy gain, consistent with the cytogenetic observation of 3 copies of chromosome 22 (fig. 6). No further changes were marked that could be categorized as major gains or losses of chromosomal regions. Furthermore, a SNP array analysis was carried out to validate the duplication of chromosome 22 through its B allele frequency profile (data not shown).
Fig. 6.

a Array-CGH profile of chromosome 22 showing whole chromosome duplication. The y-axis represents the log-ratio of the test/reference DNA intensities and the x-axis displays the genomic profile of chromosome 22. Note that the signal intensities of probes across the chromosome 22 are ≥0.5, signifying a gain of chromosome. b Whole genome array CGH profile revealing copy number changes for each chromosome. Besides the contiguously present copy gain of chromosome 22, copy number changes which are reasonably small (<200 kb) or known as benign variants are also found for other chromosomes.
Discussion
Analyzing the incidence and spectrum of chromosome abnormalities in 2,180 spontaneous abortions, Menasha et al. [2005] confirmed trisomy 16 as the most common trisomy in spontaneous abortions (18% of all trisomies), followed closely by trisomy 21 and trisomy 22 (16% of trisomies and ∼5% of all spontaneous abortions, respectively).
Prior to the era of molecular cytogenetics, Schinzel [1981] questioned whether full, non-mosaic trisomy 22 would be compatible with intrauterine survival up to term. He suspected that many cases published by that time might rather represent partial trisomies or unbalanced 11;22 translocations. Indeed, a number of published karyotypes depict one of the 3 putative chromosomes 22 to be smaller than the other two, drawing the diagnosis of complete trisomy 22 into question. Since then, additional techniques (FISH, array technologies) demonstrated that full, non-mosaic trisomy 22 can be compatible with late gestational age, including survival until birth and even beyond. Karyotyping in our case did in fact first raise the suspicion that one of the 3 chromosomes 22 could be incomplete (fig. 4; right karyotype). Yet, SNP array and array CGH analysis confirmed that, regardless of the subtle size difference, all 3 chromosomes 22 were complete (fig. 6).
The majority of trisomy 22 errors (>96%) occur during oogenesis, predominantly during the first meiotic division [Hall et al., 2007]. Our family declined investigations aiming at determination of the parental origin of the additional chromosome 22. In addition to free trisomy 22, the literature provides examples of putative isochromosomes and translocations involving chromosome 22 [Voiculescu et al., 1987; Manasse et al., 2000]. Presence of an isochromosome 22 has been described for a trisomy 22 mosaic child [Guze et al., 2004]. An early report by Lalchev et al. [1978] on a possible 21;22 Robertsonian translocation has been considered as ‘not convincing in [its] cytogenetic documentation’ by Schinzel [1981]. According to Schinzel [1981, 2001], lack of trisomy 22 offspring in families with Robertsonian translocations involving chromosome 22 argues against viability of full trisomy 22. Statistically, however, the number of such families is relatively small, so rare exceptions may still be possible.
Today, the phenotype of children with trisomy 22 appears well-defined, as reviewed by several authors [Bacino et al., 1995; Crowe et al., 1997; Tinkle et al., 2003]. Common features include midface hypoplasia with flat/broad nasal bridge, dysplastic ears with pits, tags, or poruses, cleft palate, hypertelorism, microcephaly/cranial abnormalities, congenital heart disease, genital abnormalities, and IUGR (table 1). Additional frequent findings are single umbilical artery, micrognathia, abnormal anus/anal atresia, and malformed kidneys. In individual cases, thyroid isthmus agenesis and absent gall bladder (fetocide at 27 weeks gestation [Gangbo et al., 2004]), distinct histopathological features of the temporal bone [Miura et al., 2000; Ohtani et al., 2001], agenesis of the ductus venosus [Barseghyan et al., 2009] or overlapping phenotypical features with disorders such as Fryns syndrome [Ladonne et al., 1996] or Goldenhar sequence [Kobrynski et al., 1993] have been described. An exceptional report concerns a stillborn child (birth induced at 39 weeks gestation) with alleged trisomy 22 in fetal blood cells (cordocentesis) and skin culture with prenatal IUGR but surprising lack of developmental defects [Morrison et al., 1998].
Table 1.
Synopsis of clinical findings in live-born non-mosaic trisomy 22 patients
| Reference | Survival | Sex | IUGR | Cranial anomalies | Dysplastic ears | Hypertelorism | Epicanthal fold | High-arched/cleft palate | Flat/broad nasal bridge | Micrognathia | Short neck | Cardiac anomalies | Renal anomalies | Genital anomalies | Abnormal anus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Petersen et al. [1987] | 10 weeks | m | – | + | + | – | + | + | + | + | + | ||||
| Vohra et al. [1987] | 22 hours | m | – | + | + | + | + | + | + | + | – | + | + | – | |
| Voiculesco et al. [1987] | 10 min | f | + | + | + | + | + | – | + | + | + | + | + | + | + |
| Kukolich et al. [1989] | 3 years | m | + | + | + | + | + | + | + | + | − | + | + | – | |
| Antle et al. [1990] | 2 days | f | + | + | + | + | + | + | + | ||||||
| Antle et al. [1990] | 2 days | f | + | + | + | + | + | + | + | + | + | ||||
| McPherson and Stetka [1990] | 33 days | m | + | + | + | + | + | + | + | + | + | + | + | + | |
| Phillipson et al. [1990] | 20 hours | m | + | + | + | + | + | + | + | + | + | + | + | ||
| Phillipson et al. [1990] | 4 hours | m | + | + | + | + | + | + | + | + | + | + | + | ||
| Sundareshan et al. [1990] | 4 days | f | + | + | + | + | + | + | + | + | + | – | + | ||
| Feretet al. [1991] | 40 hours | f | + | + | + | + | + | + | + | + | + | ||||
| Kim et al. [1992] | 12 hours | m | + | + | + | + | + | + | + | + | |||||
| Lean et al. [1992] | 2 months | f | – | + | + | + | + | + | – | + | – | + | |||
| Kobrynski et al. [1993] | 11 hours | f | + | + | + | + | + | + | + | + | + | + | – | ||
| Slater et al. [1993] | 6 days | + | + | + | + | + | + | + | + | + | + | ||||
| Stratton et al. [1993] | 4 months | f | + | + | + | + | + | + | + | + | + | + | – | – | – |
| Golombek and Shaw [1994] | 65 min | m | – | + | + | + | + | + | – | ||||||
| Fahmi et al. [1994] | 4 months | f | – | + | + | + | + | + | + | + | + | + | + | + | |
| Nicholl et al. [1994] | 3 months | m | + | + | + | + | + | + | + | – | + | ||||
| Bacino et al. [1995] | 2 months | f | + | + | + | + | + | – | + | + | + | + | – | – | |
| Ladonne et al. [1996] | few min | f | + | + | + | + | + | + | + | + | + | + | |||
| Manasse et al. [2000] | 18 months | f | + | + | + | + | + | + | + | + | + | ||||
| Tinkle et al. [2003] | 11 weeks | f | + | + | + | + | + | + | + | + | + | + | + | – | – |
| Miura et al. [2000] | 3.5 months | f | + | + | + | + | + | + | + | + | + | + | |||
| Rao et al. [2003] | alive at 3 years | m | – | + | + | + | + | + | – | – | |||||
| Stressig et al. [2005] | 1 day | f | + | + | + | + | + | ||||||||
| Mihci et al. [2007] | 1 day | f | + | + | + | + | + | + | high/large | + | + | – | + | – | |
| Fruhman et al. [2011]a | 3 days | f | + | + | + | + | – | + | + | + | + | – | – | ||
| This case [2012] | 29 days | m | + | + | + | + | + | + | + | + | + | + | + | + | + |
Following Tinkle et al. [2003], entries are sorted by publication year and include the 6 most recently published cases (italics). Further patient details are given in the original publications and in the synopses provided by, e.g., Tinkle et al. [2003] or Crowe et al. [1997]. + = Present; – = absent; no symbol = unknown.
a The karyotype of this patient was 47,XX,inv(9)(pllql3),+22, considered as ‘trisomy 22 and a pericentric inversion of one chromosome 9, the latter being a normal variant’.
Several reports deal with prenatal sonographic findings [Sepulveda et al., 2003; Stressig et al., 2005; Sifakis et al., 2008; Schwendemann et al., 2009] and maternal serum biochemical markers in trisomy 22 pregnancies [Sifakis et al., 2008]. Schwendemann's report also mentions a live-born child with trisomy 22, but no further clinical details are given. Sonographically identified anomalies include IUGR (cf. fig. 1), limb hypoplasia, craniofacial malformations, cardiac anomalies and increased nuchal translucency/cystic hygroma. The plethora of features observed might in part be due to different screening times. In addition, karyotype analysis based on chorionic villus sampling carries the well-known risk of missing low level mosaicism, such as proven by follow-up amniocentesis in some of the recently reported cases [Wolstenholme et al., 2001; Sifakis et al., 2008]. Differentiation between mosaic and non-mosaic trisomy 22 is important, particularly with respect to counseling issues regarding life-expectancy and possible complications which are usually milder in mosaic cases [Crowe et al., 1997; Leclercq et al., 2010].
As illustrated in figure 7, survival of live-born children with non-mosaic trisomy 22 is severely limited, ranging from hours post partum to a maximum of 3 years (observed in 2 cases) [Kukolich et al., 1989; Rao et al., 2003]. Median survival of the 30 cases summarized in figure 7 was 3.5 days. Common causes of death are respiratory/cardiorespiratory failure and infection [Tinkle et al., 2003].
Fig. 7.

Survival times of live-born children with trisomy 22. Data are taken from the references given in the figure. References in italics denote cases that were mainly reported after 2003 and therefore are not included in the review by Tinkle [Tinkle et al., 2003]. Inset: Data for children with trisomy 22 living no longer than 48 h (enlarged time scale).
In summary, the vast majority of trisomy 22 zygotes end up as spontaneous abortions. Only a minority of fetuses survive until term. Medium postpartum survival amounts to 3–4 days, and maximum survival reported is 3 years. There is a rather consistent pattern of IUGR combined with multiple and severe malformations. The question why a small proportion of trisomy 22 fetuses survive until late gestation or even beyond birth remains unsolved. Undetected confined placental mosaicism [Robinson and Kalousek, 1996] or mosaicism in fetal tissues other than blood or skin tested might explain some of the surviving cases. Even reports proving trisomy 22 not only in blood cells or fibroblasts but also in cytotrophoblast cells [Bacino et al., 1995; Hengstschlager et al., 2001] do not exclude the possibility of undetected somatic mosaicism. Most cases of trisomy 22 are of maternal origin, but as to survival the parental origin of the extra chromosome 22 does not seem to play a crucial role since there have been examples for both maternal [Petersen et al., 1987; Mihci et al., 2007] and paternal origin [Feret et al., 1991].
References
- Aljanabi SM, Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997;25:4692–4693. doi: 10.1093/nar/25.22.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antle CM, Pantzar JT, White VA. The ocular pathology of trisomy 22: report of two cases and review. J Pediatr Ophthalmol Strabismus. 1990;27:310–314. doi: 10.3928/0191-3913-19901101-09. [DOI] [PubMed] [Google Scholar]
- Bacino CA, Schreck R, Fischel-Ghodsian N, Pepkowitz S, Prezant TR, et al. Clinical and molecular studies in full trisomy 22: further delineation of the phenotype and review of the literature. Am J Med Genet. 1995;56:359–365. doi: 10.1002/ajmg.1320560404. [DOI] [PubMed] [Google Scholar]
- Barseghyan K, Sklansky MS, Paquette LB, Randolph LM, Miller DA. Agenesis of the ductus venosus in a fetus with nonmosaic trisomy 22. Prenat Diagn. 2009;29:901–902. doi: 10.1002/pd.2309. [DOI] [PubMed] [Google Scholar]
- Crowe CA, Schwartz S, Black CJ, Jaswaney V. Mosaic trisomy 22: a case presentation and literature review of trisomy 22 phenotypes. Am J Med Genet. 1997;71:406–413. [PubMed] [Google Scholar]
- Fahmi F, Schmerler S, Hutcheon RG. Hydrocephalus in an infant with trisomy 22. J Med Genet. 1994;31:141–144. doi: 10.1136/jmg.31.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feret MA, Galan F, Aguilar MS, Serrano JL, Cidras M, et al. Full trisomy 22 in a malformed newborn female. Ann Genet. 1991;34:44–46. [PubMed] [Google Scholar]
- Ford JH, Wilkin HZ, Thomas P, McCarthy C. A 13-year cytogenetic study of spontaneous abortion: clinical applications of testing. Aust N Z J Obstet Gynaecol. 1996;36:314–318. doi: 10.1111/j.1479-828x.1996.tb02719.x. [DOI] [PubMed] [Google Scholar]
- Fruhman G, El-Hattab AW, Belmont JW, Patel A, Cheung SW, et al. Suspected trisomy 22: modification, clarification, or confirmation of the diagnosis by aCGH. Am J Med Genet. 2011;155:434–438. doi: 10.1002/ajmg.a.33792. [DOI] [PubMed] [Google Scholar]
- Gangbo E, Lacombe D, Alberti EM, Taine L, Saura R, et al. Trisomy 22 with thyroid isthmus agenesis and absent gall bladder. Genet Couns. 2004;15:311–315. [PubMed] [Google Scholar]
- Golombek S, Shaw R. Trisomy 22 in an Iowa newborn. Iowa Med. 1994;84:31–33. [PubMed] [Google Scholar]
- Guze C, Qin N, Kelly J, Yang X, Bruni R, et al. Isochromosome 22 in trisomy 22 mosaic with five cell lines. Am J Med Genet A. 2004;124:79–84. doi: 10.1002/ajmg.a.20365. [DOI] [PubMed] [Google Scholar]
- Hall HE, Surti U, Hoffner L, Shirley S, Feingold E, et al. The origin of trisomy 22: evidence for acrocentric chromosome-specific patterns of nondisjunction. Am J Med Genet A. 2007;143A:2249–2255. doi: 10.1002/ajmg.a.31918. [DOI] [PubMed] [Google Scholar]
- Hassold T, Chen N, Funkhouser J, Jooss T, Manuel B, et al. A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet. 1980;44:151–178. doi: 10.1111/j.1469-1809.1980.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Hengstschlager M, Bettelheim D, Rosner M, Repa C, Deutinger J, et al. Extended prenatal survival of a non-mosaic trisomy 22 with aneuploid cytotrophoblasts. Prenat Diagn. 2001;21:897–899. doi: 10.1002/pd.154. [DOI] [PubMed] [Google Scholar]
- Kim EH, Cohen RS, Ramachandran P, Mineta AK, Babu VR. Trisomy 22 with congenital diaphragmatic hernia and absence of corpus callosum in a liveborn premature infant. Am J Med Genet. 1992;44:437–438. doi: 10.1002/ajmg.1320440410. [DOI] [PubMed] [Google Scholar]
- Kobrynski L, Chitayat D, Zahed L, McGregor D, Rochon L, et al. Trisomy 22 and facioauriculovertebral (Goldenhar) sequence. Am J Med Genet. 1993;46:68–71. doi: 10.1002/ajmg.1320460111. [DOI] [PubMed] [Google Scholar]
- Kukolich MK, Kulharya A, Jalal SM, Drummond-Borg M. Trisomy 22: no longer an enigma. Am J Med Genet. 1989;34:541–544. doi: 10.1002/ajmg.1320340417. [DOI] [PubMed] [Google Scholar]
- Ladonne J, Gaillard D, Carré-Pigeon F, Gabriel R. Fryns syndrome phenotype and trisomy 22. Am J Med Genet. 1996;61:68–70. doi: 10.1002/(SICI)1096-8628(19960102)61:1<68::AID-AJMG13>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lalchev S, Tzancheva M, Markova R. A case of trisomy 22 with a probable Robertsonian translocation 21/22. Hum Genet. 1978;45:219–223. doi: 10.1007/BF00286967. [DOI] [PubMed] [Google Scholar]
- Lean SF, Lin SP, Shen EY, Ho MY, Yang SY. Liveborn trisomy 22: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1992;33:226–230. [PubMed] [Google Scholar]
- Leclercq S, Baron X, Jacquemont ML, Cuillier F, Cartault F. Mosaic trisomy 22: five new cases with variable outcomes. Implications for genetic counselling and clinical management. Prenat Diagn. 2010;30:168–172. doi: 10.1002/pd.2427. [DOI] [PubMed] [Google Scholar]
- Manasse BF, Pfaffenzeller WM, Gurtunca N, de Ravel TJ. Possible isochromosome 22 leading to trisomy 22. Am J Med Genet. 2000;95:411–414. doi: 10.1002/1096-8628(20001218)95:5<411::aid-ajmg1>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- McPherson E, Stetka DG. Trisomy 22 in a liveborn infant with multiple congenital anomalies. Am J Med Genet. 1990;36:11–14. doi: 10.1002/ajmg.1320360104. [DOI] [PubMed] [Google Scholar]
- Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Genet Med. 2005;7:251–263. doi: 10.1097/01.gim.0000160075.96707.04. [DOI] [PubMed] [Google Scholar]
- Mihci E, Tacoy S, Yakut S, Ongun H, Keser I, et al. Maternal origin and clinical findings in a case with trisomy 22. Turk J Pediatr. 2007;49:322–326. [PubMed] [Google Scholar]
- Miura M, Sando I, Haginomori S, Casselbrant ML. Histopathological study on temporal bone and eustachian tube in trisomy 22. Int J Pediatr Otorhinolaryngol. 2000;56:191–198. doi: 10.1016/s0165-5876(00)00433-x. [DOI] [PubMed] [Google Scholar]
- Morrison JJ, Hastings R, Jauniaux E. Trisomy 22: a cause of isolated fetal growth restriction. Ultrasound Obstet Gynecol. 1998;11:295–297. doi: 10.1046/j.1469-0705.1998.11040295.x. [DOI] [PubMed] [Google Scholar]
- Nicholl RM, Grimsley L, Butler L, Palmer RW, Rees HC, et al. Trisomy 22 and intersex. Arch Dis Child Fetal Neonatal Ed. 1994;71:F57–58. doi: 10.1136/fn.71.1.f57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani I, Kano M, Sagawa Y, Ogawa H, Suzuki C. Temporal bone histopathology in trisomy 22. Int J Pediatr Otorhinolaryngol. 2001;59:137–141. doi: 10.1016/s0165-5876(01)00459-1. [DOI] [PubMed] [Google Scholar]
- Petersen MB, Hansen M, Djernes BW. Full trisomy 22 in a newborn infant. Ann Genet. 1987;30:101–104. [PubMed] [Google Scholar]
- Phillipson J, Benirschke K, Bogart M. Two live-born infants with trisomy 22. Pediatr Pathol. 1990;10:1001–1005. doi: 10.3109/15513819009064734. [DOI] [PubMed] [Google Scholar]
- Rao VB, Seema K, Lily K, Ghosh K, Mohanty D. Trisomy 22 with unusual phenotype. Indian Pediatr. 2003;40:371–372. [PubMed] [Google Scholar]
- Robinson WP, Kalousek DK. Mosaicism most likely accounts for extended survival of trisomy 22. Am J Med Genet. 1996;62:100–101. doi: 10.1002/(SICI)1096-8628(19960301)62:1<100::AID-AJMG21>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Schinzel A. Incomplete trisomy 22. III. Mosaic-trisomy 22 and the problem of full trisomy 22. Hum Genet. 1981;56:269–273. doi: 10.1007/BF00274677. [DOI] [PubMed] [Google Scholar]
- Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. ed 2. Berlin: Walter de Gruyter; 2001. [Google Scholar]
- Schwendemann WD, Contag SA, Koty PP, Miller RC, Devers P, et al. Ultrasound findings in trisomy 22. Am J Perinatol. 2009;26:135–137. doi: 10.1055/s-0028-1091399. [DOI] [PubMed] [Google Scholar]
- Sepulveda W, Be C, Schnapp C, Roy M, Wimalasundera R. Second-trimester sonographic findings in trisomy 22: report of 3 cases and review of the literature. J Ultrasound Med. 2003;22:1271–1275. doi: 10.7863/jum.2003.22.11.1271. [DOI] [PubMed] [Google Scholar]
- Sifakis S, Karkaletsi M, Christopoulou S, Donoghue J, Kaminopetros P, et al. Distinctive pattern of first trimester maternal serum biochemical markers in trisomy 22 pregnancies. Prenat Diagn. 2008;28:1174–1176. doi: 10.1002/pd.2134. [DOI] [PubMed] [Google Scholar]
- Slater HR, Voullaire LE, Vaux CE, Bankier A, Pertile M, et al. Confirmation of trisomy 22 in two cases using chromosome painting: comparison with t(11;22) Am J Med Genet. 1993;46:434–437. doi: 10.1002/ajmg.1320460417. [DOI] [PubMed] [Google Scholar]
- Stratton RF, DuPont BR, Mattern VL, Young RS, McCourt JW, et al. Trisomy 22 confirmed by fluorescent in situ hybridization. Am J Med Genet. 1993;46:109–112. doi: 10.1002/ajmg.1320460119. [DOI] [PubMed] [Google Scholar]
- Stressig R, Kortge-Jung S, Hickmann G, Kozlowski P. Prenatal sonographic findings in trisomy 22: five case reports and review of the literature. J Ultrasound Med. 2005;24:1547–1553. doi: 10.7863/jum.2005.24.11.1547. [DOI] [PubMed] [Google Scholar]
- Sundareshan TS, Naguib KK, Al-Awadi SA, Redha MA, Hamoud MS. Apparently nonmosaic trisomy 22: clinical report and review. Am J Med Genet. 1990;36:7–10. doi: 10.1002/ajmg.1320360103. [DOI] [PubMed] [Google Scholar]
- Tinkle BT, Walker ME, Blough-Pfau RI, Saal HM, Hopkin RJ. Unexpected survival in a case of prenatally diagnosed non-mosaic trisomy 22: clinical report and review of the natural history. Am J Med Genet A. 2003;118A:90–95. doi: 10.1002/ajmg.a.10216. [DOI] [PubMed] [Google Scholar]
- Vohra K, Verma RS, Concepcion L. Trisomy 22: report of a patient diagnosed as a neonate. Dis Markers. 1987;5:13–18. [PubMed] [Google Scholar]
- Voiculescu I, Back E, Duncan AM, Schwaibold H, Schempp W. Trisomy 22 in a newborn with multiple malformations. Hum Genet. 1987;76:298–301. doi: 10.1007/BF00283629. [DOI] [PubMed] [Google Scholar]
- Warburton D, Byrne JM, Canki N. Oxford Monographs on Medical Genetics. vol 21. New York: Oxford University Press; 1991. Chromosome Anomalies and Prenatal Development. An Atlas. [Google Scholar]
- Wolstenholme J, Webb A, English C, Evans J. Prenatal diagnosis of chromosome 22 mosaicism with extensive investigation of placental and fetal tissues. Abstracts of the 3rd European Cytogenetics Conference. Paris, France, July 7–10, 2001. Ann Genet 44 Suppl. 2001;1:s142. [Google Scholar]