

Abstract
Physicists have studied the aggregation of adhesive proteins, giving a central role to the elastic properties of membranes, whereas cell biologists have put the emphasis on the cytoskeleton. However, there is a dramatic lack of experimental studies probing both contributions on cellular systems. Here, we tested both mechanisms on living cells. We compared, for the same cell line, the growth of cadherin-GFP patterns on recombinant cadherin-coated surfaces, with the growth of vinculin-GFP patterns on extracellular matrix protein-coated surfaces by using evanescent wave microscopy. In our setup, cadherins are not linked to actin, whereas vinculins are. This property allows us to compare formation of clusters with proteins linked or not to the cytoskeleton and thus study the role of membrane versus cytoskeleton in protein aggregation. Strikingly, the motifs we obtained on both surfaces share common features: they are both elongated and located at the cell edges. We showed that a local force application can impose this symmetry breaking in both cases. However, the origin of the force is different as demonstrated by drug treatment (butanedione monoxime) and hypotonic swelling. Cadherins aggregate when membrane tension is increased, whereas vinculins (cytoplasmic proteins of focal contacts) aggregate when acto-myosin stress fibers are pulling. We propose a mechanism by which membrane tension is localized at cell edges, imposing flattening of membrane and enabling aggregation of cadherins by diffusion. In contrast, cytoplasmic proteins of focal contacts aggregate by opening cryptic sites in focal contacts under acto-myosin contractility.
Cell adhesion has been approached theoretically by the study of membrane-mediated protein aggregation (1–3). Entropic fluctuations and elastic properties of membranes were predicted to promote clustering of proteins. Experimentally, the principle of membranes inducing aggregation was tentatively probed on vesicles coated with proteins or beads (4, 5). Although there was no evidence for a contribution of membrane in protein aggregation in cells, the above mechanisms were suggested to be involved in cell adhesion. In contrast, adhesive formation in cells was shown by cell biologists to be intimately connected with actin cytoskeleton organization. The transmembrane protein integrin binds to extracellular matrix (ECM) proteins. This phenomenon leads to the assembly of a molecular complex, named focal contact, which is connected to acto-myosin stress fibers. The transmembrane protein cadherin binds to cadherins on neighboring cells, promoting the formation of adherens junctions, also connected to the actin cytoskeleton (for a review, see refs. 6 and 7). For focal contacts, acto-myosin force was shown to promote their growth. For adherens junctions, actin polymerization was shown to be the driving force for their assembly (8). The possible importance of the implication of the membrane was usually neglected. To take into account both contributions, we designed an experiment on living cells. We studied adhesion of a cell on substrates covered with either ECM proteins or cadherin. To follow the adhesive patterns in both cases, we transfected the same cell line with constructs encoding a C-terminal vinculin GFP fusion for ECM surfaces and a C-terminal cadherin GFP fusion for cadherin surfaces. In our setup, integrin was connected to actin through a molecular complex, but cadherin was not. Because these events occur in the vicinity of the surface, we mounted an evanescent wave microscope that enabled us to image the cadherin surface clusters. In our study, only cadherin–cadherin interactions were observed, without adherens junction assembly. So the cadherin cluster under study is different from a mature adherens junction and should be viewed as a pattern formed by molecules free to diffuse in a membrane interacting with the grafted partners on the surface. Moreover, cadherin patterns seem to behave as glue, escaping cell regulation. This type of aggregation is highly interesting: it behaves as a passive system while being in a real cell and it enables the study of the role of membrane in the aggregation process. For both integrin–ECM and cadherin–cadherin interactions, elongated patterns were formed at the cell periphery along the tension lines. However, we show in this study that the origin of the clustering is different for ECM and cadherin surfaces. The former is caused by acto-myosin contractility, whereas the latter is promoted by an increase in membrane tension.
Materials and Methods
Cell Culture and Protocol for Cell Detachment Before Replating on Cadherin Substrate. We used EAhy cells that form adherens junctions and focal contacts. This line was derived by fusing human umbilical vein endothelial cells with the permanent human cell line A549 (9). Cells were maintained in DMEM (41965-039, GIBCO) supplemented with 10% bovine calf serum (SH30072.03, HyClone). They were transfected by using ExGen 500 (ET0250, Euromedex, Mundolsheim, France) with constructs encoding a C-terminal vascular endothelial (VE)-cadherin GFP fusion, a truncated C-terminal VE-cadherin GFP fusion (10), and a C-terminal vinculin GFP fusion (11) following the protocol of the manufacturer. For experiments on cadherin surfaces, cells were grown in standard conditions until confluency. EAhy cells were incubated for 1 min with 0.25% trypsin/1 mM EDTA (25200-072, GIBCO) before centrifugation. They were washed once with DMEM with 0.1% trypsin inhibitor (T9003, Sigma) and replated in L15 medium (21083-027, GIBCO) on cadherin surfaces. For experiments with ECM proteins, cells were grown on coverslips with serum and serum-starved for 3 h before the experiment. Incubation with serum leads to a coating of ECM proteins (among which fibronectin and vitronectin). We alternatively grafted fibronectin (F1145, Sigma) on surfaces following the protocols of ref. 12.
Force Application and Drug Treatment. Glass capillaries (GC100F-10, Clark Electromedical Instruments, Pangbourne, U.K.) were pulled with a micropipette puller (caP-97, Sutter Instruments, Novato, CA), yielding micropipettes with 1 nN/μm spring constant. They were not coated. As in ref. 13, pipettes were imposed a motion by using a motorized micromanipulator (DC3001, World Precision Instruments, Hertfordshire, U.K.). Cells were treated with butanedione monoxime (BDM, B0753, Sigma) at 30 mM for 30 min and with latrunculin A (L12370, Molecular Probes) at 1 μM for 5 min. For membrane solubilization, we used 0.1% SDS (161-0416, Bio-Rad) for 5–60 min and 1% Triton X-100 (T9284, Sigma) for 5–60 min. Hypotonic swelling was made by exchanging L15 medium for L15 diluted in water (between 30% and 70%). The results obtained with drugs and force application were reproduced on at least four different cells.
Fixation and Staining. Cells were fixed with 3% paraformaldehyde (P6148, Sigma) in PBS for 20 min, permeabilized with 0.5% Triton X-100 in the same solution of paraformaldehyde for 3 min, and washed twice for 5 min in PBS. Cells were then incubated with primary antibodies [anti-β-catenin (C2206, Sigma), anti-p120 (610133, Transduction Laboratories, Lexington, KY), anti-paxillin (610193, Transduction Laboratories), anti-pan cadherin (C1821, Sigma), and anti-fibronectin (F3648, Sigma)] for 45 min. They were washed three times for 5 min in PBS, incubated for 45 min with the appropriate secondary antibody [Cy3 (Jackson ImmunoResarch), 115-165-146 (mouse), 111-166-047 (rabbit); Alexa Fluor 488 (Molecular Probes), A11045 (mouse), A11046 (rabbit); Alexa Fluor 350 (Molecular Probes), A11029 (mouse), A 11034 (rabbit)] or with fluorescently labeled phalloidin (A12379 and A22283, Molecular Probes) for actin staining. They were finally washed three times in PBS. The coverslips were then mounted with Mowiol (32,459-0, Aldrich).
Preparation of Cadherin Surfaces. To guarantee a proper orientation of the cadherin molecule on the surface, we used a recombinant cadherin expressed in Escherichia coli. It consisted of the four first domains of the extracellular part of VE cadherin with a six-histidine tag at the C terminus (VE cad1–4 HIS). Cadherins could therefore be properly oriented on Ni-functionalized glass surfaces. The protocol for grafting Ni covalently on glass surfaces, inspired by Noren et al. (14), was performed as follows. Coverslips were first washed once with water then incubated on a shaking table in one part H2O2 (21,676-3, Aldrich), nine parts H2SO4 for 10 min. Then they were washed once with distilled water, once with 1 M NaOH, and finally 20 times with distilled water. The coverslips were heated 60 min at 100°C and immediately incubated in a solution of 94% ethanol, 5% H2O, and 1% 3-aminopropyltriethoxysilane (A3648, Sigma) for 30 s, then washed three times with pure ethanol, and heated for 10 min at 100°C to eliminate most of the ethanol. They were mounted on observation chambers (no. 1 coverslip-bottomed Petri dishes). The coverslips were then incubated with 0.08% glutaraldehyde (G-7526, Sigma) in distilled water for 1 h and washed three times with distilled water. Subsequently, they were incubated with 1 μg/ml nitrilotriaceticacid ligand (N-9877, Sigma) for 20 min and charged with 100 mM nickel sulfate (N-4882, Sigma) in water for 20 min. The coverslips were then washed three times with distilled water. Finally, coverslips were coated with 1 μM VE cad1–4 HIS solution by incubation for 4 h at room temperature in DMEM with 5 mM EGTA, washed once with 1% BSA (A8806, Sigma) in DMEM, and blocked with the same solution overnight. Observation chambers were exposed with UV for 5 min before replating of cells on the surface.
Microscopy and Image Analysis. For evanescent wave microscopy, we designed a setup inspired by Tokunaga et al. (15). The optical fiber coupled laser (395 mW, 177-G02, Spectra-Physics) was focused through the lens of the epifluorescence condenser of the microscope on the back focal plane of an Olympus ×60 Plan Apo TIRFM objective (numerical aperture = 1.45). To obtain an homogenous excitation, the fiber was scrambled by a homemade device. For GFP observations, we used the filter set XF100-2 (Omega Optical, Brattleboro, VT). Fluorescence recovery after photobleaching experiments was performed with the same laser by changing the optical path: it was focused on the specimen plane through a 15-mm lens, via the epifluorescence pathway of the microscope, at maximum intensity for 5 s. Reflection interference contrast microscopy (RICM) images were obtained with the Hg lamp by using a 550-nm centered interference excitation filter (13). Cells were observed with an inverted microscope (IX70, Olympus) by using the metamorph acquisition system (Universal Imaging, Media, PA) and a cooled couple-charged device camera (MicroMAX-1300YHX, Roper Scientific, Evry, France). For living-cell experiments, the temperature was maintained at 37°C by temperature control of an insulating box surrounding the microscope. Images were processed with metamorph. We extracted the density (mean intensity per pixel) of motifs Dmotif, the whole image density Dtot, and the density of the background Dback. We also measured the area, the small and large axes of the motifs viewed as ellipses. These parameters were plotted as a function of time by using origin (Microcal Software, Northampton, MA). The density was corrected against temporal excitation intensity variations and photobleaching as follows: Dcorrec = (Dmotif – Dback)/(Dtot – Dback).
Results
Adhesive Protein Patterns. On ECM surfaces, we observed normal focal contacts (Fig. 1 a–d). They were located at the cell periphery and elongated (≈4 μm long). Stress fiber ends were colocalized with these patterns (Fig. 1d). They contained tyrosine phosphorylated proteins, vinculin, and paxillin but not cadherin (Fig. 1a). On cadherin surfaces, the individual cadherin patterns shared common features with focal contacts on ECM surfaces: they are elongated structures localized at the cell edges (compare Fig. 1 c and e for length and location). However, cadherin patterns were sometimes so close that they could also completely cover the cell borders. In contrast to focal contacts, they were not associated with tensed stress fibers (Fig. 1 d and h). Cadherin patterns are stable for at least 24 h. We checked that cellular cadherins were irreversibly bound to the surface cadherins by performing fluorescence recovery after photo-bleaching experiments: the bleached motif did not reappear within 2 h (see Fig. 7, which is published as supporting information on the PNAS web site). Note that the formation of cadherin patterns is not associated with cell spreading: they do appear only ≈5 h after plating (see Fig. 8, which is published as supporting information on the PNAS web site).
Fig. 1.

Immunofluorescence images of a cell adhering on a fibronectin substrate (a–d) and a cadherin substrate (e–h). Cells were stained for cadherins (a and e), actin (b and f), and paxillin (c and g). (d) A superimposed image of paxillin and actin. (h) A superimposed image of cadherin and actin.
The Role of Actin on Cadherin Surfaces. Actin is not required for the formation of cadherin patterns. Two different experiments led us to this conclusion. First, we used cells expressing a truncated cadherin GFP that was not able to bind actin through catenins. We observed the same type of streak patterns with these truncated cadherins (see Fig. 9, which is published as supporting information on the PNAS web site). Thus actin is not necessary for the appearance of the motifs. But more strikingly, when latrunculin A (a drug that depolymerizes actin) was added, motifs already formed remain unchanged (Fig. 10, which is published as supporting information on the PNAS web site). From these two experiments, we concluded that actin is not involved in the formation of cadherin structures. We also stained for the other proteins normally present in adherens junctions: p120, β-catenin, and α-catenin. They were absent from the patterns, confirming that actin was not connected to cadherins through these complexes.
Real Cadherin–Cadherin Interactions. In evanescent wave microscopy, ≈200-nm thickness is excited above the coverslip surface. So the fluorescent signal can come from cadherins that are not really bound to grafted cadherins. We thus dissolved the membrane by adding 0.1% SDS or 1% Triton X-100. We observed no disappearance of cadherin patterns. We concluded that GFP cadherins were really linked to surface cadherins.
Force. Both vinculin and cadherin patterns share a common feature: symmetry breaking. We thus checked whether an externally applied force could generate these patterns. We applied the force locally at the cell edges centripetally, as in ref. 13. As expected for ECM surfaces, force promoted the growth of focal contacts (Fig. 2 a–e) at the cell periphery. The same kind of protein recruitment was observed on cadherin surfaces (Fig. 2 f–i), but the extension of pipette motion had to be larger. As a consequence, membrane was pulled and the motifs growth could be observed on the cell edges expected to be under tension. The motif dynamics is entirely defined by its area, density, and axis ratio, the contact being viewed as an ellipse. For vinculin motifs, the contacts grow continuously from the cell edge to the inside, the density of the motifs being constant (Fig. 2d). As opposed to focal contact formation, there is no directionality in the growth of cadherin patterns, as their final shape is formed in a few seconds (see area and axis ratio curves, Fig. 2j). The patterns then densify, recruiting new GFP cadherins (see density curve, Fig. 2i).
Fig. 2.

Force application on a cell adhering on a serum-starved ECM surface (a–c) and a cadherin surface (f–h). (b and g) Pipette location is shown by the white arrow indicating the direction of the traction. The rectangles in b and g are the zones of images a, c, f, and h, respectively. GFP vinculin images before (a) and 15 min after (c) force application are shown. GFP VE-cadherin images before (f) and 30 min after (h) force application are shown. Typical density variation of a vinculin (d) and a cadherin (i) motif under the application of a force (time 0). (e and j) Area (rectangle) and axis ratio (stars) of the corresponding contacts of d and i, respectively. For vinculin motifs, the motifs area and axis ratio increase in 100 s, while the density stays constant. These parameters then reach a saturation as shown by Riveline et al. (13). In the case of cadherins, the motif shape appears within 100 s after the application of a force and then stays constant. Measurements are done by taking a constant motif area and axis ratio. The density in the motif increases before reaching a saturation.
From this experiment, we conclude that applying a force on cells adhering on a surface with two different ligand-receptor proteins entails a recruitment of adhesive proteins. However, we have shown (see above) that whereas focal contacts were associated with acto-myosin stress fibers, cadherin motifs were not bound to the cytoskeleton. We propose two different mechanisms for force-promoted recruitment upon force application that can be extended to natural conditions. Focal contacts grow through local force application, mimicking acto-myosin action. In contrast, on cadherin surfaces, the application of the force with the pipette provoked an increase in membrane tension and hence cadherin recruitment: cadherin patterns grow because an increase in membrane tension brings the membrane closer to the substrate and allows further interactions between adhesive partners. We checked this hypothesis by acting specifically on acto-myosin contractility and membrane tension.
Acto-Myosin Contractility. When contractility was inhibited by addition of BDM, cadherin contacts remained the same (Fig. 3 d–f). This finding is in sharp contrast with ECM surfaces: after addition of BDM, focal contacts completely disappeared (Fig. 3 a–c). These results show that acto-myosin contractility is necessary for focal contacts but not for cadherin patterns.
Fig. 3.

Inhibiting contractility using BDM entails disappearance of focal contacts but not of cadherin patterns on a cadherin surface. Contacts revealed by GFP vinculin, before addition of BDM (a) and 30 min after (b) are shown. Contacts revealed by GFP VE-cadherin before addition of BDM (d) and 30 min after (e) are shown. Typical evolution of vinculin (c) and cadherin (f) motif density when BDM is added (time 0).
Swelling. We increased the membrane tension through a hypotonic swelling (16). We observed the appearance of cadherin motifs (Fig. 4 e–h). This phenomenon was similar to the one observed with the application of the force. We performed the same experiment on ECM surfaces: we did not see any vinculin recruitment, and moreover focal contacts sometimes disappeared (Fig. 4 a–d). This experiment proves that membrane tension is the key parameter for cadherin recruitment.
Fig. 4.

Increasing membrane tension through hypotonic swelling entails recruitment of cadherin on cells adhering on a cadherin surface but not recruitment of vinculin on cells adhering on a ECM surface. GFP vinculin images before swelling (a and b) and 50 min after swelling (c). (b and c) Zoomed images of the area depicted by the rectangle in a. GFP VE-cadherin images before swelling (e and f) and 1 h after swelling (g) are shown. (f and g) Zoomed pictures of the area depicted by the rectangle in e. Typical evolution of vinculin (d) and cadherin (h) motif density during hypotonic choc. The arrows indicate the time for change in medium. The percentage refers to the distilled water proportion in the medium.
Membrane Flattening and Cadherin Recruitment by Diffusion. We proposed above a mechanism of cadherin recruitment upon an increase in membrane tension. We view the increase in membrane tension as the means to force the membrane to be in close contact with the cadherin surface. Tension per se is not the motor of aggregation but it leads indirectly to the aggregation. If flattening is the key phenomenon, upon growth of cadherin contacts, two distinct regions should be observed for the membrane-to-surface distance as revealed by RICM (17). In the contact location, the image should be dark; in the vicinity of this region, the overall RICM background should become whiter because of the expected escape of the tensed membrane. This is indeed what we observed when we performed a swelling experiment acquiring GFP VE cadherin total internal-reflection fluorescence and RICM images (see Fig. 5). To show that diffusion is responsible for recruitment, we give the following estimate. The motif is formed on a typical 100-s time scale. We performed a fluorescence recovery after photobleaching experiment in our system. The extracted cadherin diffusion coeffcient as measured with evanescent wave microscopy is 0.005 μm2/s. Such a value is lower that the diffusion coefficient of free cadherin [D = 0.04 μm2/s for Adams et al. (18) and D = 0.02 μm2/s for Sako et al. (19)]. This finding is not surprising because cadherins in our system are diffusing in the proximity of the surface. With this value, we can estimate the time required to fill a 2-μm2 area by pure diffusion: we obtain the expected 100-s time scale.
Fig. 5.

Membrane-to-surface distances. (a) Fluorescence image of cadherin GFP motifs formed by a cell adhering on a cadherin surface. (b) Corresponding RICM image. Arrow 1 indicates the motif that appears white in fluorescence image and black in RICM as membrane is close to the surface. In the vicinity of the contact zone (arrow 2), fluorescence signal is black and RICM signal is white, showing that the membrane is escaping from the surface. (Scale bar: 2 μm.)
Discussion
The nature of the mechanism that leads to protein aggregation is very different between cadherin and vinculin. The cadherin motifs are irreversibly formed (no drugs were able to remove the contacts) in contrast to vinculin motifs that could be removed by adding BDM, for example. For cadherin, it is a passive response of the cell. We propose that the cell does not have any control on the fate of cadherin, because the adhesive structures are lacking the proteins implicated in the formation of adherens junctions. In contrast, in the case of vinculin, the cell can regulate the assembly of adhesive structures via a cytoplasmic protein complex. Such a regulation can also exist with cadherins as shown by experiments using beads covered with cadherins (20–23). In this setup, cadherins are not just behaving as glue. Indeed, artificial adherens junctions are assembled around the beads, linking cadherin to the cytoskeleton and activating signaling pathways such as the rac GTPase one.
There could be several explanations for the nonformation of mature adherens junctions in our system. In adherens junctions between two cells, the plane of contact is a symmetry plane. In our system, we have broken this symmetry: cadherins on the surface are not mobile and the surface is infinitely stiff. Moreover, we grafted monomers of VE cad1–4 HIS on the surface, and the binding of catenins may require clusters of cadherins. It may also require the five glycosylated domains of VE cadherin. As there is a high homology between the cytoplasmic domain of different cadherins, work studying cells adhering on different cadherin-coated surfaces can be compared. The first experiments were done with C-cadherin adsorbed on glass surfaces (24). They showed that nontransfected Chinese hamster ovary (CHO) cells poorly adhered on such surfaces, in contrast to full-length C-cadherin-transfected cells. Moreover, when the cadherin was truncated, cell adhesion strength decreased. Altogether, these results suggested that the effect of cadherin–cadherin interaction can be tested on a planar surface.
Along the same line, signaling pathways were shown to be activated when cells were adhering to various cadherin substrates. Noren et al. (14) showed in particular that Rac was activated by C-cadherin engagement. In addition, Kovacs et al. (25) plated CHO cells transfected with E-cadherin on Fc-E-cadherin-coated surfaces and showed that cadherin engagement can recruit Arp2/3 to nascent adhesive contacts. Although all of these effects actually show a role for cadherin homophilic interaction in cell signaling, there was no clear evidence that catenins and actin colocalized with cadherin clusters on surfaces. Some immunofluorescence images of clusters were shown. However, no experiment proving that these clusters were really bound to the surface was produced, as we did with the Triton experiment. The signaling response on surfaces thus can not necessarily be compared directly with the signaling pathway linked to real adherens junctions.
Adhesive junctions can be divided into three parts: the ligand–receptor interaction, the cytoplasmic adhesive complex, and the actin cytoskeleton. We compared in this study aggregation of the ligand proteins in the case of cadherin surfaces and aggregation of the cytoplasmic part in the case of ECM surfaces. We have shown that the cytoplasmic proteins of focal contacts aggregation is controlled by acto-myosin contractility. In contrast, cadherin aggregation is controlled by membrane tension. Note that even in the case of focal contacts we do not exclude that integrins are recruited in the same fashion as cadherins. Indeed, Tsuruta et al. (26) have shown that integrin and cytoplasmic proteins of focal contacts (CPFC) do not behave in the same way when actin is depolymerized, leading in particular to acto-myosin contractility inhibition: integrin motifs remained the same, whereas actinin disappeared. So care should be taken not to extend CPFC assembly properties to integrin.
Integrin-ECM binding is not sufficient for assembly of the three parts of focal contacts mentioned above. Focal contacts act as mechanosensors, i.e., growing when submitted to a force (13, 27, 28). The tension exerted by the cytoskeleton could reveal new binding sites for cytoplasmic proteins of the adhesive complex. We propose another mechanism for cadherin adhesion on cadherin surfaces. We have shown that an increase in membrane tension could provoke a cadherin aggregation. We suggest that this increase brings the actin shell-connected membrane closer to the surface (see Fig. 6). The minute time scale observed for the formation of contact suggests that cadherins are diffusing along the membrane to reach formed clusters (Fig. 6c). As mentioned above, our measured diffusion coefficient is consistent with this scenario. We do not state here that membrane tension induces an attractive interaction between cadherins, but that it only mechanically brings the membrane in close proximity with the surface, then enabling the recruitment of cadherin by diffusion.
Fig. 6.

(a) The membrane of the cell adhering on a cadherin substrate is crumpled so that free cadherins localized between pinning points cannot bind cadherin on the surface. Dashed lines represent the actin cortex that is connected to the membrane. (b) However, when membrane tension is increased, these cadherins are brought into close contact of the surface, allowing cadherin–cadherin binding. (c) The density of the cadherin motif increases by free cadherin diffusion and binding.
Typical lengths of elongated contacts and time scales of their formations should be compared in both systems: (i) in both cases, aggregates appear at the cell periphery; (ii) symmetry is broken; (iii) both have a comparable finite length of ≈4 μm; (iv) the density of patterns can be much higher on cadherin surfaces, leading even to continuous cadherin areas (see Fig. 4g); and (v) the total recruitment time scale is a few hundred of seconds for both contacts. In the case of cadherins, concerning i, the cell senses membrane tension at its periphery. The length of the contacts is determined by the screening length of the force λ, which would explain iii. This length can be estimated by taking results obtained for ribbon lying on an elastic layer: the screening length
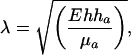 |
[1] |
where E is the cytoskeleton Young modulus, h is the cytoskeleton thickness, ha is the cadherin coating thickness, and μa is the shear modulus of this coating (29). We estimated λ by taking E ≈100 μa (F. Chamaraux and B. Fourcade, personal communication), h = 200 nm (N. Morone and A. Kusumi, personal communication), ha = 50 nm, taking into account the grafting chemistry, and obtained λ ≈1 μm. Note that this estimate should be taken only as good indications for the length, because the E over μa ratio could be smaller. Concerning ii, the symmetry breaking is caused by the direction of the force when membrane tension is increased. In the case of ECM surfaces, as stated above, integrin could be recruited with the same mechanism as cadherin, i.e., by membrane tension. However, as shown by experiments with BDM, acto-myosin force is still required for the maturation of focal contacts. The localization at the cell periphery and the symmetry breaking can be explained by the distribution and direction of stress fibers in the cell. Along the same line, the finite length of focal contacts is determined by the acto-myosin contractility (28). The 100-s time scale may be caused by a diffusion limited phenomenon in both cases. However, for vinculin, it could be also related to the time scale corresponding to the opening of cryptic site. An explanation for cadherin pattern high density requires further investigation.
It is worthwhile to ask whether the tension role observed here could be important in the first stages of adherens junctions formation. It has been established that cadherins were recruited as a consequence of actin polymerization (8). Indeed, in our case, we showed that actin was not playing this key role. However, tension could also be involved in adherens junctions formation. The corresponding observation would be as follows: between neighboring cells, cadherins would accumulate at cell edges, for example, because of membrane tension thus provoking symmetry breaking. This was indeed observed in Adams et al. (18) (see their figure 5). Moreover, we propose that in this latter case as actin was connected to cadherins it was responsible for the increase in membrane tension. The actin cytoskeleton in natural adherens junctions would play the role of our pipette in our setup.
From all of these results, one can conclude that forces definitively play a role in cells for protein clustering. We demonstrated that in contrast to focal contacts the force transduction for cadherins was not mediated by the cytoskeleton but by the membrane. Changes in the elastic properties of the membrane are certainly regulated by the cells. It has been shown, for example, that a shear stress applied on a cell, could modify membrane tension (30). It would be worthwhile to characterize these membrane-related pathways to the same extent as for the cytoskeleton.
Supplementary Material
Acknowledgments
We thank I. Grosheva, J. Zhurinsky, A. Michelot, A. Carminati, and M. Shtutman for extensive help in the first stages of this work; M. Weidenhaupt and A. Bershadsky for critical reading of our manuscript; and B. Fourcade and F. Chamaraux for helpful discussions. This work was supported by the Keshet program, Region Rhone-Alpes, Action Concertée Incitative, Institut de Physique de la Matière Condensée de Grenoble, and the Association pour la Recherche sur le Cancer.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: VE, vascular endothelial; ECM, extracellular matrix; BDM, butanedione monoxime; RICM, reflection interference contrast microscopy.
References
- 1.Weikl, T. R. & Lipowsky, R. (2001) Phys. Rev. E 64, 1–13. [DOI] [PubMed] [Google Scholar]
- 2.Menes, R. & Safran, S. A. (1997) Phys. Rev. E 56, 1891–1899. [Google Scholar]
- 3.Zuckerman, D. M. & Bruinsma, R. F. (1998) Phys. Rev. E 57, 964–977. [Google Scholar]
- 4.Bruinsma, R., Behrisch, A. & Sackmann, E. (2000) Phys. Rev. E 61, 4253–4267. [DOI] [PubMed] [Google Scholar]
- 5.Koltover, I., Radler, J. O. & Safinya, C. R. (1999) Phys. Rev. Lett. 82, 1991–1994. [Google Scholar]
- 6.Bershadsky, A. & Geiger, B. (1999) in Guidebook to the Extracellular Matrix, Anchor, and Adhesion Proteins, eds. Kreis, T. & Vale, R. (Oxford Univ. Press, Oxford), pp. 3–11.
- 7.Gumbiner, B. M. (1996) Cell 84, 345–357. [DOI] [PubMed] [Google Scholar]
- 8.Vasioukhin, V., Bauer, C., Yin, M. & Fuchs, E. (2000) Cell 100, 209–219. [DOI] [PubMed] [Google Scholar]
- 9.Edgell, C. J., McDonald, C. C. & Graham, J. B. (1983) Proc. Natl. Acad. Sci. USA 80, 3734–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navarro, P., Caveda, L., Breviario, F., Mandoteanu, I., Lampugnani, M. G. & Dejana, E. (1995) J. Biol. Chem. 270, 30965–30972. [DOI] [PubMed] [Google Scholar]
- 11.Zamir, E., Katz, B. Z., Aota, S., Yamada, K. M., Geiger, B. & Kam, Z. (1999) J. Cell Sci. 112, 1655–1669. [DOI] [PubMed] [Google Scholar]
- 12.Bershadsky, A., Chausovsky, A., Becker, E., Lyubimova, A. & Geiger, B. (1996) Curr. Biol. 6, 1279–1289. [DOI] [PubMed] [Google Scholar]
- 13.Riveline, D., Zamir, E., Balaban, N. Q., Schwarz, U. S., Ishizaki, T., Narumiya, S., Kam, Z., Geiger, B. & Bershadsky, A. D. (2001) J. Cell Biol. 153, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noren, N. K., Niessen, C. M., Gumbiner, B. M. & Burridge, K. (2001) J. Biol. Chem. 276, 33305–33308. [DOI] [PubMed] [Google Scholar]
- 15.Tokunaga, M., Kitamura, K., Saito, K., Iwane, A. H. & Yanagida, T. (1997) Biochem. Biophys. Res. Commun. 235, 47–53. [DOI] [PubMed] [Google Scholar]
- 16.Raucher, D. & Sheetz, M. P. (2000) J. Cell Biol. 148, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rädler, J. & Sackmann, E. (1993) J. Phys. II [French] 3, 727–748. [Google Scholar]
- 18.Adams, C. L., Chen, Y. T., Smith, S. J. & Nelson, W. J. (1998) J. Cell Biol. 142, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sako, Y., Nagafuchi, A., Tsukita, S., Takeichi, M. & Kusumi, A. (1998) J. Cell Biol. 140, 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levenberg, S., Katz, B. Z., Yamada, K. M. & Geiger, B. (1998) J. Cell Sci. 111, 347–357. [DOI] [PubMed] [Google Scholar]
- 21.Lambert, M., Choquet, D. & Mege, R. M. (2002) J. Cell Biol. 157, 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baumgartner, W., Schutz, G. J., Wiegand, J., Golenhofen, N. & Drenckhahn, D. (2003) J. Cell Sci. 116, 1001–1011. [DOI] [PubMed] [Google Scholar]
- 23.Kusumi, A., Suzuki, K. & Koyasako, K. (1999) Curr. Opin. Cell Biol. 11, 582–590. [DOI] [PubMed] [Google Scholar]
- 24.Yap, A. S., Brieher, W. M., Pruschy, M. & Gumbiner, B. M. (1997) Curr. Biol. 7, 308–315. [DOI] [PubMed] [Google Scholar]
- 25.Kovacs, E. M., Goodwin, M., Ali, R. G., Paterson, A. D. & Yap, A. S. (2002) Curr. Biol. 12, 379–382. [DOI] [PubMed] [Google Scholar]
- 26.Tsuruta, D., Gonzales, M., Hopkinson, S. B., Otey, C., Khuon, S., Goldman, R. D. & Jones, J. C. (2002) FASEB J. 16, 866–868. [DOI] [PubMed] [Google Scholar]
- 27.Choquet, D., Felsenfeld, D. P. & Sheetz, M. P. (1997) Cell 88, 39–48. [DOI] [PubMed] [Google Scholar]
- 28.Balaban, N. Q., Schwarz, U. S., Riveline, D., Goichberg, P., Tzur, G., Sabanay, I., Mahalu, D., Safran, S., Bershadsky, A., Addadi, L. & Geiger, B. (2001) Nat. Cell Biol. 3, 466–472. [DOI] [PubMed] [Google Scholar]
- 29.Maugis, D. (2000) Contact, Adhesion, and Rupture of Elastic Solids (Springer, New York).
- 30.Haidekker, M. A., L'Heureux, N. & Frangos, J. A. (2000) Am. J. Physiol. 278, H1401–H1406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}