

Abstract
Carpenter syndrome is caused by mutations in the RAB23 gene that encodes a small GTPase of the Rab subfamily of proteins. Rab proteins are known to be involved in the regulation of cellular trafficking and signal transduction. Currently, only few mutations in RAB23 have been reported in patients with Carpenter syndrome. In this paper, we report the clinical features, molecular and functional analysis of 2 children from an Emirati consanguineous family with this syndrome. The affected children exhibit the typical features including craniosynostosis, typical facial appearance, polysyndactyly, and obesity. Molecular analysis of the RAB23 gene revealed a homozygous mutation affecting the first nucleotide of the acceptor splice site of exon 5 (c.482-1G>A). This mutation affects the authentic mRNA splicing and activates a cryptic acceptor site within exon 5. Thus, the erroneous splicing results in an eight nucleotide deletion, followed by a frameshift and premature termination codon at position 161 (p.V161fsX3). Due to the loss of the C-terminally prenylatable cysteine residue, the truncated protein will probably fail to associate with the target cellular membranes due to the absence of the necessary lipid modification. The p.V161fsX3 extends the spectrum of RAB23 mutations and points to the crucial role of prenylation in the pathogenesis of Carpenter syndrome within this family.
Key Words: Carpenter syndrome, GTPase, Mutation, Prenylation, RAB23
Carpenter syndrome (MIM 201000) is a rare autosomal recessive pleiotropic disorder characterized by combination of acrocephaly, brachydactyly and syndactyly in the hands as well as preaxial polydactyly and syndactyly of the toes. Other variable features include obesity, mental retardation and hypogonadism [Carpenter, 1909; Temtamy, 1966; Robinson et al., 1985; Cohen et al., 1987]. It has an incidence of 1 in a million live births (http://rarediseases.about.com/od/acps/a/carpenter.htm?). The most common and evident physical manifestation at birth is acrocephaly due to early fusion of the cranial sutures. Facial abnormalities include flat nasal bridge, broad cheeks and malformed and unevenly set ears. In addition, it includes a wide upturned nose with abnormally large nares and usually late eruption of small and widely spaced teeth. Mild mental retardation is present in 75% of affected children, and roughly half of them are born with some types of heart defects [Frias et al., 1978; Perlyn and Marsh, 2008] (http://www.rarediseases.org/rare-disease-information/rare-diseases/byID/612/viewAbstract).
Using homozygosity mapping, Jenkins et al. [2007] mapped the gene for this syndrome to chromosome 6p12.1q12 and identified several mutations in the RAB23 gene. This gene consists of 1 noncoding and 6 coding exons, spanning a region of 35.43 kb, which encodes a small GTPase protein belonging to the Ras superfamily. This protein plays an essential regulator role in the sonic hedgehog signaling pathway and vesicular trafficking [Jenkins et al., 2007]. Rab23 has been identified earlier to act as a negative regulator of sonic hedgehog signaling [Eggenschwiler et al., 2006]. More recently, Boehlke et al. [2010] showed that Rab23 is involved in the protein turnover within the cilium by increasing the recycling of Smo, a downstream effector smoothened, in the sonic hedgehog signaling pathway.
To date, 11 mutations have been reported in the RAB23 gene responsible for Carpenter syndrome [Jenkins et al., 2007; Alessandri et al., 2010; Jenkins et al., 2011]. In the present study, we report the identification of a novel splicing mutation (c.482-1G>A) in the RAB23 gene causing Carpenter syndrome in a consanguineous Emirati family. This mutation abolished the acceptor splice site of exon 5, which led to an eight nucleotide deletion in the RAB23 mRNA followed by a stop codon.
Subjects and Methods
Subjects
Blood samples were collected from the 2 affected children, parents and one unaffected sibling.
Mutation Screening
To identify the mutation(s) causing this syndrome, PCR amplification of the 6 coding exons of the RAB23 gene were performed on 2720 thermal cycler (Applied Biosystems, USA). Primers were designed using Primer3 software version 0.4.0 (http://frodo.wi.mit.edu/) (online suppl. table 1, for all supplementary material see www.karger.com/doi/10.1159/000345653). A total volume of 20 μl of PCR reactions were prepared containing 1× PCR buffers (Qiagen Gmbh, Germany), 0.2 mM dNTPs, 5 μM of each forward and reverse primers, 100 ng of template DNA, and 0.5 U Taq DNA polymerase (Qiagen Gmbh). The PCR products were purified using ExoSAP-IT (USB Inc.) followed by DNA Sanger cycle sequencing using the BigDye Terminator kit v3.1 (Applied Biosystems) and were run on the 3130xl Genetic Analyzer System (Applied Biosystems). The results were analyzed using Sequencing Analysis® 5.3 software (Applied Biosystems).
In silico Prediction of the Cryptic Splice Site Mutation c.482-1G>A
To evaluate the potential influence of c.482-1G>A mutation on splicing signals, in silico prediction was carried out using the scan program (https://splice.uwo.ca/) [Schneider, 1997a, b] along with the Human Splicing Finder software version 2.4.1 (http://www.umd.be/HSF/) [Desmet et al., 2009]. The prediction was performed based on the following reference sequence: ENST00000317483 transcript.
Effect of c.482-1G>A Mutation on mRNA Splicing
To elucidate the effect of this mutation, total RNA was extracted from blood using QiAamp RNA isolation Mini kit (Qiagen Gmbh). The cDNA was prepared by reverse transcription (RT-PCR) using Omni Script RT kit (Qiagen Gmbh) according to the manufacturer's instructions. A PCR amplification of RAB23 cDNA was performed in patient IV-2 and control samples. PCR products were purified, followed by Sanger cycle sequencing reactions and screened by 3130xl automated sequencer (Applied Biosystems).
Relative Quantification of RAB23 mRNA Transcripts
The expression levels of RAB23 mRNA were analyzed in one affected (IV-2), carrier (III-1) and healthy control samples using TaqMan assays using the 7500 Real Time PCR system (Applied Biosystems). GAPDH was used as an internal control, and all experiments were run in duplicates. A 270 bp RAB23 product, spanning exon 1 and exon 3 (primers listed in online suppl. table 1), was amplified and quantified in a total volume of 25 μl containing 12.5 μl of 2X TaqMan gene expression Master Mix (Applied Biosystems), 0.5 μl of each forward and reverse primers, 0.625 μl of fluorescent probe, and 2 μl of the cDNA samples. The amplification condition is as following: 10 min activation at 95°C, followed by 40 cycles amplification for 95°C for 45 s and 57°C for 45 s. Data analysis was performed using 7500 System Software (Applied Biosystems).
Ethics Statement
This study has been approved by Al-Ain Medical Human Research Ethics Committee according to the national regulations (protocol number 10/09). The parents of the patients provided an informed written consent form prior to research, publication and agreed to use the photographs of their children for medical publication.
Results
Clinical Description
The parents are first cousins of United Arab Emirates origin and have 4 children (fig. 1). Two of the children are affected by Carpenter syndrome.
Fig. 1.
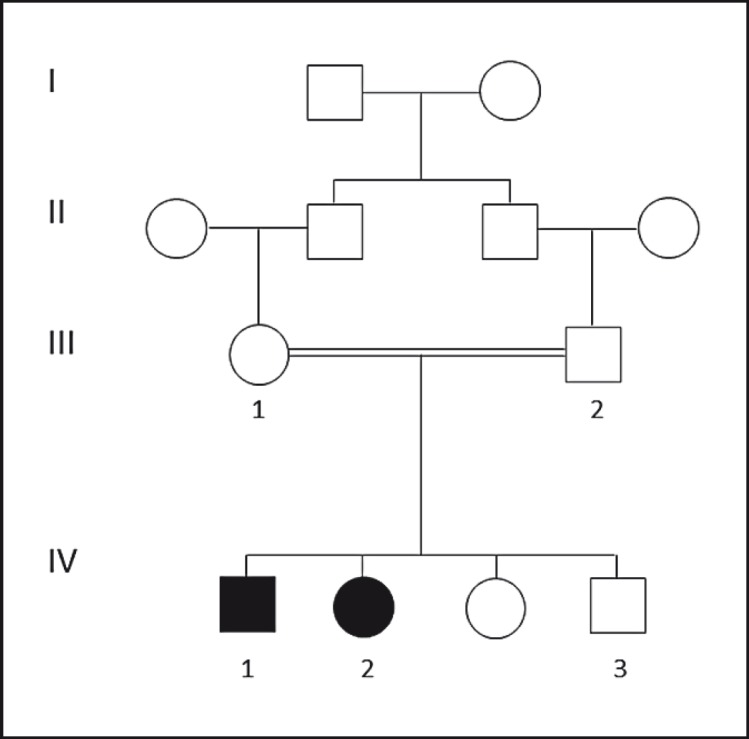
Pedigree of Emirati family with Carpenter syndrome. Pedigree shows the Emirati family with 2 patients with Carpenter syndrome. Circles and squares denote females and males, respectively; filled symbols represent affected members; double lines denote consanguineous marriage. Arabic numbers represent tested individuals; Roman numbers indicate the first generation until their offspring.
The first child (IV-1) was the product of normal pregnancy. Prenatal ultrasound showed abnormal head shape, and, therefore, the baby was delivered by low cesarean section at 38 weeks. His birth weight was 4,080 g (>90%), length 48 cm (<50%) and head circumference 37 cm (>90%). The Apgar scores were 8 and 9 at 1 and 5 min, respectively. Clinical examination revealed acrocephaly with closed anterior fontanelle, upward slanting of the palpebral fissures, prominent eyes, and depressed nasal bridge with low set ears. The boy also had brachydactyly with contractures of the proximal interphalangeal joints and complete cutaneous syndactyly between 3rd and 4th fingers bilaterally (fig. 2A). There was preaxial polydactyly of both feet and syndactyly of all the other toes. There was bilateral metatarsus varus (fig. 2B). Cranial CT scan confirmed the presence of synostosis of all sutures. There was dilatation of the lateral ventricles. Ophthalmological examination was normal. Craniosynostosis was operated on successfully. This child is 7 years old now and has mild developmental delay. He is attending school and is at the appropriate grade level; however, he needs extra help.
Fig. 2.

Clinical features of 2 Emirati patients with Carpenter syndrome. A Appearance of patient IV-1 at birth. Note acrocephaly, upslanting palpebral fissures, wide nasal bridge and tip, and polysyndactyly of the toes. B Appearance of the hand at birth in patient IV-1 showing syndactyly of the fingers. C Appearance of patient IV-2 at 6 years. The craniosynostosis has been operated on. Note prominent metopic ridge, widely spaced eyes with epicanthic folds and depressed nasal bridge. D Appearance of the hand in patient IV-2. Note brachydactyly and scar of the operation for syndactyly.
The second child (IV-2) was a girl of normal pregnancy. Prenatal ultrasound at the third trimester showed immobile fingers and abnormal skull shape. Delivery was by low cesarean section. Her birth weight was 3,375 g (50%), length 45 cm (<3%) and head circumference 36 cm (90%). At birth, she was noted to have clover-leaf appearance of the cranium with all sutures; the anterior and posterior fontanelles were closed. The eyes were prominent, furthermore there was a depressed nasal bridge, high arched palate and low set ears (fig. 2C). She also had cutaneous syndactyly and brachydactyly of all fingers of both hands with limited extension of the proximal interphalangeal joints of all fingers (fig. 2D) as well as bilateral preaxial polydactyl of feet and cutaneous syndactyly of all toes. Cranial CT scan showed closure of all sutures, with absence of bone structure in the temporal-parietal region on both sides. Moreover, the affected child also had dilatation of the lateral ventricles which required a ventriculo-peritoneal shunt. Hand X-rays showed absence of the middle phalanges of all the fingers. Craniosynostosis was operated on successfully at the age of 3 months. She is 6 years old now and has mild delay in speech. She is attending school and she is in the appropriate grade level.
Identification of G to A Substitution in the First Nucleotide of the Acceptor Splicing Site of Exon 5
DNA sequencing of RAB23 identified a homozygous c.482-1G>A substitution affecting the first nucleotide of the acceptor splicing site of exon 5 (fig. 3A). This novel mutation segregated well with the phenotype in the studied family (fig. 3B). Only the affected individuals (IV-1 and IV-2) were homozygous for this mutation, while the parents (III-1 and III-2) and the child VI-3 were carriers for this mutation.
Fig. 3.

Schematic representation of RAB23 and molecular analysis of c.482-1G>A on splicing and protein translation. A Top: genomic organization of exons and introns of RAB23. Middle: wild-type pre-mRNA representation and functional domain in the 237 amino acid protein. Bottom: the truncated pre-mRNA and mutant protein. B Chromatograms of healthy control, homozygous and heterozygous individuals, respectively. Chromatograms show a novel substitution of G to A in the acceptor splice site of exon 5 (c.482-1G>A) depicted by a small arrow. The beginning of exon 5 is underlined. C Chromatograms of cDNA sequence analysis from patient (IV-2) and healthy control. The substitution alters the 3′ consensus splice site and activates a cryptic acceptor splice site downstream in exon 5 at position c.490. This results in the deletion of 8 nucleotides from the beginning of exon 5, causing a frameshift followed by a premature termination codon. X = Represents the premature termination codon. Nucleotide sequences of exon 4 and 5 are underlined.
Computational Analysis of the c.482-1G>A Effect on Splicing
Several studies have shed light on the role of the splice site variants that are disease causing. Mutations affecting the highly conserved sequences of the acceptor and donor splice sites were expected to cause splicing aberrations [Burset et al., 2000]. Both computational studies used in the present study predicted the loss of the canonical acceptor splice site and the activation of cryptic splice site 8 nucleotides upstream of exon 5. The c.482-1G>A mutation is expected to cause the deletion of TTTTTAAG at the beginning of exon 5, leading to a frameshift and thus, creating a stop codon (online suppl. fig. 1 and suppl. table 2).
Sanger Sequencing of the RAB23 cDNA
Bidirectional sequencing of RAB23 transcripts were performed in one of the patients (IV-2) and healthy control samples. Analysis of the chromatograms, as shown in figure 3C, confirmed the presence of the deletion of 8 nucleotides (TTTTTAAG) at the beginning of exon 5. The out-of-frame sequences would allow the translation of only 3 amino acids (p.V161fsX3) in exon 5 before encountering a stop codon (fig. 3).
Quantitative Expression of RAB23 Transcripts
Relative quantification of RAB23 mRNA was carried out in cDNA samples from one affected (IV-2), one carrier (III-1) and a healthy individual using qReal-Time PCR. Data analysis showed no significant difference in the level of expression of RAB23 mRNA between the 3 studied samples normalized to an internal control GAPDH (online suppl. fig. 2).
Discussion
We report here the identification of a novel mutation in the RAB23 gene in a consanguineous family from the United Arab Emirates. The 2 affected siblings had the typical features of Carpenter syndrome. There was no intrafamilial phenotypic variability in this family. Both had craniosynostosis leading to acrocephaly with clover-leaf shape of the skull in one of them. There was brachydactyly with absence of the middle phalanges and syndactyly of the hands, with preaxial polydactyly and syndactyly of the toes in both children. Both had high birth weight, developed obesity in childhood and had mild developmental delay. Carpenter syndrome is known to be clinically heterogeneous, in particular, the severity of craniosynostosis, mental retardation and obesity with high birth weight. However, molecular studies have shown that at the molecular level it is a homogenous condition [Jenkins et al., 2007, 2011; Alessandri et al., 2010].
All Rab proteins display a similar distribution within their functional domains. Rab23 proteins encompasses 4 GTP/GDP pockets, 2 ‘switch’ domains interacting with the Rab effector proteins, which experience conformational changes based on the presence of either GDP or GTP. In addition, it comprises a magnesium binding residue as well as cysteine residues in the C-terminus which is subjected to prenylation. Cysteine residues undergo lipid modification followed by geranylgeranylation during post regulation. This step is crucial for the activation and targeting of Rabs proteins to their specific membrane [Ali and Seabra, 2005; Ali et al., 2010; Jenkins et al., 2011]. Rab13 and 23 represent an exception, since they contain only a single cysteine residue in their C-terminal compare to other Rabs proteins. This single cysteine distinguishes Rab23 by allowing it to escape from trafficking and pass through the secretory pathway [Leung et al., 2007]. So far, the contribution of different RAB23 domains in the pathogenesis of Carpenter syndrome is not well understood [Jenkins et al., 2011].
To date, 30% of the inherited genetic disorders and different forms of cancer are modulated by nonsense-mediated decay (NMD) due to frameshifts or nonsense point mutations [Frischmeyer and Dietz, 1999; Khajavi et al., 2006]. The c.482-1G>A is the first homozygous splice site mutation described affecting the 3′ end of RAB23. This mutation led to a frameshift which would allow the translation of only 3 amino acids of exon 5 before encountering a stop codon. Thus, the truncated proteins, if produced, will lack 76 amino acids in their C-terminus (fig. 2C). From a total number of 11 mutations, reported to date in the RAB23 gene, 8 nonsense mutations have been identified (table 1) [Jenkins et al., 2007, 2011; Alessandri et al., 2010]. Accordingly, the truncation mutations are the major cause of Carpenter syndrome due to the loss of function of RAB23 proteins. Screening a total of 10 unrelated families of Northern Europe origin with Carpenter syndrome revealed the presence of one mutation (c.434T>A (p.L145X)), suggesting a founder effect [Jenkins et al., 2007, 2011]. Functional analysis has demonstrated that the nonsense p.L145X mutation encodes transcripts which undergo NMD [Jenkins et al., 2011]. To investigate whether the c.482-1G>A will produce truncated transcripts subject to NMD, we performed a quantitative expression of RAB23 mRNA. Notably, relative quantification of RAB23 transcripts showed no significant difference in the level of expression of RAB23 mRNA in patient (IV-2), carrier (III-1) and control samples using TaqMan assays (online suppl. fig. 2). Thus, the c.482-1G>A mutation will give rise most probably to a stable truncated mRNA, since the position of this mutation is very close to the end of RAB23 transcript. Consequently, the mRNAs most likely escape from the NMD survey of the cell. As a result, the truncated protein will probably be mislocalized within the cell due to the lack of its lipid modifications. Subsequently, the mutation leads to loss of its biological function(s) which explains the pathogenicity of this mutation in this family. This finding highlights the crucial role of lipid modification as alternative machinery of cellular control and is a vital step for the targeting of functional RAB23 proteins.
Table 1.
Summary of RAB23 mutations identified in patients with Carpenter syndrome
| Exons | DNA | Protein | Country/region of origin | References |
|---|---|---|---|---|
| Exon 1 | c.35T>A | p.M12K | Mexico | Jenkins el al., 2011 |
| Exon 1 | c.82C>T | p.R28X | Australia | Jenkins el al., 2011 |
| c.434T>A | p.L145X | |||
| Exon 1 | c.86dupA | p.Y29X | Comoros Islands | Alessandri et al., 2010 |
| Exon 1 | c.140_141insA | p.E48fsX7 | UK | Jenkins el al., 2007 |
| Exon 1 | c.156-3T>G | p.V53fsX13 | Belgium | Jenkins el al., 2011 |
| Exon 2 | c.232delT | p.Y78fsX30 | Brazil | Jenkins el al., 2007 |
| Exon 2 | c.234_236delCTA | p.Y79del | Turkey | Jenkins el al., 2011 |
| Exon 3 | c.253T>C | p.C85R | UK | Jenkins el al., 2007 |
| Exon 3 | c.362_363insG | p.N121fsX4 | Brazil | Jenkins el al., 2011 |
| Exon 4 | c.408_409insT | p.E137X | Brazil | Jenkins el al., 2007 |
| Exon 4 | c.434T>A | p.L145X | Northern Europe | Jenkins el al., 2007 |
| Exon 5 | c.482-1G>A | p.V161fsX3 | United Arab Emirates | this study |
The numeration of exons was restricted to the coding exons only. The numbering for the nucleotide and protein changes are based on cDNA sequence in accordance with the GenBank entries NM_183227.1 and NP_899050.1. ins = Insertion; dup = duplication; del = deletion.
In conclusion, we show that the c.482-1G>A splice site mutation, found in the RAB23 gene, is responsible for the typical features of Carpenter syndrome in a consanguineous Emirati family. Molecular and bioinformatics analyses revealed that the single base substitution led to an activation of cryptic splice site downstream in exon 5. The truncated transcripts produced nonfunctional proteins due to loss of lipid modification. This finding extends the spectrum of RAB23 mutations and highlights the important role of this gene as major cause of Carpenter syndrome worldwide. Finding disease causing mutations of recessive disorders is crucial for adopting effective prevention strategies, especially in highly consanguineous populations like the United Arab Emirates [Al-Gazali and Ali, 2010].
Supplementary Material
Supplemental Figures
Supplemental Tables
Acknowledgements
The authors are grateful to the family members and patients for their participation in this research study. The laboratories of L.A. and B.R.A. are funded by the Dubai Harvard Foundation for Medical Research (http://www.dhfmr.hms.harvard.edu/) and the United Arab Emirates University (http://www.uaeu.ac.ae/).
References
- Alessandri JL, Dagoneau N, Laville JM, Baruteau J, Hébert JC, Cormier-Daire V. RAB23 mutation in a large family from Comoros Islands with Carpenter syndrome. Am J Med Genet A. 2010;152A:982–986. doi: 10.1002/ajmg.a.33327. [DOI] [PubMed] [Google Scholar]
- Al-Gazali L, Ali BR. Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE) Hum Mutat. 2010;31:505–520. doi: 10.1002/humu.21232. [DOI] [PubMed] [Google Scholar]
- Ali BR, Seabra MC. Targeting of Rab GTPases to cellular membranes. Biochem Soc Trans. 2005;33:652–656. doi: 10.1042/BST0330652. [DOI] [PubMed] [Google Scholar]
- Ali BR, Nouvel I, Leung KF, Hume AN, Seabra MC. A novel statin-mediated ‘prenylation block-and-release’ assay provides insight into the membrane targeting mechanisms of small GTPases. Biochem Biophys Res Commun. 2010;397:34–41. doi: 10.1016/j.bbrc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlke C, Bashkurov M, Buescher A, Krick T, John AK, et al. Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J Cell Sci. 2010;123:1460–1467. doi: 10.1242/jcs.058883. [DOI] [PubMed] [Google Scholar]
- Boyer J, Crosnier C, Driancourt C, Raynaud N, Gonzales M, et al. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet. 2005;116:445–453. doi: 10.1007/s00439-005-1262-7. [DOI] [PubMed] [Google Scholar]
- Burset M, Seledtsov IA, Soloyvev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G. Acrocephaly, with other Congenital Malformations. Proc R Soc Med. 1909;2:45–53. doi: 10.1177/003591570900201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DM, Green JG, Miller J, Gorlin RJ, Reed JA. Acrocephalopolysyndactyly type II – Carpenter syndrome: clinical spectrum and an attempt at unification with Goodman and Summitt syndromes. Am J Med Genet. 1987;28:311–324. doi: 10.1002/ajmg.1320280208. [DOI] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev Biol. 2006;290:1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Frias JL, Felman AH, Rosenbloom AL, Finkelstein SN, Hoyt WF, Hall BD. Normal intelligence in two children with Carpenter syndrome. Am J Med Genet. 1978;2:191–199. doi: 10.1002/ajmg.1320020210. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Jenkins D, Seelow D, Jehee FS, Perlyn CA, Alonso LG, et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins D, Baynam G, De Catte L, Elcioglu N, Gabbett MT, et al. Carpenter syndrome: extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay. Hum Mutat. 2011;32:E2069–E2078. doi: 10.1002/humu.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- Leung KF, Baron R, Ali BR, Magee AI, Seabra MC. Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J Biol Chem. 2007;282:1487–1497. doi: 10.1074/jbc.M605557200. [DOI] [PubMed] [Google Scholar]
- Perlyn CA, Marsh JL. Craniofacial dysmorphology of Carpenter syndrome: lessons from three affected siblings. Plast Reconstr Surg. 2008;121:971–981. doi: 10.1097/01.prs.0000299284.92862.6c. [DOI] [PubMed] [Google Scholar]
- Robinson LK, James HE, Mubarak SJ, Allen EJ, Jones KL. Carpenter syndrome: natural history and clinical spectrum. Am J Med Genet. 1985;20:461–469. doi: 10.1002/ajmg.1320200307. [DOI] [PubMed] [Google Scholar]
- Schneider TD. Information content of individual genetic sequences. J Theor Biol. 1997a;189:427–441. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- Schneider TD. Sequence Walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences. Nucleic Acids Res. 1997b;25:4408–4415. doi: 10.1093/nar/25.21.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temtamy SA. Carpenter's syndrome: acrocephalopolysyndactyly. An autosomal recessive syndrome. J Pediat. 1966;69:111–120. doi: 10.1016/s0022-3476(66)80368-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures
Supplemental Tables