

Abstract
Liberibacter crescens BT-1, a Gram-negative, rod-shaped bacterial isolate, was previously recovered from mountain papaya to gain insight on Huanglongbing (HLB) and Zebra Chip (ZC) diseases. The genome of BT-1 was sequenced at the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. A finished assembly and annotation yielded one chromosome with a length of 1,504,659 bp and a G+C content of 35.4%. Comparison to other species in the Liberibacter genus, L. crescens has many more genes in thiamine and essential amino acid biosynthesis. This likely explains why L. crescens BT-1 is culturable while the known Liberibacter strains have not yet been cultured. Similar to Candidatus L. asiaticus psy62, the L. crescens BT-1 genome contains two prophage regions.
Keywords: Huanglongbing, citrus greening, reduced genome, fastidious, hybrid assembly
Introduction
Huanglongbing (HLB), also known as citrus greening, is a disease that poses a major economic threat to the worldwide citrus industry [1,2]. The disease was discovered to be present in Florida in 2005 and is characterized by yellowing of citrus tree leaves, premature defoliation, small bitter fruit, and a pale green fruit color after ripening. No known cure for the disease has been discovered, but preventative measures include chemical treatment against insect vectors and removal of infected trees to prevent the spread of disease [3,4].
The causal agents of HLB are believed to be Candidatus Liberibacter asiaticus, Candidatus L. africanus, and Candidatus L. americanus, named according to the regions of where the organism was first identified [5,6]. Similar diseases have been found to occur in potatoes (Solanum tuberosum) and other solanaceous crops infected with Candidatus L. solanacearum [7]. Additionally, the Liberibacter genus contains the plant endophyte Candidatus L. europaeus [8], signifying that virulence in the Liberibacter genus is not universal. None of these organisms have been cultured but a metagenomic analysis of phloem suggests that this is the only bacterium present in the phloem of symptomatic trees [9].
Due to the highly fastidious nature of the genus Liberibacter, research on these organisms has traditionally been limited to electron microscopy and genomic analysis [3,7,10]. However, one species of the genus, Liberibacter crescens, has recently been cultured and characterized [11], and the relationship between its genome and close relatives will be the focus here.
In order to gain insight on both the virulence and metabolism of the genus Liberibacter, all available genomes of the Liberibacter spp. were compared to Liberibacter crescens. To date, the genomes of Candidatus L. asiaticus and Candidatus L. solanacearum are publicly available. The differences between these species may be responsible for the fastidious nature of the Liberibacter spp. Sequencing, assembly, and annotation of L. crescens were performed in order to proceed with the investigation.
Classification and features
Figure 1 and Table 1 summarize the phylogenetic position and characteristics of Liberibacter crescens BT-1, respectively. Figure 2 shows transmission electron microscopy of L. crescens BT-1.
Figure 1.
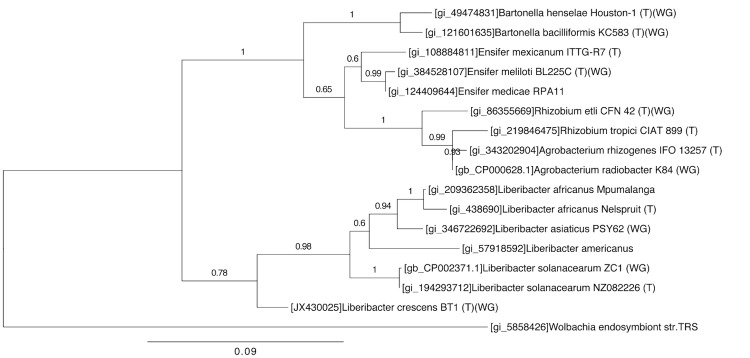
Maximum likelihood phylogenetic tree constructed using 16S rRNA genes of Liberibacter crescens BT-1 and related members of the Alphaproteobacteria. Branch supports are provided above branches. Sequences were aligned using MUSCLE [12]. Overhanging regions were removed, and the alignment was curated using Gblocks [13]. Phylogeny was determined with PhyML [14] using the GTR substitution model [15] with 500 bootstraps. These tools were accessed through phylogeny.fr [16]. The resultant phylogenetic tree was generated with FigTree [17].
Table 1. Classification and general features of Liberibacter crescens BT-1 according to the MIGS recommendations [18].
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [11] | |
| Phylum Proteobacteria | TAS [19] | ||
| Class Alphaproteobacteria | TAS [20,21] | ||
| Order Rhizobiales | TAS [22] | ||
| Family Rhizobiaceae | TAS [23,24] | ||
| Genus Liberibacter | TAS [25,26] | ||
| Species Liberibacter crescens | |||
| Type strain BT-1 | |||
| Gram stain | negative | TAS [11] | |
| Cell shape | rod-shaped | TAS [11] | |
| Motility | nonmotile | IDA | |
| Sporulation | nonsporulating | IDA | |
| Temperature range | mesophile | IDA | |
| Optimum temperature | 27° C | TAS [11] | |
| Carbon source | unknown | NAS | |
| Energy source | unknown | NAS | |
| MIGS-6 | Habitat | mountain papaya | TAS [11] |
| MIGS-6.3 | Salinity | unknown | NAS |
| MIGS-22 | Oxygen | aerobic | TAS [11] |
| MIGS-15 | Biotic relationship | endophyte | TAS [11] |
| MIGS-14 | Pathogenicity | none | TAS [11] |
| MIGS-4 | Geographic location | Puerto Rico, USA | NAS |
| MIGS-5 | Sample collection time | 1995 | NAS |
| MIGS-4.1 | Latitude | 18.051944N | NAS |
| MIGS-4.2 | Longitude | 67.059722W | NAS |
| MIGS-4.3 | Depth | surface | NAS |
| MIGS-4.4 | Altitude | 12 m | NAS |
a) Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [27].
Figure 2.

Transmission electron microscopy of L. crescens BT-1. Negative stain. Scale bar represents 500 nm.
Genome sequencing and annotation
Three sequencing platforms were used to obtain the data necessary to close the genome sequence (Table 2). In addition, other project information and its association with MIGS version 2.0 compliance [32] is provided (Table 2).
Table 2. Project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three libraries: one illumina library, two 454 paired-end libraries (3kb and 8kb insert size), one PacBio standard library |
| MIGS-29 | Sequencing platforms | Illumina GAIIx, 454 GS FLX Titanium, PacBio RS |
| MIGS-31.2 | Fold coverage | 6121.2x illumina, 166.9× 454, 31.7× PacBio |
| MIGS-30 | Assemblers | Mira v3.4.0.1 [28]; Amos v3.0.0 [29]; WGS v7.0.1.22 [30]; Velvet v1.2.06 [31] |
| MIGS-32 | Gene calling method | Glimmer [31] |
| Genome Database release | ||
| Genbank ID | CP003789 | |
| Genbank Date of Release | On publication, or August 31, 2013 | |
| GOLD ID | ||
| Project relevance | Agriculture |
Growth conditions and DNA isolation
The initial culture of BT-1 was obtained in 1995 and was isolated from the peduncle of the tropical Babaco plant, also known as the hybrid mountain papaya (Carica stipulata x C. pubescens). Babaco was provided by the Lajas Experiment station in Puerto Rico because it showed signs of Papaya bunchy top (PBT), a disease of papaya in the American tropics. The sap of Babaco expressed an extremely high titer of small, rod-shaped bacteria [1].
Despite being fastidious, the bacterium was able to be grown on BM7 media, a modified form of BBM [1,11]. Cells were grown in BM7 liquid culture at 27°C for 4 days in a shaking incubator with a speed of 120 rpm. DNA was extracted using the UltraClean Microbial DNA Isolation Kit and the manufacturer’s protocol (M0-BIO, Carlsbad, CA).
Genome sequencing and assembly
Sequencing was performed by the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida using the PacBio RS, Illumina GaIIx, and Roche/454 GS/FLX Titanium platforms. The initial draft assembly yielded 5 large (>1,500 bp), non-redundant contigs with an N50 of 379,608bp by combing 831,945 Roche/454 reads (3kb and 8kb insert libraries) at 166.93× coverage, 3,514,850 normalized Illumina reads [33] at 107.95× coverage, and 10,798 corrected PacBio reads [34] at 7.81× coverage by hybrid assembly through the Mira assembler [28]. The resulting maximal base-error rate (<Q40) of the initial assembly was 45 in 50,000.
Contigs were subject to an NcoI restriction digest (in silico) and mapped to an OpGen optical map of BT-1 using the same enzyme [35]. Optical mapping yielded a circular map of approximately 1.5 Mbp. Misjoined contigs and contig redundancy were indicated by comparison of the scaffold to the optical map of L. crescens, and were manually corrected with the CLC Genomics Workbench (CLCbio, Katrinebjerg, Denmark).
Intrascaffold gaps were closed by further passes of the Mira hybrid assembly combining the current scaffold with varying combinations of read data. Omitting certain read technologies at further hybrid assembly iterations allowed more successful assemblies at different points of the genome. Pseudo 454-like paired-end reads were generated from the scaffold to allow very large contigs to be employed in further iterations of Mira hybrid assembly. Pseudo 454-like reads conformed to the 19 kb upper limit of Mira read length and consisted of a 34 kb insert size. Additionally, subsets of the original Illumina paired-end reads and normalized Roche/454 reads were entered into the read pool to avoid problematic reads. Contigs of each hybrid assembly pass were manually corrected for misjoined contigs and combined by Minimus2 [29] to yield a circular genomic sequence.
Genome annotation
Genome annotation was performed by the Rapid Annotation using Subsystem Technology (RAST) pipeline [36]. RAST employs tRNAscan-SE [37] to identify tRNA genes, Niels Larsen’s "search_for_rnas" (available from the author) to identify rRNA encoding genes, and GLIMMER [38] to identify candidate protein-encoding genes. RAST compares the set of candidate protein-encoding genes to a collection of protein families, referred to as FIGfams [36], in order to correct CDS starting positions and place the genome in a phylogenic context. The candidate protein set was compared to the National Center for Biotechnology Information (NCBI) non-redundant (nr) database, SwissProt database, European Bioinformatics Institute (EBI) phage database, and COG subset of the NCBI Conserved Domain Database (CDD) through the NCBI BLAST suite.
Additionally, predicted proteins were annotated through the Kyoto Encyclopedia of Genes and Genomes (KEGG) automatic annotation server (KAAS). KAAS employs NCBI BLAST to search the KEGG Orthology database [39].
Genome properties
The genome consists of one circular chromosome of 1,504,659 bp (35.35% GC content). 1,433 genes were predicted, 1,379 of which are protein-coding genes. 1,039 of protein coding genes were assigned to a putative function with the remaining being annotated as hypothetical proteins. The properties and the statistics of the genome are summarized in Tables 3 and 4.
Table 3. Nucleotide content and gene count levels of the genome.
| Attribute | Value | % of totala |
|---|---|---|
| Genome size (bp) | 1,504,659 | 100 |
| DNA coding region (bp) | 1,264,794 | 84.05 |
| DNA G+C content (bp) | 531,980 | 35.35 |
| Total genesb | 1433 | 100 |
| RNA genes | 54 | 3.77 |
| Protein-coding genes | 1379 | 96.23 |
| Genes in paralog clusters | 870 | 63.08 |
| Genes assigned to COGs | 857 | 62.14 |
| Genes assigned Pfam domains | 1057 | 76.65 |
| Genes with signal peptides | 84 | 6.09 |
| Genes with transmembrane helices | 327 | 23.71 |
a) The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
b) Does not include pseudogenes or other genes.
Table 4. Number of genes associated with the 25 general COG functional categories.
| Code | Value | %agea | Description |
|---|---|---|---|
| J | 123 | 14.35 | Translation |
| A | 0 | 0.00 | RNA processing and modification |
| K | 21 | 2.45 | Transcription |
| L | 66 | 7.70 | Replication, recombination and repair |
| B | 0 | 0.00 | Chromatin structure and dynamics |
| D | 14 | 1.63 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.00 | Nuclear structure |
| V | 12 | 1.40 | Defense mechanisms |
| T | 26 | 3.03 | Signal transduction mechanisms |
| M | 57 | 6.65 | Cell wall/membrane biogenesis |
| N | 31 | 3.62 | Cell motility |
| Z | 0 | 0.00 | Cytoskeleton |
| W | 0 | 0.00 | Extracellular structures |
| U | 19 | 2.22 | Intracellular trafficking and secretion |
| O | 45 | 5.25 | Posttranslational modification, protein turnover, chaperones |
| C | 65 | 7.58 | Energy production and conversion |
| G | 30 | 3.50 | Carbohydrate transport and metabolism |
| E | 96 | 11.20 | Amino acid transport and metabolism |
| F | 41 | 4.78 | Nucleotide transport and metabolism |
| H | 56 | 6.53 | Coenzyme transport and metabolism |
| I | 33 | 3.85 | Lipid transport and metabolism |
| P | 29 | 3.38 | Inorganic ion transport and metabolism |
| Q | 5 | 0.58 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 57 | 6.65 | General function prediction only |
| S | 31 | 3.62 | Function unknown |
| - | 522 | Not in COGs |
a) The total is based on the total number of COGs in the annotated genome.
Insights from the genome sequence and comparative genomics
Sequencing of Liberibacter crescens BT-1 was conducted to learn why this strain can be cultured while the other Liberibacter strains cannot. Also, as BT-1 is not a pathogen of citrus, the BT-1 genome may suggest how Candidatus L. asiaticus causes symptoms on citrus while BT-1 does not. Members of the Liberibacter genus (Candidatus. L. asiaticus, Candidatus L. africanus, and Candidatus. L. americanus) are known to be the causative agent of Huanglongbing (HLB), commonly called citrus greening, and other HLB-like diseases (Candidatus L. solanacearum) [5-7]. However, some members of the Liberibacter genus are non-pathogenic, Candidatus L. europeaus [8] and L. crescens [11].
Although L. crescens is currently the only member of the Liberibacter genus to be cultured, the sequences of Candidatus L. asiaticus and Candidatus L. solanacearum are available through NCBI. Comparison of gene function and sequence in BT-1 to Candidatus L. asiaticus and Candidatus L. solanacearum provided insight to both the virulence and the fastidious nature of the Liberibacter genus. Additionally, the Liberibacter genus is predicted to be susceptible to bacteriophage insertions, which were also analyzed between the known genomes.
Sequence comparison of L. crescens to Ca. L. asiaticus and Ca. L. solanacearum
KEGG orthology and RAST automated annotation were the basis of functional comparison of the genes in L. crescens to the genes in Candidatus L. asiaticus and Candidatus L. solanacearum.
Analysis of KEGG orthology uncovered the complete inability of Candidatus L. asiaticus and Candidatus L. solanacearum to synthesize histidine, tryptophan, and thiamine, as well as a severely reduced ability to produce phenylalanine and tyrosine when compared to L. crescens. Candidatus L. asiaticus and Candidatus L. solanacearum both possess 2 out of the 12 enzymes required for phenylalanine and tyrosine biosynthesis. To compensate, all three species possess a general L-amino acid ATP-binding cassette (ABC) transporter. ABC transporters are known to be associated with nutrient uptake, drug resistance, and virulence [40,41]. Also, Candidatus L. asiaticus and Candidatus L. solanacearum possess a thiamine ABC transporter not found in L. crescens, presumably to compensate for the inability to synthesize thiamine. These deficiencies provide insight into the metabolic requirements of the uncultured Liberibacter species.
Furthermore, KEGG orthology and RAST annotation indicate the presence of a zinc ABC transporter in all three species. Transporters of metal ions have been shown to play a role in bacterial virulence, including ABC transporters of iron, zinc, and manganese [42,43]. Although the zinc transporter was located in L. crescens through RAST annotation, it was not detected by KEGG orthology. This discrepancy is attributed to a low sequence similarity between the protein components of the zinc ABC transporter (ZnuA, ZnuB, ZnuC) in L. crescens compared to Candidatus L. asiaticus and Ca. L. solanacearum, at 43.6%, 55.3%, and 48.5% average similarity for each component, respectively (Table 5). In contrast, the similarity of each component between Candidatus L. asiaticus and Candidatus L. solanacearum is 78.6%, 93.0%, and 92.2% respectively (Table 5). Sequence similarity was determined through sequence alignment using the EMBOSS Water tool [44] and the EBLOSUM62 scoring matrix. This variation in zinc ABC transport proteins may contribute to the virulence of the Liberibacter genus.
Table 5. Species similarity of zinc ABC transporter components.
| ZnuA | ZnuB | ZnuC | |
|---|---|---|---|
| L. crescens to Candidatus L. asiaticus | 43.1% | 55.2% | 46.9% |
| L. crescens to Candidatus L. solanacearum | 44.0% | 55.4% | 50.0% |
| Average | 43.6% | 55.3% | 48.5% |
| Ca. L. asiaticus to Ca. L. solanacearum | 78.6% | 93.0% | 92.2% |
Also present in L. crescens, but not in Candidatus L. asiaticus and Candidatus L. solanacearum, is a twin-arginine translocation (Tat) protein export pathway and an additional iron ABC transporter. The significance of these two transporters is not currently known, but their existence may explain why L. crescens, is less fastidious than Candidatus L. asiaticus and Candidatus L. solanacearum.
Present in Candidatus L. asiaticus and Candidatus L. solanacearum, but not in L. crescens, are several components of a fimbrial low-molecular-weight protein (flp) pilus system. These pili are involved in tight adherence and are encoded by the Tad family proteins [7]. Diversity in the flp pilus operon is predicted to contribute to variation in virulence among pathogenic species [45-48], and provides further insight to the virulence of the Liberibacter genus.
Phages in the genomes of Candidatus L. asiaticus and L. crescens
Recently, two prophages, SC1 and SC2, were found to exist in tandem in Candidatus L. asiaticus through DNA isolation from diseased citrus phloem and an insect vector of the family Psyllidae [10]. Candidatus L. solanacearum is known to host two prophage regions as well, not in tandem, with one region maintaining a high degree of similarity with the prophage regions in Candidatus L. asiaticus and the other containing a small segment with lower similarity [7]. Two putative prophages were found in the L. crescens genome through the use of the Prophage Finder tool [49], the Phage_Finder [50] tool, and the methods described in Casjens et al (2003).
Prophage boundary identification is an inexact process due to the diversity of bacteriophages, and is made even more difficult by the possibility of evolutionary decay of prophages that do not enter a lytic cycle. Additionally, prophage boundaries are indicated by a multitude of factors, but not defined by any particular criteria. Position of nearby tRNAs close to the predicted prophage region may be indicative of a boundary, as tRNAs are often sites of phage insertion [50]. A sharp shift in G+C content at the predicted prophage region may also indicate the range of phage insertion, but only if the phage G+C content differs dramatically from the host. Certain genes are unique to phage genomes, and non-phage genes were not typically found to be present between phage genes in an inserted phage. From a genomic standpoint, prophage regions are also indicated by regions not present in closely related species, as well as long strings of unidentified proteins in similar orientation [51].
From the above criteria, the locations and boundaries of two prophages in L. crescens were predicted to extend from base pair 523,789-564,039 in prophage LC1 and from base pair 848,435-886,798 in prophage LC2. Unlike the two prophages in Candidatus L. asiaticus, the prophages in L. crescens were not homologues, sharing only short (<1,000 bp) regions of moderate similarity, determined through Wise2 alignment [52]. Additionally, the prophages in L. crescens were not found in Candidatus L. asiaticus. Homology was inferred through alignment by the progressiveMauve algorithm [53] (Figures 3-5). While the SC1 phage in Candidatus L. asiaticus is known to enter a lytic cycle in the phloem of citrus, the lifecycles of the prophages in L. crescens have yet to be explored experimentally [10].
Figure 3.

Circular genomic map of L. crescens BT-1. From outside to the center: Genes on forward strand (colored by labeled COG categories), genes on reverse strand (colored by labeled COG categories), RNA genes (tRNA green, rRNA red), putative prophage regions, GC content, GC skew.
Figure 5.
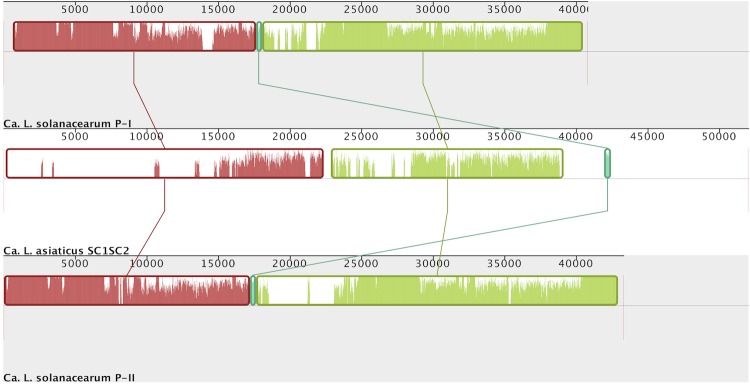
Candidatus L. solanacearum and Candidatus L. asiaticus. Signifies that prophages in L. crescens are not homologous to each other or to the tandem prophage region in Candidatus L. asiaticus.
Figure 4.

Whole sequence alignment of phage regions between L. crescens and Candidatus L. asiaticus. The two prophage regions Candidatus L. solanacearum are homologous, and both share higher similarity with the prophage region in Candidatus L. asiaticus. Graphical representation obtained through Mauve [53].
Interestingly, the same zinc ABC transporter mentioned above is present in the LC2 region. Prophage insertions have been known to add functions to hosts, making the host more competitive [54]. In addition to metabolic variation, the differences in extra-chromosomal genomic content between species of the Liberibacter genus may also be indicative of the virulence and fastidious nature of the genus.
Conclusion
Liberibacter crescens BT-1 is the first member of the Liberibacter genus to be cultured. The complete genome sequences of Candidatus L. asiaticus and Candidatus L. solanacearum have been determined through isolation from the disease vectors [7,9], but any attempt to culture these species typically depends on employing a co-culture with insect or plant host cells [5]. Genomic sequencing of L. crescens BT-1 was performed in an attempt to find possible indications for virulence in Candidatus L. asiaticus and Candidatus L. solanacearum, as well as an explanation for the fastidious nature of these pathogens.
Assembly of L. crescens yielded a complete genome containing two predicted prophages. Sequence comparison of Candidatus L. asiaticus to L. crescens indicated that the species are 75.5% similar [11]. However, the prophage regions are not homologous. Sequencing and analysis of the L. crescens genome provided insight to the metabolic requirements of Candidatus L. asiaticus, which appears to lack the ability to synthesize thiamine and several essential amino acids. Less is known about the virulence of Candidatus L. asiaticus, although bacteriophages have also been known to add virulence to an otherwise non-pathogenic bacterium [54]. Further genomic analysis indicated that virulence in Candidatus L. asiaticus could also be due to a zinc ABC transporter. While the sequencing of L. crescens gave much insight into the Liberibacter genus, further experiments must be conducted to verify these predictions.
Abbreviations:
- EMBL- European Molecular Biology Laboratory
NCBI- National Center for Biotechnology Information (Bethesda, MD, USA), HLB- Huanglongbing, ZC- Zebra Chip, PBT- Papaya bunchy top, ICBR- Interdisciplinary Center for Biotechnology Research, RAST- Rapid Annotation using Subsystem Technology, NR- non-redundant, CDD- Conserved Domain Database, KEGG- Kyoto Encyclopedia of Genes and Genomes, KASS- automatic annotation server, ABC- ATP-binding cassette, Tat- twin-arginine translocation, flp- fimbrial low-molecular-weight protein
References
- 1.Davis MJ, Mondal SN, Chen H, Rogers ME, Brlansky RH. Co-cultivation of ‘ Candidatus Liberibacter asiaticus ’ with Actinobacteria from Citrus with Huanglongbing. Plant Dis 2008; 92:1547-1550 10.1094/PDIS-92-11-1547 [DOI] [PubMed] [Google Scholar]
- 2.Kapur SP, Kapoor SK, Cheema SS, Dhillon RS. Effect of greening disease on tree and fruit characters of Kinnow mandarin. Punjab Horticultural Journal 1978; 50:76-79 [Google Scholar]
- 3.Bové J. Huanglongbing: A Destructive, Newly-Emerging, Century-Old Disease of Citrus. J Plant Pathol 2006; 88:7-37 [Google Scholar]
- 4.Zhang M, Powell CA, Guo Y, Doud MS, Duan Y. A Graft-Based Chemotherapy Method for Screening Effective Molecules and Rescuing Huanglongbing-Affected Citrus Plants. Phytopathology 2012; 102:567-574 10.1094/PHYTO-09-11-0265 [DOI] [PubMed] [Google Scholar]
- 5.Sechler A, Schuenzel EL, Cooke P, Donnua S, Thaveechai N, Postnikova E. Stone aL, Schneider WL, Damsteegt VD, Schaad NW. Cultivation of 'Candidatus Liberibacter asiaticus', 'Ca. L. africanus', and 'Ca. L. americanus' associated with huanglongbing. Phytopathology 2009; 99:480-486 10.1094/PHYTO-99-5-0480 [DOI] [PubMed] [Google Scholar]
- 6.Teixeira DC. 'Candidatus Liberibacter americanus', associated with citrus huanglongbing (greening disease) in Sao Paulo State, Brazil. Int J Syst Evol Microbiol 2005; 55:1857-1862 10.1099/ijs.0.63677-0 [DOI] [PubMed] [Google Scholar]
- 7.Lin H, Lou B, Glynn JM, Doddapaneni H, Civerolo EL, Chen C, Duan Y, Zhou L, Vahling CM. The Complete Genome Sequence of ‘Candidatus Liberibacter solanacearum’, the Bacterium Associated with Potato Zebra Chip Disease. PLoS ONE 2011; 6:e19135 10.1371/journal.pone.0019135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raddadi N, Gonella E, Camerota C, Pizzinat A, Tedeschi R, Crotti E, Mandrioli M, Attilio Bianco P, Daffonchio D, Alma A. ‘Candidatus Liberibacter europaeus’ sp. nov. that is associated with and transmitted by the psyllid Cacopsylla pyri apparently behaves as an endophyte rather than a pathogen. Environ Microbiol 2011; 13:414-426 10.1111/j.1462-2920.2010.02347.x [DOI] [PubMed] [Google Scholar]
- 9.Tyler HL, Roesch LF, Gowda S, Dawson WO, Triplett EW. Confirmation of the sequence of 'Candidatus Liberibacter asiaticus' and assessment of microbial diversity in Huanglongbing-infected citrus phloem using a metagenomic approach. Mol Plant Microbe Interact 2009; 22:1624-1634 10.1094/MPMI-22-12-1624 [DOI] [PubMed] [Google Scholar]
- 10.Zhang S, Flores-Cruz Z, Zhou L, Kang BH. Fleites La, Gooch MD, Wulff Na, Davis MJ, Duan Y-P, Gabriel DW. 'Ca. Liberibacter asiaticus' carries an excision plasmid prophage and a chromosomally integrated prophage that becomes lytic in plant infections. Molecular plant-microbe interactions. Mol Plant Microbe Interact 2011; 24:458-468 10.1094/MPMI-11-10-0256 [DOI] [PubMed] [Google Scholar]
- 11.Fagen J, Leonard MT, McCullough CM, Triplett EW, Davis MJ. Liberibacter crescens gen.nov; sp. nov. first cultured member of the Liberibacter genus. International Journal of Systematic and Evolutionary Bacteriology 2012. [DOI] [PubMed]
- 12.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792-1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- 14.Guindon S, Gascuel OA. Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst Biol 2003; 52:696-704 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- 15.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol 2006; 55:539-552 10.1080/10635150600755453 [DOI] [PubMed] [Google Scholar]
- 16.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 2008; 36:W465-W469 10.1093/nar/gkn180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rambaut A. FigTree. <http://tree.bio.ed.ac.uk/software/figtree/%3E
- 18.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1. [Google Scholar]
- 20.Validation List No 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1-6 10.1099/ijs.0.64188-0 [DOI] [PubMed] [Google Scholar]
- 21.Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1. [Google Scholar]
- 22.Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 324. [Google Scholar]
- 23.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 24.Conn HJ. Taxonomic relationships of certain non-sporeforming rods in soil. J Bacteriol 1938; 36:320-321 [Google Scholar]
- 25.Jagoueix S, Bové JM, Garnier M. The Phloem-Limited Bacterium of Greening Disease of Citrus Is a Member of the {alpha} Subdivision of the Proteobacteria. Int J Syst Bacteriol 1994; 44:379-386 10.1099/00207713-44-3-379 [DOI] [PubMed] [Google Scholar]
- 26.Garnier M, Jagoueix-Eveillard S, Cronje PR, Le Roux HF, Bové JM. Genomic characterization of a liberibacter present in an ornamental rutaceous tree, Calodendrum capense, in the Western Cape Province of South Africa. Proposal of 'Candidatus Liberibacter africanus subsp. capensis'. Int J Syst Evol Microbiol 2000; 50:2119-2125 10.1099/00207713-50-6-2119 [DOI] [PubMed] [Google Scholar]
- 27.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information. … Science and Biology: Proceedings of the … 1999. [Google Scholar]
- 29.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 2007; 8:64 10.1186/1471-2105-8-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myers EW. A Whole-Genome Assembly of Drosophila. Science 2000; 287:2196-2204 10.1126/science.287.5461.2196 [DOI] [PubMed] [Google Scholar]
- 31.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH. A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. eprint arXiv 2012;1203:1-18.
- 34.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z. Rasko Da, McCombie WR, Jarvis ED and others. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 2012; 30:693-700 10.1038/nbt.2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, Bode HB, Darby C, Du Z, Forst S, et al. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics 2007; 8:321 10.1186/1471-2164-8-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008; 9:75 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955-964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salzberg SL. Delcher aL, Kasif S, White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res 1998; 26:544-548 10.1093/nar/26.2.544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 2007; 35:W182-W185 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garmory HS, Titball RW. MINIREVIEW ATP-Binding Cassette Transporters Are Targets for the Development of Antibacterial Vaccines and Therapies. Infect Immun 2004; 72:6757-6763 10.1128/IAI.72.12.6757-6763.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Testerman TL, Conn PB, Mobley HLT, Mcgee DJ. Nutritional Requirements and Antibiotic Resistance Patterns of Helicobacter Species in Chemically Defined Media Nutritional Requirements and Antibiotic Resistance Patterns of Helicobacter Species in Chemically Defined Media. Infect Immun. 2006 doi: 10.1128/JCM.44.5.1650-1658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hantke K. Bacterial zinc uptake and regulators. Curr Opin Microbiol 2005; 8:196-202 10.1016/j.mib.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 43.Panina EM. Comparative genomics of bacterial zinc regulons: Enhanced ion transport, pathogenesis, and rearrangement of ribosomal proteins. Proc Natl Acad Sci USA 2003; 100:9912-9917 10.1073/pnas.1733691100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 2000; 16:276-277 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- 45.Boyd JM, Dacanay A, Knickle LC, Touhami A, Brown LL, Jericho MH, Johnson SC, Reith M. Contribution of Type IV Pili to the Virulence of Aeromonas salmonicida subsp. salmonicida in Atlantic Salmon (Salmo salar L.). Infect Immun 2008; 76:1445-1455 10.1128/IAI.01019-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clock SA, Planet PJ, Perez BA, Figurski DH. Outer Membrane Components of the Tad (Tight Adherence) Secreton of Aggregatibacter actinomycetemcomitans. J Bacteriol 2007; 190:980-990 10.1128/JB.01347-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li T, Xu Z, Zhang T, Li L, Chen H, Zhou R. The genetic analysis of the flp locus of Actinobacillus pleuropneumoniae. Arch Microbiol 2011; 194:167-176 10.1007/s00203-011-0741-6 [DOI] [PubMed] [Google Scholar]
- 48.Wairuri CK, van der Waals JE, van Schalkwyk A, Theron J. Ralstonia solanacearum Needs Flp Pili for Virulence on Potato. Mol Plant Microbe Interact 2011; 25:546-556 10.1094/MPMI-06-11-0166 [DOI] [PubMed] [Google Scholar]
- 49.Bose M, Barber RD. Prophage Finder: a prophage loci prediction tool for prokaryotic genome sequences. In Silico Biol 2006; 6:223-227 [PubMed] [Google Scholar]
- 50.Fouts DE. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res 2006; 34:5839-5851 10.1093/nar/gkl732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol 2003; 49:277-300 10.1046/j.1365-2958.2003.03580.x [DOI] [PubMed] [Google Scholar]
- 52.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res 2000; 28:263-266 10.1093/nar/28.1.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darling AE, Mau B, Perna NT. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010; 5:e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winstanley C, Langille MGI, Fothergill JL, Kukavica-ibrulj I, Paradis-bleau C, Sanschagrin F, Thomson NR, Winsor GL, Quail MA, Lennard N, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res 2009; 19:12-23 10.1101/gr.086082.108 [DOI] [PMC free article] [PubMed] [Google Scholar]