

Abstract
Macrophages are centrally involved during atherosclerosis development and are the predominant cell type that accumulates cholesterol in the plaque. Macrophages however, are heterogeneous in nature reflecting a variety of microenvironments and different phenotypes may be more prone to contribute towards atherosclerosis progression. Using primary human monocyte-derived macrophages, we sought to evaluate one aspect of atherogenic potential of different macrophage phenotypes by determining their propensity to associate with and accumulate oxidized low density lipoprotein (oxLDL). Classically-activated macrophages treated simultaneously with interferon γ (IFNγ) and tumour necrosis factor α (TNFα) associated with less oxLDL and accumulated less cholesterol compared to untreated controls. The combined treatment of IFNγ and TNFα reduced the mRNA expression of CD36 and the expression of both cell surface CD36 and macrophage scavenger receptor 1 (MSR1) protein. Under oxLDL loaded conditions, IFNγ and TNFα did not reduce macrophage protein expression of the transcription factor peroxisome proliferator-actived receptor γ (PPARγ) which is known to positively regulate CD36 expression. However, macrophages treated with IFNγ did prevent the PPARγ-specific agonist rosiglitazone from upregulating cell surface CD36 protein expression. Our results demonstrate that the observed reduction of cholesterol accumulation in macrophages treated with IFNγ and TNFα following oxLDL treatment was due at least in part to reduced cell surface CD36 and MSR1 protein expression.
Keywords: Macrophage, cholesterol, interferon γ, CD36, macrophage scavenger receptor I, peroxisome proliferator-activated receptor γ
1. INTRODUCTION
Atherosclerosis is a chronic inflammatory condition characterized by the formation of sub-endothelial lesions composed of immune cells, connective tissue and lipids [1]. Although various cell types participate in the development of the atherosclerotic plaque, the monocyte-macrophage lineage has been identified as one of the main contributors to the initiation and progression of atherosclerotic disease. Circulating monocytes have been shown to bind to the vascular endothelium and infiltrate into the intima where they differentiate into macrophages, eventually becoming lipid-engorged foam cells. Foam cells are present at all stages of atherosclerosis; their appearance in the arterial intima marks the initial stage of atherosclerosis while during late stages, foam cells perpetuate the destabilization of the plaque through their secretion of inflammatory mediators and matrix-degrading enzymes, leading to a stream of events that culminate in plaque rupture and often fatal ischemic attacks [2–4]. It is generally accepted that receptor-mediated uptake of modified forms of LDL is a common pathway implicated in foam cell formation. Unlike the uptake of native forms of LDL, macrophages have been shown to take up modified LDL in an unregulated fashion through a family of receptors known as scavenger receptors [5–7]. Of the known scavenger receptors, the combined contribution of two in particular have been shown to be responsible for up to 90% of modified LDL uptake in vitro: CD36 and macrophage scavenger receptor I (MSR1, also known as SR-A) [8].
Macrophages within the lesion are heterogeneous in nature and exhibit a variety of phenotypic states ranging from pro-inflammatory to anti-inflammatory[9]. These phenotypes are thought to be dynamic responses to their environment as their ability to switch from one phenotype to another has been demonstrated in vitro [10]. Classically-activated pro-inflammatory macrophages can be induced by incubation with the Th1 cytokines interferon (IFN) γ and tumour necrosis factor (TNF) α which work synergistically together and are both required to induce maximal macrophage activation [11–14]. These macrophages are characterized by their secretion of pro-inflammatory cytokines, nitric oxide and high capacity to present antigen [15]. Evidence suggests that classically-activated macrophages are pro-atherogenic: In vivo, administration of exogenous IFNγ increased lesion sizes in the atherosclerosis-prone Apoe−/− model while mice deficient in the IFNγ receptor show smaller lesions [16, 17]. Furthermore, mouse models such as C57BL/6 mice that demonstrate a dominant Th1 response are more prone to developing atherosclerosis [18].
By contrast, alternatively-activated anti-inflammatory macrophages can be induced with the Th2 cytokines interleukin (IL)-4 and IL-13 and are characterized by their secretion of anti-inflammatory cytokines such as IL-10 and TGF-β [15]. IL-4 stimulates the activity of the nuclear receptor peroxisome proliferator-activated receptor (PPARγ) which has been shown to mediate alternative macrophage activation as well as the transcriptional repression of several pro-inflammatory transcription factors [9, 19–21]. PPARγ activation is partially responsible for the antagonization of several IFNγ-mediated activities such as the upregulation of TNFα, IL-12(p40), IL-1β, NO, and promoting T helper cell differentiation towards a Th1 phenotype [22–26].
Treatment of macrophages with IL-10 also induces an alternatively-activated macrophage phenotype albeit distinct from those induced by IL-4 and IL-13. Like IL-4, IL-10 has antagonizing effects on pro-inflammatory responses; it can attenuate inflammation by inhibiting IL-1α, IL-1β, IL-6, IL-12, and TNFα production and also prevent STAT1 activation by IFNγ [27, 28]. IL-10 deactivates macrophages by downregulating the MHC class II molecule and preventing endocytosis which is conversely stimulated by IL-4 and IL-13 [29, 30]. In part due to their anti-inflammatory profile, alternatively activated macrophages are believed to be protective against atherosclerosis although of these two alternatively activated sub-phenotypes, only IL-10 has shown consistent in vivo evidence of atheroprotection [31–33].
We have found that there are few studies that make direct comparisons of the atherogenic potential among different macrophage phenotypes. In the present study, we have compared the propensity of different macrophage sub-phenotypes to accumulate cholesterol following treatment with oxidized LDL. Our results indicate that pro-inflammatory primary human monocyte-derived macrophages (MDMs) accumulate less cholesterol following oxLDL treatment than untreated controls and are associated with reduced expression of the scavenger receptors MSR1 and CD36.
2. METHODS
2.1 Cell culture
CD14+ human peripheral blood monocytes (AllCells) were cultured in RPMI 1640 complete media (Hyclone) containing 10% FBS (Hyclone), 2% sodium bicarbonate, 1% sodium pyruvate, and 1%penicillin-streptomycin (Invitrogen). Cells were plated at a density of 106/ml in 24, 12 or 6 well CellBind plates (Corning) depending on the assay. Recombinant human M-CSF (R&D Systems) was added for the initial 4 days of culture at a concentration of 10 ng/ml. A media change was performed at day 4 of culture in the absence of M-CSF. Macrophages were treated with specific cytokines (R&D Systems) for 72 hours starting at day 8 of culture including human interleukin-4 and interleukin-13 (IL-4/13, 10 ng/ml each), interferon γ and tumor necrosis factor α (IFNγ/TNFα, 10 ng/ml each), interleukin-10 (IL-10, 20 ng/ml), or an untreated control containing complete media with no additional cytokines. Where loading with oxLDL was applicable, macrophages were washed twice with plain RPMI 1640 and loaded in serum-free RPMI 1640 containing 50 μg/ml of oxLDL on day 10 for 24 hours. Cytokines were maintained during the entire loading period. Rosiglitazone was added to wells at a final concentration of 5 uM on day 9 for 48 hours where appropriate.
2.2 Isolation and oxidation of low density lipoprotein (LDL)
Pooled normal human plasma using K3 EDTA as an anticoagulant (Innovative Research) was adjusted to a density of 1.019 g/ml using sodium bromide (Sigma Aldrich) and spun at 50,000 RPM using a L8-55M Ultracentrifuge (Beckman Coulter) for 24 hours at 8°C. The top layer containing chylomicrons and VLDL was removed and the density of the remaining solution was adjusted to 1.063 g/ml using sodium bromide and spun again at 50,000RPM for 24 hours at 8°C. The top LDL layer was collected and dialyzed using 7K MWCO Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific) in 4 L of 1 × PBS for 48 hours. PBS was changed twice per day.
LDL protein concentration was determined using a bicinchoninic acid protein assay (Thermo Scientific). LDL was oxidized using 5 uM copper sulfate per 200 ug/ml of LDL for a period of 18 hours at 37°C. Oxidation was verified by determining the electrophoretic mobility using a Paragon Lipo Kit (Beckman) and by measuring the fluorescence of Shiff base imine products at excitation/emission 360 nm/430 nm on a Safire fluorescence plate reader (Tecan) [34].
2.3 oxLDL cellular association
Oxidized LDL (oxLDL) was labeled with the lipophilic dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate ((DiI, Sigma) based on the protocol developed by Stephan and Yurachek [35] by adding 300 ug of DiI dissolved in DMSO per 1 mg of LDL and incubating at 37°C for 18 hours followed by filtering with a 0.22 um filter (Millipore). DiI-labeled oxidized LDL from here on in will be referred to as DiI-oxLDL.
Polarized macrophages were washed twice with sterile 1 x PBS and subsequently treated with serum-free RPMI containing 10 ug/ml of DiI-oxLDL for 4 hours. Cytokine treatments were maintained during the loading period. The wells were washed twice with 1 x PBS and the cells were lysed with 100 ul of RIPA buffer. The wells were scraped with a cell scraper and collected in a 1.5 ml microcentrifuge tube prior to reading with a Safire fluorescent plate reader (Tecan) at excitation wavelength 520 nm and emission at 580 nm. Fluorescence was compared with a DiI-oxLDL standard curve and normalized by cellular protein levels.
2.4 Cellular cholesterol accumulation
Macrophages were washed twice with 1 x PBS and lysed with 100 ul of RIPA buffer. The wells were scraped with a cell scraper and collected in 1.5 ml microcentrifuge tubes. Endogenous peroxides in the cellular lysates were removed by treating with 1U of bovine catalase and incubating at 37°C for 15 minutes. Total cholesterol was then determined by using the Amplex Red Kit (Invitrogen) and measured using a Safire fluorescent plate reader (Tecan) at the excitation wavelength 545 nm and emission at 590 nm. Cellular cholesterol was normalized by cellular protein levels.
2.5 RNA extraction and analysis
Total RNA was isolated using an RNeasy mini kit (Qiagen). RNA quality and concentration was assessed using the NanoDrop 8000 (Thermo Scientific). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was conducted using a one step method using the qScript One-step Fast MGB qRT-PCR Kit, ROX (Quantas Biosciences) combined with Taqman gene expression assays (Applied Biosystems) containing the probe and primers for the targets of interest. qRT-PCR was conducted on an ABI 7900 and mRNA levels were normalized to peptidylprolyl isomerase B. A list of the Taqman gene expression assays used in this study are provided in supplementary material Table 1.
2.6 Protein extraction and Western blot analysis
Polarized macrophages in 6-well plates were washed twice with 1 x PBS and lysed with RIPA buffer containing protease and phosphatase inhibitors (Pierce) on ice. Wells were scraped with a cell scraper and collected into 1.5 ml microcentrifuge tubes. Lysates were left on ice for 30 minutes, vortexing briefly every 10 minutes. Lysates were then spun at 4°C at 20,800×g for 10 minutes and the supernatant was collected and stored at −80°C.
Western blots were performed by first adding 2 x laemmli buffer to 15 ug of total cellular protein and boiled at 90°C for 10 minutes. Samples were vortexed, briefly spun down and added to lanes of a 4–15% gradient polyacrylamide gel (Biorad Laboratories). Gels were run at 100V for 1.5 hours and a wet transfer was performed to a PVDF membrane (Millipore) overnight at 35V, in a cold room set to 4°C. After transfer was completed, membranes were blocked for 1 hour at room temperature with SuperBlock (Pierce). Membranes were incubated with primary antibodies followed by 5 × 5-minute washes in 1 x TBST. Primary antibodies include mouse anti-β-actin (Sigma), mouse anti-MSR1 (Cosmo Bio) and rabbit anti-PPARγ (Santa Cruz Biotechnology). Membranes were then incubated with the either HRP-conjugated mouse or rabbit TrueBlot (eBioscience) where appropriate followed by another set of 5 × 5-minute wash. Protein-antibody interactions were visualized using Super Signal West Femto enhanced chemiluminescence reagent (Pierce) on a Chemigenius (Syngene). Densitometric analysis of protein bands was performed using ImageJ Software.
2.7 Flow cytometry
Polarized macrophages in 24-well plates were washed with plain RPMI 1640 and subsequently incubated with 1 ml of Accutase (Innovative Cell Technologies) at 37°C for 15 minutes. Adherent cells were removed by pipetting up and down. Accutase was neutralized with an equal volume of complete RPMI 1640 and transferred to 5 ml polystyrene tubes. Cells were spun down at 2000 RPM at 4 °C for 5 minutes and washed 1 ml of PBS +0.5% BSA. Samples were incubated with either phycoerythrin (PE)-conjugated, mouse anti-CD36 antibody, (BD Pharmingen) or a PE-conjugated mouse IgM κ isotype control (BD Pharmingen) for 30 minutes on ice, in the dark. Cells were washed twice with PBS + 0.5% BSA and analyzed on a Beckman Coulter EpicsXL-MCL flow cytometer using Coulter System II Software v3.0.
2.8 Statistical analysis
Statistical differences among groups for functional assays including oxLDL cellular association and cholesterol accumulation which tested our hypothesis that differences among our macrophage sub-phenotypes would exist were analyzed using a repeated measures one-way analysis of variance followed with a Tukey’s post-hoc test.
Mechanistic studies investigating changes in mRNA and protein expression levels were expressed as values relative to the untreated control. These values were log transformed in order to obtain symmetrical ratios and a one sample t-test was performed on each treatment condition to evaluate if their means were significantly different from the untreated control. In scenarios where two different treatment conditions were compared with one another, a student’s t-test was performed.
In order to test the effect of rosiglitazone in each of the cytokine conditions, a paired t-test was performed. All statistical tests were performed using GraphPad Prism 5.04. Values of p<0.05 were considered significant.
3. RESULTS
3.1 Macrophages polarized with IFNγ/TNFα associate with less oxLDL and have reduced CD36 and MSR1 expression
To evaluate the ability of different macrophage sub-phenotypes to associate with oxLDL, macrophages were treated with IL-4/13, IFNγ/TNFα, IL-10 or left untreated and subsequently exposed to DiI-labeled oxLDL for 4 hours at 37°C. A one-way analysis of variance showed that very significant differences in oxLDL cellular association were detected among our macrophage sub-phenotypes (p=0.003). Post hoc analyses using Tukey’s multiple comparisons test revealed an 86% decrease in oxLDL cellular association in our IFNγ/TNFα treated macrophages compared with untreated controls (p<0.05, Figure 1A). This significant decrease led us to investigate the expression of two of the primary scavenger receptors involved in oxLDL metabolism, CD36 and MSR1. There was a moderate 14% decrease in MSR1 mRNA in macrophages treated with IL-10 (p=0.02, Figure 1B), and a 30% reduction in MSR1 protein for both IL-4/13 and IL-10 treated macrophages (p=0.03, p=0.03). IFNγ/TNFα treated macrophages also had a 57% decrease in MSR1 protein as determined by Western blotting (p=0.0004, Figure 1C). CD36 mRNA was reduced by 51% and 49% in IL-4/13 and IFNγ/TNFα treated macrophages, respectively (p=0.006, p=0.003 Figure 1D). A corresponding 46% decrease in cell surface CD36 protein expression was observed for IFNγ/TNFα treated macrophages when evaluated by flow cytometry (p=0.002, Figure 1E).
Figure 1. Macrophages polarized with IFNγ/TNFα associate with less oxLDL and have reduced CD36 and MSR1 expression.

(A) Association of DiI-labeled oxLDL with different macrophage sub-phenotypes was measured after loading with 10ug/ml of oxLDL for 4 hours (n=4). mRNA levels of MSR1 (B), and CD36 (D) were measured by qRT-PCR and normalized to PPIB (n=7,3). Protein levels of MSR1 were determined by Western blotting (C) and shown with a representative blot (n=5). Cell surface CD36 protein expression was determined by flow cytometry (E), shown with a representative histogram (n=6). Results are expressed as means ± SEM of at least 3 different donors. OxLDL cellular association was analyzed by one-way ANOVA followed by Tukey’s post-hoc test compared with untreated. mRNA and protein data was analyzed with a one-sample t-test. *p<0.05, **p<0.01, ***p<0.001.
3.2 Macrophages treated with IFNγ/TNFα accumulate less total cholesterol when treated with oxLDL
To determine if differences in oxLDL cellular association were related to changes in the concentration of cellular cholesterol over a longer incubation time, we measured the amount of cholesterol accumulated by different macrophage sub-phenotypes over a 24-hour time period following treatment with 50 μg/ml of oxLDL. Differences were detected among our macrophage sub-phenotypes (p<0.0001). Post-hoc analysis revealed that IFNγ/TNFα treatment led to a 30% reduction in accumulated cholesterol compared to untreated controls (p<0.01) while IL-10 treatment caused a modest but significant 1.2-fold increase (p<0.05, Figure 2A). We analyzed CD36 and MSR1 mRNA and protein once again to address whether changes in scavenger receptor expression were responsible for the observed differences using different cholesterol loading conditions. No changes were seen in MSR1 mRNA (Figure 2B), but IL-4/13 and IFNγ/TNFα caused a 21% and 54% reduction in MSR1 protein, respectively, as assessed by Western blot (p=0.007, p=0.0001, Figure 2C). CD36 mRNA and cell surface protein expression were reduced by 50% and 81% in IFNγ/TNFα treated macrophages respectively (p=0.01, p<0.0001), corresponding to a reduction in cholesterol accumulation (Figure 2D and E). Cholesterol accumulation was measured in macrophages treated with either IFNγ or TNFα individually in order to determine if both cytokines were capable of affecting cholesterol homeostasis. Both IFNγ and TNFα decreased total cholesterol accumulated to a similar degree (33%, p=0.012, and 28%, p=0.027, respectively, Figure 3A). We found that TNFα caused a 69% reduction in MSR1 mRNA (p=0.0006) while IFNγ and TNFα also caused a 30% and 68% reduction in MSR1 protein levels, respectively (p=0.003, p=0.03, Figure 3C). We also found that both IFNγ and TNFα were capable of causing a reduction in CD36 mRNA levels (27%, p<0.05 and 40%, p<0.05, respectively) as well a corresponding reduction in cell surface CD36 protein levels (35%, p<0.05, and 44%, p=0.006 respectively (Figure 3E).
Figure 2. Macrophages treated with IFNγ and TNFα accumulate less cholesterol and are associated with a decrease in CD36 and MSR1 expression.

(A) Total cholesterol levels of different macrophage sub-phenotypes were measured after loading with 50ug/ml of oxLDL for 24 hours (n=6). mRNA levels of MSR1 (B), and CD36 (D) were measured by qRT-PCR and normalized to PPIB (n=4). Protein levels of MSR1 were determined by Western blotting (C) and shown with a representative blot (n=10). Cell surface CD36 protein expression was determined by flow cytometry (E), shown with a representative histogram (n=4). Results are expressed as means ± SEM of at least 3 different donors. Cholesterol accumulation was analyzed by one-way ANOVA followed by Tukey’s post-hoc test compared with untreated. mRNA and protein data was analyzed with a one-sample t-test. *p<0.05, **p<0.01, ***p<0.001.
Figure 3. Both IFNγ and TNFα are able to reduce cholesterol accumulation and are associated with either a decrease in CD36 or MSR1 expression.

(A) Total cholesterol levels of different macrophage sub-phenotypes were measured after polarizing with individual cytokines and subsequently loading with 50ug/ml of oxLDL for 24 hours (n=5). mRNA levels of MSR1 (B) and CD36 (D) were measured by qRT-PCR and normalized to PPIB and beta actin (n=3,4). Protein levels of MSR1 were determined by Western blotting (C) and shown with a representative blot (n=5). Cell surface CD36 protein expression was determined by flow cytometry (E), shown with a representative histogram (n=6).. Results are expressed as means ± SEM of at least 3 different donors. Cholesterol accumulation was analyzed by a paired t-test compared with untreated. mRNA and protein data was analyzed with a one-sample t-test. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.
3.3 IL-4/13 treatment increases PPARγ expression levels
As the transcription factor PPARγ has been directly linked to modulating CD36 expression, we decided to determine if its expression was affected by any of the cytokine treatments [36]. Both mRNA and protein levels were determined in cytokine treated macrophages that were either unloaded or loaded with 50 μg/ml of oxLDL. IL-4/13 treatment increased PPARγ mRNA expression 1.9-fold in unloaded macrophages (p=0.001) and 2.5-fold under loaded conditions (p=0.001, Figure 4A, C). IL-4/13 treatment also increased PPARγ protein levels 1.3-fold in unloaded macrophages (p=0.04, Figure 4B) and 1.6-fold under loaded conditions (p=0.04, Figure 4D). Conversely, IFNγ/TNFα treatment decreased PPARγ mRNA by 42% in unloaded macrophages (p=0.02, Figure 4A) and 55% under loaded conditions (p=0.0006, Figure 4C). Despite the decrease in PPARγ mRNA with IFNγ/TNFα treatment, only a minor 16% decrease in PPARγ protein was observed in unloaded macrophages (p=0.005, Figure 4B) no significant decrease was observed under loaded conditions (Figure 4D). When treated with IFNγ or TNFα individually, a 56% and 49% reduction in PPARγ mRNA was observed under loaded conditions (p=0.01, p=0.04, Figure 4C) although no differences in PPARγ protein were observed.
Figure 4. IL-4/13 treatment increases PPARγ expression levels.

mRNA levels of PPARγ were measured with qRT-PCR and normalized to PPIB mRNA in polarized macrophages that were either unloaded (A) or loaded (C) with 50ug/ml of oxLDL (n=6). PPARγ protein levels were measured by Western blotting in unloaded (B) and loaded (D) macrophages and shown with representative blots (n=8). Results are expressed as means ± SEM of at least 3 different donors. mRNA and protein data was analyzed with a one-sample t-test. *p<0.05, **p<0.01, ***p<0.001.
3.4 IFNγ prevents the upregulation of CD36 by rosiglitazone
Despite the lack of change in PPARγ protein, it was still possible that IFNγ was affecting cell surface CD36 protein expression through a PPARγ–dependent pathway. To test this, macrophages were treated with IFNγ, TNFα, both IFNγ and TNFα or left untreated and subsequently exposed to the PPARγ specific agonist rosiglitazone to evaluate its ability to stimulate CD36 expression. Rosiglitazone caused a 1.5-fold increase in cell surface CD36 protein expression in untreated macrophages, however treatment with either IFNγ in combination with TNFα or IFNγ alone inhibited this increase by 42% and 37%, respectively (p=0.002 and p= 0.036, Figure 5 A). Interestingly, although TNFα causes a reduction in CD36 expression, addition of rosiglitazone is capable of upregulating its expression 1.4-fold suggesting that the reduction in cell surface CD36 by IFNγ is PPARγ-dependent while TNFα operates in a PPARγ-independent pathway. In order to confirm that PPARγ activity was being altered by IFNγ, we evaluated the fold change in mRNA expression of two PPARγ target genes, CD36 and FABP4 upon the addition of rosiglitazone. IFNγ attenuated the response of CD36 mRNA to rosiglitazone by 42% compared to untreated macrophages (p=0.01, Figure 5 B). IFNγ also caused a 57% reduction in the fold induction of FABP4 although this difference did not reach statistical significance (Figure 5C).
Figure 5. Rosiglitazone increases CD36 expression in control untreated macrophages and macrophages treated with TNFα but not with IFNγ.
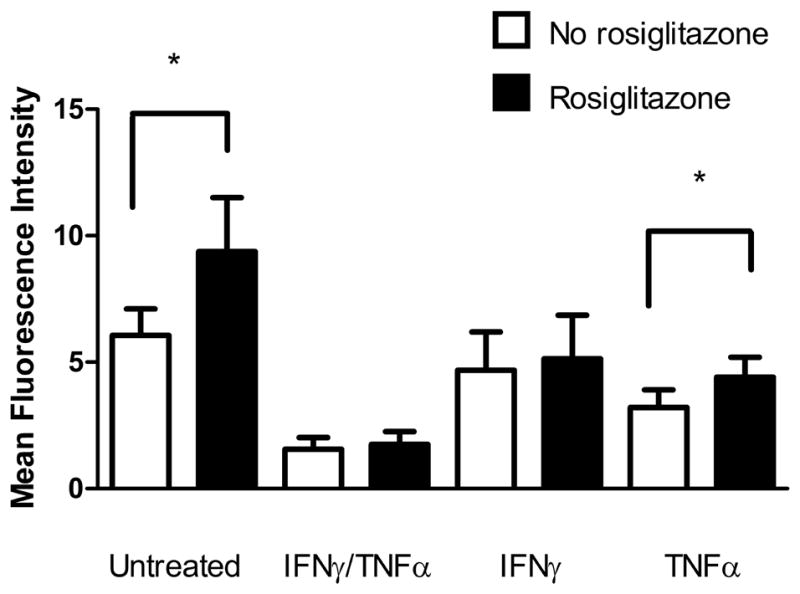
Macrophages were polarized with specific cytokine treatments and subsequently treated with 5uM rosiglitzone for 24 hours prior to loading with 50ug/ml of oxLDL. (A) Cell surface CD36 protein expression was determined by flow cytometry (n=5) and analyzed with a one-sample t-test. Fold change in (B) CD36 and (C) FABP4 mRNA upon rosiglitazone treatment were measured by qRT-PCR and normalized to PPIB and beta actin (n=3 and 4, respectively). Results are expressed as means ± SEM of at least 3 different donors. *p<0.05, **p<0.01 paired t-test compared with untreated.
4. DISCUSSION
Macrophage heterogeneity has been observed to exist in several human pathologies. For example, classically activated macrophages exposed to IFNγ and microbial products such as LPS during bacterial infection are critical for efficient immune responses while alternatively activated macrophages have been shown to be important in the resolution of inflammation [37, 38]. Both macrophage sub-phenotypes have been identified in human atherosclerosis and have been found to localize to unique areas of the plaque suggesting that they are involved in different processes [9, 39, 40]. Because of these differences in function, we sought to compare the ability of established macrophage sub-phenotypes to maintain cholesterol homeostasis.
In this study, we demonstrated that human MDMs polarized towards a pro-inflammatory sub-phenotype with either IFNγ or TNFα associate with less oxLDL and accumulate less cholesterol than untreated macrophages or macrophages polarized with either IL-4/13 or IL-10. We hypothesized that this may have been due to a decrease in the expression of CD36 and/or MSR1 so we measured both mRNA and protein levels for these receptors. We discovered that treatment with IFNγ and TNFα was indeed reducing MSR1 protein levels and both mRNA and protein levels of CD36. When comparing our results with the limited number of other studies employing primary human MDMs to compare different macrophage sub-phenotypes, we find that the results are similar to our own. Van Tits et al. used GM-CSF and M-CSF in a different model of macrophage polarization and also found that pro-inflammatory macrophages accumulate less cholesterol [41]. To our knowledge, there have not been any previous studies investigating the effects of TNFα on cholesterol accumulation upon exposure to oxLDL. Studies investigating the effect of IFNγ on macrophage cholesterol metabolism have revealed inconsistent results which appear to be model dependent. Reports using the human monocyte cell line THP-1 indicate that IFNγ and the transcription factor that it signals through, STAT1, are responsible for increased foam cell formation and CD36 expression[42, 43]. By contrast, similar studies performed using primary human MDMs however are in agreement with our own [44–46]. Geng and Hansson, for example, found that IFNγ caused a reduction in acetylated LDL cellular association and cholesterol accumulation [44]. Likewise, Nakagawa et al. found that IFNγ caused a decrease in CD36 expression in primary human MDMs [45]. A recent publication by Oh et al. using human MDMs from diabetic patients treated with IFNγ and LPS showed reduced cholesterol accumulation when compared with alternatively activated macrophages [46]. We have verified in our own studies that, unlike primary human MDMs, IFNγ does not decrease CD36 expression in THP-1-derived macrophages, and in the presence of oxLDL, causes an increase in CD36 expression (data not shown). These differences reinforce the need to exercise caution when comparing results obtained from different macrophage cell models.
We investigated the transcription factor PPARγ, a known regulator of CD36 expression as a possible mechanism by which IFNγ and TNFα were exerting their effects. Although IFNγ and TNFα did not cause significant reductions in PPARγ protein under loaded conditions, it was still possible that IFNγ and TNFα were reducing CD36 expression in a PPARγ-dependent manner. We tested this possibility by treating macrophages with the PPARγ-specific agonist rosiglitazone and looked for changes in CD36 expression. We found that rosiglitazone was unable to upregulate CD36 expression when macrophages were treated with IFNγ, but this inhibition was not present in untreated macrophages or macrophages treated with TNFα suggesting that IFNγ and TNFα regulate CD36 through independent mechanisms. Our results suggest that IFNγ downregulates CD36 primarily by preventing PPARγ activation as opposed to reducing PPARγ expression. It is possible that IFNγ signaling through STAT1 causes a sequestering of the coactivator proteins CREB binding protein (CBP) and p300 which are in limiting amounts in the cell [47–50]. Since PPARγ and STAT1 are in competition for cellular CBP and p300, this would prevent PPARγ activation by rosiglitazone. MSR1 protein expression may be being regulated in a similar manner. MSR1 transcription is positively regulated by AP-1/ets – two transcription factors which can be inhibited by IFNγ treatment [51, 52]. This inhibition is dependent on STAT1 binding to CBP and can be relieved by overexpressing CBP [51]. Our results are consistent with this model of transcriptional regulation although further studies would be required to confirm the functionality of this pathway in primary human MDMs.
W observed that despite the fact that IL-4/IL-13 treatment caused an increase in PPARγ protein, there was no corresponding increase in the PPARγ downstream target CD36. In contrast to expectations, IL-4/IL-13 treatment caused a decrease in CD36 mRNA during the oxLDL cellular association assay. This observation supports the notion demonstrated with IFNγ treatment that merely measuring the protein levels of a transcription factor alone does not provide enough information to make predictions about transcription factor activity. While we are not entirely sure of the mechanism behind this phenomenon, it is possible that IL-4/IL-13 signaling through STAT6 is influencing downstream targets that prevent the activation of PPARγ while simultaneously increasing its expression. Further investigation will be required to understand the mechanism in more detail.
Our results show that TNFα is also capable of causing a decrease in both CD36 and MSR1 which has been confirmed by several in vitro studies in different models [53–56]. MSR1 expression appears to be regulated both transcriptionally and post-transcriptionally and likely involves the phosphatidylinositol 3-kinase/Rac1/PAK/JNK and phosphatidylinositol 3-kinase/Rac1/PAK/p38 pathways [53, 54]. Boyer et al. also found that CD36 expression is downregulated by TNFα in primary human MDMs, but in contrast to our results reported that TNFα treatment was sufficient to prevent rosiglitazone-mediated PPARγ activation [56]. The reason for this discrepancy could be due to the shorter duration of treatment with the cytokine (5–30 minute), whereas our study was focused on prolonged chronic TNFα treatment over a 72 hour period.
Due to the established roles of CD36 and MSR1 in foam cell formation, we believe that their reduced expression is at least in part responsible for the decrease in oxLDL cellular association and cholesterol accumulation in IFNγ treated macrophages. This can be exemplified by the fact that blocking CD36 with specific antibodies decreases oxLDL binding, association and cholesterol accumulation in multiple cell models [57–60]. MSR1 has also been demonstrated to play a prominent role in oxLDL uptake as MSR1 deficient murine peritoneal macrophages demonstrate reduced oxLDL binding and uptake [8]. While our results indicate that a combined treatment of IFNγ and TNFα cannot reduce cholesterol accumulation beyond what is seen from either cytokine alone, we have demonstrated that they can work additively to further reduce CD36 expression. It is unclear why we did not observe a further reduction in cholesterol accumulation in our IFNγ/TNFα combination treatment although it is possible that other scavenger receptors capable of binding oxLDL, such as LOX-1, are upregulated to compensate for the decrease in CD36 expression.
Although IFNγ and TNFα have been associated with reduced CD36 expression and cholesterol accumulation in this study, this may not necessarily translate to a net anti-atherogenic effect. IFNγ for example, has also been shown to inhibit collagen synthesis and reduce ABCA1-mediated cholesterol efflux, and both IFNγ and TNFα can increase the expression of several MMPs that could potentially lead to plaque destabilization [61–63]. Reduced CD36 expression may not be athero-protective – while macrophages from CD36 deficient humans do show reduced oxLDL binding, patients are also more likely to exhibit hypertriglyceridemia, impaired glucose metabolism, and have mild hypertension10. Furthermore, the frequency of CD36 deficiency was three times higher in patients with coronary heart disease than control subjects [64].
In conclusion, we have characterized the propensity of different macrophage sub-phenotypes to uptake and accumulate cholesterol when exposed to oxLDL. Furthermore, we have identified changes in expression of CD36 and MSR1 as likely reasons for the differences in cholesterol homeostasis. Additional studies will be required to fully describe the underlying mechanisms behind how each cytokine treatment affects scavenger receptor expression.
Supplementary Material
Acknowledgments
The work was funded by grants from Pfizer Canada Inc. and the Canadian Institutes of Health Research (MOP-93527). Eugene Chu and Daven Tai were supported by an internship with MITACS accelerate BC and Pfizer Canada. We would like to thank the members of the UBC James Hogg Research Centre for their resources, support and helpful suggestions.
Abbreviations
- oxLDL
oxidized low density lipoprotein
- DiI
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DiI-oxLDL
DiI-labeled oxLDL
- STAT
Signal Transducer and Activator of Transcription
- MSR1
Macrophage scavenger receptor I
- IFNγ
interferon γ
- TNFα
Tumour necrosis factor α
- IL
interleukin
- PPAR
peroxisome proliferator-activated receptor
- MDMs
monocyte-derived macrophages
- qRT-PCR
Quantitative reverse transcription polymerase chain reaction
- CBP
CREB binding protein
References
- 1.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nature Immunology. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 2.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis. American Heart Association. 1995;15:1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 3.Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1177–1178. doi: 10.1161/01.atv.20.5.1177. [DOI] [PubMed] [Google Scholar]
- 4.Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009;20:370–378. doi: 10.1097/MOL.0b013e3283309848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 6.Edwards PA, Tabor D, Kast HR, Venkateswaran A. Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta. 2000;1529:103–113. doi: 10.1016/s1388-1981(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys. 2002;397:139–148. doi: 10.1006/abbi.2001.2615. [DOI] [PubMed] [Google Scholar]
- 8.Kunjathoor VV, Febbraio M, Podrez Ea, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. The Journal of biological chemistry. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 9.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, Dormont D, Gras G. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vila-del Sol V, Díaz-Muñoz MD, Fresno M. Requirement of tumor necrosis factor alpha and nuclear factor-kappaB in the induction by IFN-gamma of inducible nitric oxide synthase in macrophages. Journal of leukocyte biology. 2007;81:272–283. doi: 10.1189/jlb.0905529. [DOI] [PubMed] [Google Scholar]
- 13.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz-Fernandez MA, Fernandez MA, Fresno M. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur J Immunol. 1992;22:301–307. doi: 10.1002/eji.1830220203. [DOI] [PubMed] [Google Scholar]
- 15.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 16.Gupta S, Pablo AM, Jiang Xc, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. The Journal of clinical investigation. 1997;99:2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. The American journal of pathology. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huber SA, Sakkinen P, David C, Newell MK, Tracy RP. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation. 2001;103:2610–2616. doi: 10.1161/01.cir.103.21.2610. [DOI] [PubMed] [Google Scholar]
- 19.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflammation research : Official Journal of: The International Association of Inflammation Societies + The European Histamine Research Society. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 20.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mijatovic T, Kruys V, Caput D, Defrance P, Huez G. Interleukin-4 and -13 inhibit tumor necrosis factor-alpha mRNA translational activation in lipopolysaccharide-induced mouse macrophages. J Biol Chem. 1997;272:14394–14398. doi: 10.1074/jbc.272.22.14394. [DOI] [PubMed] [Google Scholar]
- 23.te Velde AA, Huijbens RJ, Heije K, de Vries JE, Figdor CG. Interleukin-4 (IL-4) inhibits secretion of IL-1 beta, tumor necrosis factor alpha, and IL-6 by human monocytes. Blood. 1990;76:1392–1397. [PubMed] [Google Scholar]
- 24.Bogdan C, Vodovotz Y, Paik J, Xie QW, Nathan C. Mechanism of suppression of nitric oxide synthase expression by interleukin-4 in primary mouse macrophages. J Leukoc Biol. 1994;55:227–233. doi: 10.1002/jlb.55.2.227. [DOI] [PubMed] [Google Scholar]
- 25.Gautam S, Tebo JM, Hamilton TA. IL-4 suppresses cytokine gene expression induced by IFN-gamma and/or IL-2 in murine peritoneal macrophages. J Immunol. 1992;148:1725–1730. [PubMed] [Google Scholar]
- 26.Nakamura T, Lee RK, Nam SY, Podack ER, Bottomly K, Flavell Ra. Roles of 11–4 and IFN-y in Stabilizing the T Helper Cell Type 1 and 2 Phenotype. The Journal of Immunology. 1997;158:2648–2653. [PubMed] [Google Scholar]
- 27.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, Larner AC, Finbloom DS. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma-induced genes by suppressing tyrosine phosphorylation of STAT1. Blood. 1999;93:1456–1463. [PubMed] [Google Scholar]
- 29.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annual review of immunology. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 30.Montaner LJ, da Silva RP, Sun J, Sutterwala S, Hollinshead M, Vaux D, Gordon S. Type 1 and type 2 cytokine regulation of macrophage endocytosis: differential activation by IL-4/IL-13 as opposed to IFN-gamma or IL-10. J Immunol. 1999;162:4606–4613. [PubMed] [Google Scholar]
- 31.Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C, Staels B, Duverger N, Scherman D, Tedgui A. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 32.Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, Curtiss LK, Berliner JA, Boisvert WA. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circulation research. 2002;90:1064–1071. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
- 33.Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869–2880. doi: 10.1096/fj.09-148155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinbrecher UP. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. The Journal of biological chemistry. 1987;262:3603–3608. [PubMed] [Google Scholar]
- 35.Stephan ZF, Yurachek EC. Rapid fluorometric assay of LDL receptor activity by DiI-labeled LDL. J Lipid Res. 1993;34:325–330. [PubMed] [Google Scholar]
- 36.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 37.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 38.Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, Holland SM, Schreiber RD, Casanova JL. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–303. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 39.Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–1126. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Reiss AB, Patel CA, Rahman MM, Chan ES, Hasneen K, Montesinos MC, Trachman JD, Cronstein BN. Interferon-gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP-1 human monocytes/macrophages. Med Sci Monit. 2004;10:BR420–425. [PubMed] [Google Scholar]
- 43.Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation. 2007;115:2939–2947. doi: 10.1161/CIRCULATIONAHA.107.696922. [DOI] [PubMed] [Google Scholar]
- 44.Geng YJ, Hansson GK. Interferon-gamma inhibits scavenger receptor expression and foam cell formation in human monocyte-derived macrophages. J Clin Invest. 1992;89:1322–1330. doi: 10.1172/JCI115718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakagawa T, Nozaki S, Nishida M, Yakub JM, Tomiyama Y, Nakata A, Matsumoto K, Funahashi T, Kameda-Takemura K, Kurata Y, Yamashita S, Matsuzawa Y. Oxidized LDL increases and interferon-gamma decreases expression of CD36 in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1350–1357. doi: 10.1161/01.atv.18.8.1350. [DOI] [PubMed] [Google Scholar]
- 46.Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C. Endoplasmic reticulum stress controls m2 macrophage differentiation and foam cell formation. J Biol Chem. 2012;287:11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J. p300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J Biol Chem. 1999;274:7681–7688. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- 48.Konstantinopoulos PA, Vandoros GP, Sotiropoulou-Bonikou G, Kominea A, Papavassiliou AG. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma ‘on/off’ switches and induction of CBP in colon adenocarcinomas: correlation with COX-2 expression. Int J Colorectal Dis. 2007;22:57–68. doi: 10.1007/s00384-006-0112-y. [DOI] [PubMed] [Google Scholar]
- 49.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 51.Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen TM, Rose DW, Rosenfeld MG, Glass CK. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu H, Moulton K, Horvai A, Parik S, Glass CK. Combinatorial interactions between AP-1 and ets domain proteins contribute to the developmental regulation of the macrophage scavenger receptor gene. Mol Cell Biol. 1994;14:2129–2139. doi: 10.1128/mcb.14.3.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsu HY, Twu YC. Tumor necrosis factor-alpha -mediated protein kinases in regulation of scavenger receptor and foam cell formation on macrophage. J Biol Chem. 2000;275:41035–41048. doi: 10.1074/jbc.M003464200. [DOI] [PubMed] [Google Scholar]
- 54.Hsu HY, Nicholson AC, Hajjar DP. Inhibition of macrophage scavenger receptor activity by tumor necrosis factor-alpha is transcriptionally and post-transcriptionally regulated. J Biol Chem. 1996;271:7767–7773. doi: 10.1074/jbc.271.13.7767. [DOI] [PubMed] [Google Scholar]
- 55.Olagnier D, Lavergne R-A, Meunier E, Lefèvre L, Dardenne C, Aubouy A, Benoit-Vical F, Ryffel B, Coste A, Berry A, Pipy B. Nrf2, a PPARγ alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS pathogens. 2011;7:e1002254. doi: 10.1371/journal.ppat.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyer JF, Balard P, Authier H, Faucon B, Bernad J, Mazières B, Davignon J-L, Cantagrel A, Pipy B, Constantin A. Tumor necrosis factor alpha and adalimumab differentially regulate CD36 expression in human monocytes. Arthritis research & therapy. 2007;9:R22. doi: 10.1186/ar2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 58.Kar NS, Ashraf MZ, Valiyaveettil M, Podrez EA. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoprotein of scavenger receptor CD36. J Biol Chem. 2008;283:8765–8771. doi: 10.1074/jbc.M709195200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katayama I, Hotokezaka Y, Matsuyama T, Sumi T, Nakamura T. Ionizing radiation induces macrophage foam cell formation and aggregation through JNK-dependent activation of CD36 scavenger receptors. Int J Radiat Oncol Biol Phys. 2008;70:835–846. doi: 10.1016/j.ijrobp.2007.10.058. [DOI] [PubMed] [Google Scholar]
- 60.Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann N Y Acad Sci. 2001;947:350–355. doi: 10.1111/j.1749-6632.2001.tb03961.x. [DOI] [PubMed] [Google Scholar]
- 61.Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb. 1991;11:1223–1230. doi: 10.1161/01.atv.11.5.1223. [DOI] [PubMed] [Google Scholar]
- 62.Wang X-q, Panousis CG, Alfaro ML, Evans GF, Zuckerman SH. Interferon-gamma-mediated downregulation of cholesterol efflux and ABC1 expression is by the Stat1 pathway. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:e5–9. doi: 10.1161/01.atv.0000018287.03856.dd. [DOI] [PubMed] [Google Scholar]
- 63.Nareika A, Sundararaj KP, Im Y-B, Game Ba, Lopes-Virella MF, Huang Y. High glucose and interferon gamma synergistically stimulate MMP-1 expression in U937 macrophages by increasing transcription factor STAT1 activity. Atherosclerosis. 2009;202:363–371. doi: 10.1016/j.atherosclerosis.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamashita S, Hirano K, Kuwasako T, Janabi M, Toyama Y, Ishigami M, Sakai N. Physiological and pathological roles of a multi-ligand receptor CD36 in atherogenesis; insights from CD36-deficient patients. Mol Cell Biochem. 2007;299:19–22. doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.