

Abstract
Substantial evidence supports a role for the renin-angiotensin system (RAS) in the regulation of metabolic function, but an apparent paradox exists where genetic or pharmacological inhibition of the RAS occasionally have similar physiological effects as chronic angiotensin infusion. Similarly, while RAS targeting in animal models has robust metabolic consequences, effects in humans are more subtle. Here we review the data supporting a role for the RAS in metabolic rate regulation and propose a model where the local brain RAS works in opposition to the peripheral RAS, thus helping to explain the paradoxically similar effects of RAS supplementation and inhibition. Selectively modulating the peripheral RAS or brain RAS may thus provide a more effective treatment paradigm for obesity and obesity-related disorders.
Keywords: Obesity, Metabolism, Energy Homeostasis, Thermogenesis
Metabolic disease: impact and therapeutics
Metabolic diseases are among the fastest growing health and economic concerns for industrialized nations. Disease states including obesity, diabetes, the metabolic syndrome, and even the less-recognized ‘underweight’ status are all associated with enormous health, economic, and social costs. BMI of less than 18.5, or greater than 30, are both associated with increased mortality relative to normal weight subjects [24]. Lean hypertensive subjects of both genders are at greater risk of ischemic heart disease and cardiovascular disease than obese hypertensive subjects [9]. Hammond and Levine estimated that in 2010 the annual aggregated costs to the United States from obesity were in excess of $215 billion [34]. The American Diabetes Association estimated that the annual costs of diabetes to the United States in 2007 were over $174 billion [4].
Only one therapeutic compound is currently approved for the long-term treatment of obesity in the United States. Orlistat (Xenical), a pancreatic lipase inhibitor, reduces intestinal fat absorption efficiency and thereby reduces caloric intake [35, 53]. This drug was first approved for use by the Food and Drug Administration in April 1999, and when combined with dietary and physical activity interventions induces around a 10% reduction in body weight over 1–2 years, and modest reductions in fasting blood glucose and serum cholesterol are also observed [35, 73]. Compared to placebo (dietary and physical activity alone), most studies have demonstrated the relatively small average weight loss of approximately 3 kilograms over 12 months of orlistat treatment [35, 73]. Other therapeutic compounds have been utilized for human obesity, and most of these drugs target the suppression of appetite. Compounds such as desoxyephedrine (methamphetamine), phenmetrazine, phentermine, diethylpropion (amfepramone), phendimetrazine, benzphetamine, mazindol, fenfluramine, fenfluramine/phentermine (Fen-Phen), sibutramine, and rimonabant have all been previously approved by the FDA, and have subsequently been removed from the market or are only used for short-term (around 12-week) weight loss objectives due to the recognition of serious and harmful side-effects, and the potential for addiction and/or abuse [35, 53]. Interestingly, the side effects that have resulted in the discontinuation of many of these anti-obesity medications have frequently been related to an increased risk of cardiovascular complications, underscoring the overlap in physiological mechanisms regulating metabolic and cardiovascular function.
Why the Renin-Angiotensin System?
The renin-angiotensin system (RAS) has long been studied for its involvement in cardiovascular regulation. This peptide hormone system is present both in the circulation, where it acts as a traditional hormone system, but also within individual organs and tissues where it acts in a paracrine/autocrine fashion. The kidneys, adipose, pancreas, liver, brain, heart, and many other tissues express many or all of the components of the RAS [25, 45].
In addition to its importance for blood pressure control, the RAS is suspected to contribute to the regulation of resting metabolic rate, glucose homeostasis, and other key metabolic parameters [50]. Adipose mass positively correlates with RAS activity, and RAS gene expression correlates with the severity of obesity in both humans and animal models [22, 28, 47]. Circulating angiotensinogen (AGT), renin, angiotensin converting enzyme (ACE), and aldosterone levels are increased in obese humans [6, 15, 19, 54], and weight loss reduces or normalizes them [19]. Men fed high-fat diets for two months exhibited increased ACE, AGT, aminopeptidase A, and neprilysin expression in subcutaneous white adipose (WAT) [3]. Weight loss in humans is associated with reduced serum ACE activity, and body weight regain is lower for individuals with greater reductions in ACE [74]. Further, adipose-specific overexpression of AGT in mice leads to obesity [47], and AGT knockout prevents diet-induced obesity [48]. These observations generally support the concept that adipose produces or stimulates the RAS. Indeed, adipose tissue expresses all components of the RAS [41, 65, 84]. AGT is expressed by adipocytes in animals and humans, and contributes to circulating levels [47]. Renin, ACE, angiotensin converting enzyme 2 (ACE2), and the various RAS receptors AT1, AT2, AT4 (insulin regulated aminopeptidase, IRAP), and Mas are also all expressed by preadipocytes and mature adipocytes, though the relative expression of each changes during adipocyte differentiation [16, 17, 75].
While cardiac tissue levels of renin protein strongly correlate with plasma levels, adipose renin levels do not correlate with plasma levels, and adipose renin levels are maintained at a higher concentration than in plasma [26]. This suggests specific, plasma-independent control of adipose renin concentrations, perhaps through an adipose-specific mechanism of renin gene expression. An alternative hypothesis that would need experimental validation is that the expression of the (pro)renin receptor (P)RR in the stromal fraction of adipose tissue acts to concentrate renin protein within the adipose tissue [1]. Interestingly, the expression level of the (P)RR varies by adipose bed – with visceral adipose expressing higher levels of (P)RR than subcutaneous adipose [1]. Perivascular brown adipose (BAT) expresses more (P)RR than mesenteric WAT, but expresses lower levels of AT1A, AT2, chymase, (possibly ACE), and exhibits lower angiotensin (Ang) II concentrations compared to mesenteric WAT [27]. What remains unclear is whether the abundance of (P)RR within adipose tissue reflects (or determines) a differential dependence of adipose RAS activity on plasma renin abundance.
Angiotensinergic signaling within adipose tissue has multiple consequences, depending on the cell type and receptor type that is examined. Endogenous Ang II enhances evoked release of norepinephrine from nerves innervating BAT [12], and chronic exogenous Ang II alters BAT sympathetic nerve kinetics to increase norepinephrine release [20], and these effects are mediated through the AT1 receptor. The effects of AT1 versus AT2 receptor stimulation on adipocytes is extremely dynamic, with effects differing based upon the setting (in vitro vs. ex vivo vs. in vivo), the developmental phase (preadipocytes vs. adipocytes), and the relative expression of various RAS receptors (AT1 and AT2, at least, are strongly regulated in adipocytes by various hormones and stimuli) (reviewed in [21, 29, 50, 72, 84]). Thus it is reasonable to conclude that adipocytes in vivo are sensitive to Ang II through both AT1 and AT2 receptors, but our understanding of the exact function(s) of each is currently rather muddled. Moreover, given the well established link between sympathetic discharge to BAT and thermogenesis, one could reasonably hypothesize that one of the mechanisms by which Ang II modulates BAT thermogenesis is by facilitating neurotransmission.
To determine the directionality of the RAS/obesity correlation and to examine the functions of RAS signaling in vivo, many knockout, transgenic, and pharmacological studies have been performed to either interfere with, or stimulate, the RAS. These studies have consistently demonstrated the involvement of the RAS in the regulation of adipose morphology and function, and whole-body metabolism.
RAS interference
A. Angiotensin Production
Reduced production of angiotensin peptides tends to decrease body mass in rodents. Takahashi [71] determined that global genetic knockout of renin resulted in lean mice that were resistant to weight gain on a high fat diet, due to both an increase in metabolic rate and reduced intestinal absorption of fat. This was reversed by exogenous angiotensin delivery demonstrating the lean phenotype was due to a loss of Ang peptides and not just renin per se. This finding is supported by pharmacological studies showing that chronic renin inhibition with aliskiren reduced body weight gain, adiposity, and plasma leptin levels in a C57BL/6J mouse model of diet-induced obesity [69]. Similarly, aliskiren prevented selected features of the metabolic syndrome that are induced by a high-fructose diet in SD rats [13].
Like renin-deficiency, global knockout of AGT resulted in small, lean mice that were resistant to weight gain on a high-fat diet [48]. Mechanistically, this was due to altered adipose development, decreased lipogenesis, and increased locomotor activity. Global knockout of ACE also resulted in small, lean mice, in this case in response to increased basal metabolic rate and lipolysis [39]. Like the renin inhibitor studies, chronic pharmacological inhibition of ACE with perindopril reduced body mass and reduced adiposity [51], and when administered from birth caused large reductions in fat mass [76]. Likewise, chronic delivery of enalapril to young rats resulted in reduced body mass regardless of diet, concomitant with a reduction in food intake [62]. The beneficial weight reducing effects of ACE inhibitors is independent of age as the delivery of enalapril to old rats resulted in retention of physical performance measures and reduced adiposity [10].
Some human studies also support a role for the RAS in metabolic regulation. Ang II infusion causes reduced lipolysis and glucose uptake in healthy young men [7]. ACE inhibition via enalapril can amplify the beneficial metabolic effects of weight loss [49], and some studies suggest that perindopril may be more effective than enalapril in regulating metabolic function in humans [43].
The studies discussed above suggest that inhibiting the production of Ang peptides could attenuate weight gain or cause weight loss associated with high fat feeding. What remains unclear, however, is whether this is caused by a reduction in circulating versus adipose Ang II. Indeed, one weakness of global knockout and pharmacological studies is that they cannot distinguish between the systemic effects of RAS inhibition and the adipose-specific effects. Complicating this analysis are findings that adipose AGT can contribute to plasma AGT which will affect both systems. Yiannikouris [80] recently demonstrated that adipose-specific knockout of AGT in mice resulted in a reduction in plasma AGT levels with a concomitant reduction in blood pressure. Although this reduction in adipose and circulating AGT demonstrates a role for adipose AGT as a contributor to the plasma pool of AGT and to blood pressure regulation, there were, perhaps surprisingly, no effects on body weight or adiposity. It remains possible that the modest (~25%) reduction in circulating AGT preserved sufficient Ang II substrate to maintain normal adipose function. As an alternative to adipose-specific AGT deficiency, Massiera [47] and Yvan-Charvet [83] reported that adipose-specific overexpression of AGT on either a global AGT-deficient or wildtype genetic background increased body mass and adiposity. This suggests that increased activity of the adipose RAS may have detrimental effects on body weight regulation. This conclusion is supported by reports that Ang II can modulate adipogenesis, though considerable controversy exists regarding the specific actions of Ang II as conflicting results are observed across species, adipose beds, and model systems [38, 40, 52, 55, 70]. Clearly, a paradox exists; Ang II can facilitate neurotransmission to BAT and thus increase thermogenesis, while (in some models) simultaneously increasing white adipose mass. Consequently, fully understanding and uncovering the relative importance and the molecular mediators of these two disparate mechanisms will be essential to translate these results into the clinic.
B. Angiotensin Actions
Interference with RAS receptors has varied effects depending on the receptor. Zanchi [85] showed that that AT1 receptor blocker (ARB) telmisartan reduced the weight gain typically induced by pioglitazone (a PPARγ agonist) treatment, most likely through a reduction in food intake. Whereas valsartan, another ARB, treatment had no protective effect against weight gain on a “western” diet, it prevented the inflammation and impaired glycemic control associated with this diet [14]. Likewise, chronic delivery of candesartan cilexetil reduced body mass and adipocyte size [86]. Global knockout of AT1A receptors resulted in the attenuation of weight gain and adipose deposition during high-fat feeding due to an increase in metabolic rate, consistent with the results of AT1 receptor inhibition [42]. In contrast, global knockout of AT2 receptors had no obvious effect on adipose pads, but greatly changed their cellular morphology toward a larger number of smaller adipocytes, and increased skeletal muscle utilization of fatty acids [82]. Iwai [37] determined that AT2 receptor knockout on the ApoE knockout background had minor baseline effects, but resulted in accelerated adipose mass gain (and hypertrophy of adipocytes) compared to ApoE knockouts when the mice were maintained on a high cholesterol diet. Interestingly, AT2 receptor knockout prevented the metabolic abnormalities of mice with adipose-specific AGT overexpression suggesting, like for blood pressure, a counter-regulatory circuit between AT1 and AT2 receptors in metabolic regulation [83]. Furthermore, Santos [63] determined that global knockout of Mas receptors (the receptor for Ang-(1–7)) has a complex effect on body composition, including an increase in abdominal fat mass, and decreased glucose tolerance and insulin sensitivity.
Stimulating or supplementing the RAS
A. Systemic Application
Supplementation of the RAS has been achieved through delivery of exogenous peptides, interventions that stimulate endogenous RAS activity, and transgenic expression of RAS components. Through the inherent site-specificity of RAS activation in these models, it has become clear that the actions of the RAS to modulate metabolic function depend not only on the RAS component targeted, but also the site of action.
The effects of chronic exogenous Ang II delivery to the periphery have been evaluated in rodents. Cassis [11] determined that chronic low-dose subcutaneous infusion of Ang II resulted in weight loss. Using a pair-feeding paradigm, the authors determined that approximately 63% of the weight loss was due to a reduction in food intake by the animals. Brink [8] also demonstrated body weight loss with Ang II infusion, and the mechanism again included both reduced food intake and increased catabolic metabolism. The suppressed food intake in mice associated with either peripheral (subcutaneous) or central (intracerebroventricular) infusion of Ang may be mediated through the suppression of hypothalamic neuropeptide Y and orexin expression [81].
Perhaps one of the most surprising and inexplicable results were obtained by Gratze who reported that transgenic rats overexpressing human renin exhibited hyperphagia and developed obesity and insulin resistance [30]. The mechanism of the hyperphagia is unclear as ACE inhibition, renin inhibition, and (P)RR blockade all failed to reverse the phenotype, and none of the known hypothalamic mechanisms for feeding control were altered. Additionally, since human renin on its own does not catalytically cleave rat AGT, an Ang II-dependent mechanism must be questioned.
B. Brain-Specific Application
The effects of chronic exogenous Ang II delivery directly to the brain have also been evaluated in rodents. Porter [59] determined that in young rats, chronic infusion of Ang II into the lateral cerebral ventricle resulted in reduced body mass compared to both ad libitum and pair-fed controls, leading to the conclusion that metabolic rate must have increased in the animals. The same researchers also demonstrated similar results in adult rats [60].
Using a unique transgenic animal model of brain-specific RAS hyperactivity previously developed to study water and electrolyte homeostasis [61], we have also investigated the role of the brain RAS in the control of metabolic rate. Mice expressing a human renin transgene under transcriptional control of the neuron-specific synapsin promoter (“sR” mice, [56]) were bred with mice expressing a precisely regulated human AGT transgene (“A” mice, [67, 68, 79]). Because of the tight species-specificity of the renin-mediated cleavage of AGT to form Ang I, single-transgenic parents and littermates are phenotypically normal but double-transgenic offspring (“sRA” mice) exhibit an elevation of Ang peptide formation anywhere the two transgenes are both expressed. We determined that this elevation was grossly limited to the hypothalamus and lamina terminalis of the brain [32]. Not only was RAS hyperactivity limited to portions of the brain, but we also observed a significant suppression of the circulating RAS. As expected based upon previous studies into the cardiovascular and hydromineral effects of brain RAS activity, sRA mice exhibit robust hypertension, polydipsia, sodium intake, and polyuria.
Surprisingly, these animals also exhibited an extremely lean phenotype that was mediated entirely through an elevation in resting metabolic rate [32]. This elevation in metabolic rate was mediated through both an elevation in sympathetic nervous activity, but also through the suppression of the peripheral RAS, as peripheral delivery of either the beta-adrenergic blocker propranolol or Ang II could both independently normalize the metabolic rate increase in sRA mice. Work from de Kloet [18] also supports a role for the brain RAS in metabolic control, as chronic infusion of Ang II into the lateral cerebral ventricle of Long-Evans rats resulted in increased thermogenesis. Interestingly, the rats also exhibited reduced food intake, which correlated with increased hypothalamic expression of agouti-related peptide, proopiomelanocortin, corticotropin, and thyroid-releasing hormone. Similar experiments from Yoshida [81] showed that central infusion of Ang II suppressed neuropeptide Y and orexin expression, which also correlated with suppressed food intake. Thus, it is clear that chronic elevations of RAS activity within the brain result in increased thermogenic energy expenditure. Variable effects are observed on food intake behavior, depending on method of delivery, time-course, etc., and further studies of the anorexic effects of brain Ang are obviously needed.
Using a different double-transgenic mouse model of global, but not brain-specific RAS hyperactivity (the “R+/A+” model), we determined that a whole-body elevation in Ang levels resulted in increased interscapular BAT sympathetic nervous activity (SNA), but no significant changes in body mass, adipose mass, or food intake. Figure 1 shows a comparison of body and adipose masses, food intake and BAT SNA in R+/A+ (elevated circulating RAS activity) versus sRA (suppressed circulating RAS activity) mice, compared with their own littermate controls. These data underscore the importance of the circulating RAS in setting resting metabolic rate, as despite the maintenance of elevated BAT SNA (as observed in sRA mice), elevated peripheral RAS activity apparently prevents the development of an increased resting metabolic rate in R+/A+ mice. This prompts an interesting hypothesis that the peripheral (circulating and/or adipose) RAS acts as a gain control on the metabolic responses to sympathetic stimulation.
Figure 1. Metabolic phenotypes of “R+/A+” mice with global transgenic hyperactivity of the RAS, compared to “sRA” mice with brain-specific hyperactivity of the RAS.
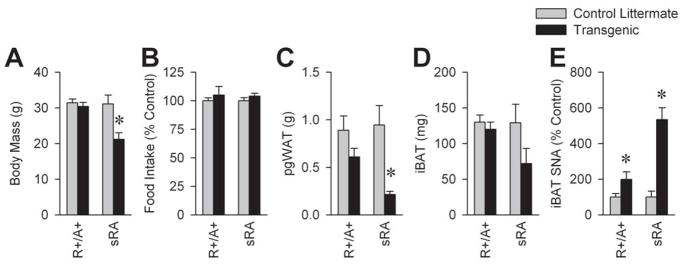
(A) Body mass, (B) food intake, (C) perigenital white adipose (pgWAT), (D) interscapular brown adipose (iBAT), and (E) sympathetic nerve activity (SNA) to iBAT. * P<0.05 vs. littermate controls.
Independent evidence for a role of the brain RAS in the regulation of metabolic rate was provided by the deoxycorticosterone (DOCA)-salt model of hypertension, which also exhibits a suppression of the circulating RAS. In this model, animals are treated with chronic delivery of deoxycorticosterone acetate and are allowed ad libitum access to 0.15 M NaCl drink solution in addition to standard chow and tap water. The development of hypertension in this model can also be accelerated if the animals are uninephrectomized, but this is not required for any of the phenotypes in the model [31]. Chronic delivery of the ACE inhibitor, captopril, to the lateral cerebral ventricle resulted in both the prevention and reversal of hypertension in the DOCA-salt model, suggesting a requirement for Ang II synthesis within the brain [36]. Kubo [44] and Park [58] also reported similar results using the AT1 receptor antagonist, losartan, implicating a requirement for Ang II receptor activation. Together, these data highlight the dependence upon brain RAS activity for the cardiovascular effects of the DOCA-salt model. We subsequently determined that DOCA-salt treated mice exhibit a robust elevation in resting metabolic rate [31], much like the sRA transgenic model (Figure 2). Importantly, this increase was dependent upon the brain RAS as chronic delivery of losartan to the lateral cerebral ventricles significantly attenuated this elevation.
Figure 2. Major metabolic and cardiovascular phenotypic overlaps between “sRA” mice with transgenic brain-specific RAS hyperactivity and mice treated with DOCA-salt.
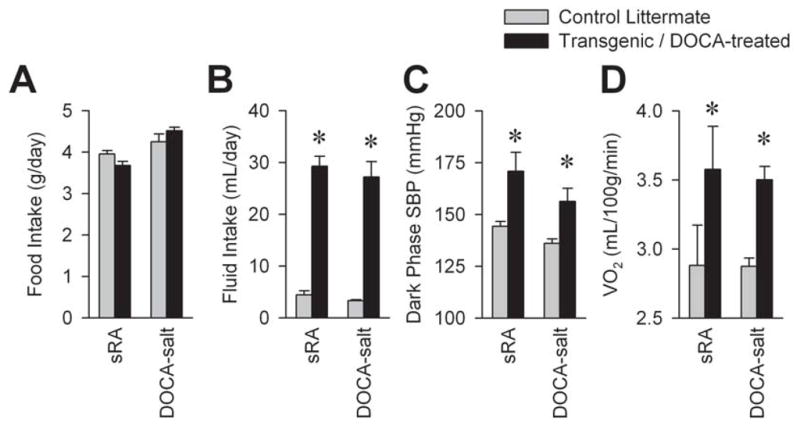
(A) Food intake, (B) total fluid intake, (C) systolic blood pressure (SBP) averaged during the dark phase of the light-dark cycle, and (D) metabolic rate as estimated by indirect calorimetry at thermoneutrality (30°C). * P<0.05 vs. littermate controls.
It is notable that the reversal of metabolic rate increases in the brain-specific sRA model through chronic replacement of peripheral Ang II required eight weeks of infusion – a stark contrast against the normalization achieved by propranolol within a few minutes of injection [32]. This leads us to hypothesize that the function of the peripheral RAS as a gain control on thermogenesis may occur through the modulation of the morphology of adipose tissue. Indeed, various groups have determined that AT1 and AT2 receptors differentially contribute to cellular functions in adipose tissue [84]. Preliminary data presented by the Cassis lab at the 2011 American Heart Association Council for High Blood Pressure Research conference demonstrates that adipose-specific knockout of AT1A receptors in C57BL/6 mice had no effect on the weight gain or glucose intolerance induced by a high fat diet [84]. Combined with the previous work from Yvan-Charvet [82, 83] and Iwai [37] more directly implicating the AT2 receptor in the regulation of adipose morphology, we hypothesize that circulating or adipose angiotensin may function as a gain control on metabolic rate through a mechanism involving adipose AT2 receptors, and experiments to directly test this in sRA mice are in progress.
Metabolic Control by the Brain RAS
Data from our group and many others support the existence of a brain RAS (reviewed in [23, 33]), and a role for this local brain RAS in the control of metabolic rate [18, 31, 32, 59, 60]. The identification of the RAS components involved, the cell types and brain regions involved, and second-messenger mechanisms are all the focus of ongoing work.
It is reasonable to hypothesize that the contributions of the brain RAS to metabolic control must simultaneously take place in regions where RAS components – especially Ang receptors – are expressed. Further, because we have previously demonstrated a causative role for the sympathetic nervous system in the metabolic consequences of brain RAS activity [31, 32], these regions will likely be known for contributing to thermoregulatory sympathetic nervous system control (reviewed in [57]). Finally, based on our observations that the circulating and/or adipose RAS is suppressed by the brain RAS [31, 32] and that normalization of blood pressure also normalizes renal renin expression [31], we hypothesize that regions of the brain which regulate blood pressure – including both premotor sympathetic regions and sympathetic-independent regions (such as vasopressin-producing regions) – may also mediate the metabolic effects of the brain RAS.
Generation of RAS peptides within the brain appears to be mediated through two different isoforms of renin. The mRNA transcript that encodes the classic renin isoform (secreted renin, or sREN) includes a signal sequence that targets the resulting protein into the cell’s secretory apparatus. We [66] and Lee-Kirsch [46] previously identified a novel mRNA variant of renin that includes an alternate first exon, and thus an alternative promoter. Translation of this alternate mRNA transcript results in a protein that lacks the signal peptide but includes all the remaining components of the renin enzyme, and thus the resulting protein should be targeted to the cytoplasm instead of the cell’s secretory apparatus. The transcript for this alternate, intracellular renin enzyme (icREN) is expressed primarily in adult brain tissue [78], and may therefore help explain the complex biochemistry of the brain RAS and a mechanism for the generation of RAS peptides for use as neurotransmitters [33]. sREN and icREN are independently regulated at the mRNA level, as DOCA-salt treatment results in a strong induction of sREN and a possible suppression of icREN expression (Figure 3). Selective knockout of sREN (while retaining icREN) throughout the body results in the same severe developmental problems that occur with the deletion of all renin isoforms [78]. Selective knockout of sREN specifically within neurons and glia, however, does not impact baseline blood pressure, heart rate, hydromineral balance, or metabolism [77]. Together with the observation that complete global knockout of all renin isoforms has potent metabolic consequences [71], these data are consistent with roles for both peripheral sREN and brain icREN in metabolic rate control. We have recently developed a selective knockout of icREN, and ongoing studies are aimed at identifying the cardiovascular, hydromineral, and metabolic consequences of this maneuver at baseline and following DOCA-salt treatment. Further, because DOCA-salt induces sREN, we are examining the effects of neuron- and glia-specific knockout of sREN on the various phenotypes induced by DOCA-salt treatment.
Figure 3. Relative expression of mRNA transcripts for secreted (sREN) and intracellular (icREN) isoforms of renin within brain tissue.
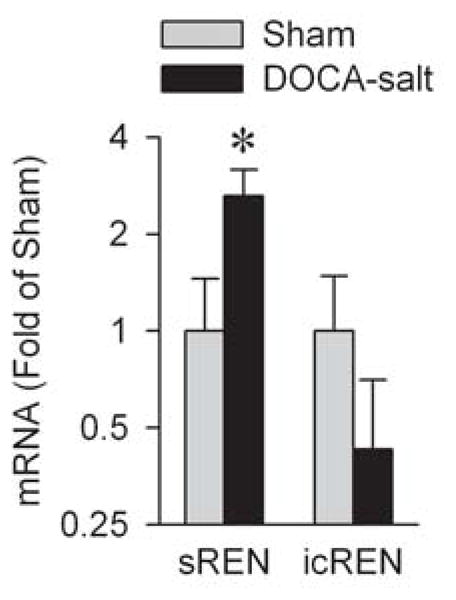
Adult male C57BL/6J mice were treated for three weeks with DOCA-salt or sham treatments before analysis of mRNA transcripts within brain homogenates. N=20 each group. * P<0.05 vs. Sham.
Summary, Proposed Model, and Perspectives
In summary, the RAS plays many roles in the regulation of metabolic function, but these actions vary widely based upon the site of action and receptor type. Therapeutic interventions that specifically target individual receptor types within specific organs may thus represent an ideal therapy for obesity and obesity-related disorders. Importantly, targeting of the RAS should result in altered thermogenic energy expenditure - a novel therapeutic paradigm when compared to the litany of failed appetite-suppressing paradigms. In Figure 4, we summarize our working model for the mechanisms by which the brain RAS regulates resting metabolic rate. Using a combination of transgenic (sRA) and pharmacological (DOCA-salt) models of elevated brain RAS activity, we have determined that brain Ang activates both a direct thermogenic stimulus mechanism through the promotion of sympathetic nervous drive to adipose tissues, and an indirect thermogenic capacity-regulating mechanism that is mediated through modulation of blood pressure and subsequently the peripheral (circulating/adipose) RAS.
Figure 4. Working model.

Increased activity of the brain RAS results in the activation of two major efferent signaling mechanisms; arginine vasopressin (AVP) and the sympathetic nervous system. Elevated sympathetic nervous activity (SNA) results in a direct stimulation of thermogenesis, and together with AVP, an elevation in blood pressure. Elevated blood pressure suppresses renal renin production and subsequently the activity of the peripheral (circulating and possibly adipose) RAS. Suppressed peripheral RAS activity results in increased recruitment of adaptive thermogenic mechanisms, resulting in increased total thermogenic capacity. Together, increased capacity for–and stimulation of–thermogenesis results in elevated catabolic metabolism.
Balancing Cardiovascular and Metabolic Risks
Americans have consumed approximately 3,500 mg of sodium per day for at least the last half-century, down from intakes approaching 20 grams per day before the widespread use of refrigeration for food storage [5, 64]. In humans, daily sodium intakes less than 3,000 mg result in significant elevations in RAS activity [2]. Despite this, the USDA and Institute of Medicine have recommended that the adult population reduce daily sodium intake to 2,300 mg (or 1,500 mg for salt-sensitive hypertensives) in an attempt to reduce hypertension and related disorders. As hypothesized by Satin [64], should we expect a robust elevation in RAS-dependent pathologies (including metabolic disorders) across our population as these dietary changes are forced upon us? Clearly, understanding the role of the RAS in metabolic regulation is more important than ever.
Acknowledgments
This work was supported by research grants from the US National Institutes of Health including HL098276 to JLG, HL014388 to KR, HL084207 to CDS and KR, and HL048058, and HL061446 to CDS. KR was also supported by a grant (1-11-BS-127) from the American Diabetes Association. The authors also gratefully acknowledge the generous support of the Roy J. Carver Trust.
References
- 1.Achard V, Boullu-Ciocca S, Desbriere R, Nguyen G, Grino M. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2007;292:R274–R282. doi: 10.1152/ajpregu.00439.2005. [DOI] [PubMed] [Google Scholar]
- 2.Alderman MH, Madhavan S, Ooi WL, Cohen H, Sealey JE, Laragh JH. Association of the renin-sodium profile with the risk of myocardial infarction in patients with hypertension. N Engl J Med. 1991;324:1098–1104. doi: 10.1056/NEJM199104183241605. [DOI] [PubMed] [Google Scholar]
- 3.Alligier M, Meugnier E, Debard C, Lambert-Porcheron S, Chanseaume E, Sothier M, Loizon E, Ait HA, Brozek J, Scoazec JY, Morio B, Vidal H, Laville M. Subcutaneous Adipose Tissue Remodeling during the Initial Phase of Weight Gain Induced by Overfeeding in Humans. J Clin Endocrinol Metab. 2011 doi: 10.1210/jc.2011-2314. [DOI] [PubMed] [Google Scholar]
- 4.American Diabates Association. Economic costs of diabetes in the U.S. In 2007. Diabetes Care. 2008;31:596–615. doi: 10.2337/dc08-9017. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein AM, Willett WC. Trends in 24-h urinary sodium excretion in the United States, 1957–2003: a systematic review. Am J Clin Nutr. 2010;92:1172–1180. doi: 10.3945/ajcn.2010.29367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloem LJ, Manatunga AK, Tewksbury DA, Pratt JH. The serum angiotensinogen concentration and variants of the angiotensinogen gene in white and black children. J Clin Invest. 1995;95:948–953. doi: 10.1172/JCI117803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boschmann M, Ringel J, Klaus S, Sharma AM. Metabolic and hemodynamic response of adipose tissue to angiotensin II. Obes Res. 2001;9:486–491. doi: 10.1038/oby.2001.63. [DOI] [PubMed] [Google Scholar]
- 8.Brink M, Price SR, Chrast J, Bailey JL, Anwar A, Mitch WE, Delafontaine P. Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology. 2001;142:1489–1496. doi: 10.1210/endo.142.4.8082. [DOI] [PubMed] [Google Scholar]
- 9.Carman WJ, Barrett-Connor E, Sowers M, Khaw KT. Higher risk of cardiovascular mortality among lean hypertensive individuals in Tecumseh, Michigan. Circulation. 1994;89:703–711. doi: 10.1161/01.cir.89.2.703. [DOI] [PubMed] [Google Scholar]
- 10.Carter CS, Cesari M, Ambrosius WT, Hu N, Diz D, Oden S, Sonntag WE, Pahor M. Angiotensin-converting enzyme inhibition, body composition, and physical performance in aged rats. J Gerontol A Biol Sci Med Sci. 2004;59:416–423. doi: 10.1093/gerona/59.5.b416. [DOI] [PubMed] [Google Scholar]
- 11.Cassis L, Helton M, English V, Burke G. Angiotensin II regulates oxygen consumption. Am J Physiol Regul Integr Comp Physiol. 2002;282:R445–R453. doi: 10.1152/ajpregu.00261.2001. [DOI] [PubMed] [Google Scholar]
- 12.Cassis LA, Dwoskin LP. Presynaptic modulation of neurotransmitter release by endogenous angiotensin II in brown adipose tissue. J Neural Transm Suppl. 1991;34:129–137. doi: 10.1007/978-3-7091-9175-0_17. [DOI] [PubMed] [Google Scholar]
- 13.Chou CL, Lai YH, Lin TY, Lee TJ, Fang TC. Aliskiren prevents and ameliorates metabolic syndrome in fructose-fed rats. Arch Med Sci. 2011;7:882–888. doi: 10.5114/aoms.2011.25566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole BK, Keller SR, Wu R, Carter JD, Nadler JL, Nunemaker CS. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension. 2010;55:715–721. doi: 10.1161/HYPERTENSIONAHA.109.148049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper R, McFarlane-Anderson N, Bennett FI, Wilks R, Puras A, Tewksbury D, Ward R, Forrester T. ACE, angiotensinogen and obesity: a potential pathway leading to hypertension. J Hum Hypertens. 1997;11:107–111. doi: 10.1038/sj.jhh.1000391. [DOI] [PubMed] [Google Scholar]
- 16.Crandall DL, Armellino DC, Busler DE, McHendry-Rinde B, Kral JG. Angiotensin II receptors in human preadipocytes: role in cell cycle regulation. Endocrinology. 1999;140:154–158. doi: 10.1210/endo.140.1.6430. [DOI] [PubMed] [Google Scholar]
- 17.Darimont C, Vassaux G, Ailhaud G, Negrel R. Differentiation of preadipose cells: paracrine role of prostacyclin upon stimulation of adipose cells by angiotensin-II. Endocrinology. 1994;135:2030–2036. doi: 10.1210/endo.135.5.7956925. [DOI] [PubMed] [Google Scholar]
- 18.de Kloet AD, Krause EG, Scott KA, Foster MT, Herman JP, Sakai RR, Seeley RJ, Woods SC. Central angiotensin II has catabolic action at white and brown adipose tissue. Am J Physiol Endocrinol Metab. 2011;301:E1081–E1091. doi: 10.1152/ajpendo.00307.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, Luft FC, Sharma AM. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45:356–362. doi: 10.1161/01.HYP.0000154361.47683.d3. [DOI] [PubMed] [Google Scholar]
- 20.English V, Cassis L. Facilitation of sympathetic neurotransmission contributes to angiotensin regulation of body weight. J Neural Transm. 1999;106:631–644. doi: 10.1007/s007020050185. [DOI] [PubMed] [Google Scholar]
- 21.Ernsberger P, Koletsky RJ. Metabolic effects of antihypertensive agents: role of sympathoadrenal and renin-angiotensin systems. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:245–258. doi: 10.1007/s00210-006-0080-3. [DOI] [PubMed] [Google Scholar]
- 22.Faloia E, Gatti C, Camilloni MA, Mariniello B, Sardu C, Garrapa GG, Mantero F, Giacchetti G. Comparison of circulating and local adipose tissue renin-angiotensin system in normotensive and hypertensive obese subjects. J Endocrinol Invest. 2002;25:309–314. doi: 10.1007/BF03344010. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson AV, Washburn DL, Latchford KJ. Hormonal and neurotransmitter roles for angiotensin in the regulation of central autonomic function. Exp Biol Med (Maywood) 2001;226:85–96. doi: 10.1177/153537020122600205. [DOI] [PubMed] [Google Scholar]
- 24.Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA. 2005;293:1861–1867. doi: 10.1001/jama.293.15.1861. [DOI] [PubMed] [Google Scholar]
- 25.Fleming I, Kohlstedt K, Busse R. The tissue renin-angiotensin system and intracellular signalling. Curr Opin Nephrol Hypertens. 2006;15:8–13. doi: 10.1097/01.mnh.0000196146.65330.ea. [DOI] [PubMed] [Google Scholar]
- 26.Fowler JD, Krueth SB, Bernlohr DA, Katz SA. Renin dynamics in adipose tissue: adipose tissue control of local renin concentrations. Am J Physiol Endocrinol Metab. 2009;296:E343–E350. doi: 10.1152/ajpendo.90693.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galvez-Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S, Ruiz-Gayo M, Huber M, Wehland M, Kreutz R, Fernandez-Alfonso MS. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol. 2008;197:55–64. doi: 10.1677/JOE-07-0284. [DOI] [PubMed] [Google Scholar]
- 28.Giacchetti G, Faloia E, Mariniello B, Sardu C, Gatti C, Camilloni MA, Guerrieri M, Mantero F. Overexpression of the renin-angiotensin system in human visceral adipose tissue in normal and overweight subjects. Am J Hypertens. 2002;15:381–388. doi: 10.1016/s0895-7061(02)02257-4. [DOI] [PubMed] [Google Scholar]
- 29.Goossens GH, Blaak EE, van Baak MA. Possible involvement of the adipose tissue renin-angiotensin system in the pathophysiology of obesity and obesity-related disorders. Obes Rev. 2003;4:43–55. doi: 10.1046/j.1467-789x.2003.00091.x. [DOI] [PubMed] [Google Scholar]
- 30.Gratze P, Boschmann M, Dechend R, Qadri F, Malchow J, Graeske S, Engeli S, Janke J, Springer J, Contrepas A, Plehm R, Klaus S, Nguyen G, Luft FC, Muller DN. Energy metabolism in human renin-gene transgenic rats: does renin contribute to obesity? Hypertension. 2009;53:516–523. doi: 10.1161/HYPERTENSIONAHA.108.124966. [DOI] [PubMed] [Google Scholar]
- 31.Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D, Sigmund CD. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension. 2011;57:600–607. doi: 10.1161/HYPERTENSIONAHA.110.165829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, de Lange WJ, Li H, Sakai K, Thedens DR, Cassis LA, Rahmouni K, Mark AL, Johnson AK, Sigmund CD. The brain Renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab. 2010;12:431–442. doi: 10.1016/j.cmet.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 2008;23:187–193. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammond RA, LeVine R. The economic impact of obesity in the United States. Diabetes Metab Syndr Obes. 2010;3:285–295. doi: 10.2147/DMSOTT.S7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Idelevich E, Kirch W, Schindler C. Current pharmacotherapeutic concepts for the treatment of obesity in adults. Ther Adv Cardiovasc Dis. 2009;3:75–90. doi: 10.1177/1753944708098226. [DOI] [PubMed] [Google Scholar]
- 36.Itaya Y, Suzuki H, Matsukawa S, Kondo K, Saruta T. Central renin-angiotensin system and the pathogenesis of DOCA-salt hypertension in rats. Am J Physiol. 1986;251:H261–H268. doi: 10.1152/ajpheart.1986.251.2.H261. [DOI] [PubMed] [Google Scholar]
- 37.Iwai M, Tomono Y, Inaba S, Kanno H, Senba I, Mogi M, Horiuchi M. AT2 receptor deficiency attenuates adipocyte differentiation and decreases adipocyte number in atherosclerotic mice. Am J Hypertens. 2009;22:784–791. doi: 10.1038/ajh.2009.85. [DOI] [PubMed] [Google Scholar]
- 38.Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Mature adipocytes inhibit in vitro differentiation of human preadipocytes via angiotensin type 1 receptors. Diabetes. 2002;51:1699–1707. doi: 10.2337/diabetes.51.6.1699. [DOI] [PubMed] [Google Scholar]
- 39.Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, Egan GF, McKinley MJ, Rodger PD, Sinclair AJ, Wark JD, Weisinger HS, Jois M, Weisinger RS. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci U S A. 2008;105:6531–6536. doi: 10.1073/pnas.0802690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karlsson C, Lindell K, Ottosson M, Sjostrom L, Carlsson B, Carlsson LM. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J Clin Endocrinol Metab. 1998;83:3925–3929. doi: 10.1210/jcem.83.11.5276. [DOI] [PubMed] [Google Scholar]
- 41.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 42.Kouyama R, Suganami T, Nishida J, Tanaka M, Toyoda T, Kiso M, Chiwata T, Miyamoto Y, Yoshimasa Y, Fukamizu A, Horiuchi M, Hirata Y, Ogawa Y. Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a receptor. Endocrinology. 2005;146:3481–3489. doi: 10.1210/en.2005-0003. [DOI] [PubMed] [Google Scholar]
- 43.Krysiak R, Sierant M, Marek B, Bienek R, Okopien B. The effect of angiotensin-converting enzyme inhibitors on plasma adipokine levels in normotensive patients with coronary artery disease. Endokrynol Pol. 2010;61:280–287. [PubMed] [Google Scholar]
- 44.Kubo T, Yamaguchi H, Tsujimura M, Hagiwara Y, Fukumori R. Blockade of angiotensin receptors in the anterior hypothalamic preoptic area lowers blood pressure in DOCA-salt hypertensive rats. Hypertens Res. 2000;23:109–118. doi: 10.1291/hypres.23.109. [DOI] [PubMed] [Google Scholar]
- 45.Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology. 2003;144:2179–2183. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- 46.Lee-Kirsch MA, Gaudet F, Cardoso MC, Lindpaintner K. Distinct renin isoforms generated by tissue-specific transcription initiation and alternative splicing. Circ Res. 1999;84:240–246. doi: 10.1161/01.res.84.2.240. [DOI] [PubMed] [Google Scholar]
- 47.Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15:2727–2729. doi: 10.1096/fj.01-0457fje. [DOI] [PubMed] [Google Scholar]
- 48.Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, Fukamizu A, Negrel R, Ailhaud G, Teboul M. Angiotensinogen-deficient mice exhibit impairment of diet-induced weight gain with alteration in adipose tissue development and increased locomotor activity. Endocrinology. 2001;142:5220–5225. doi: 10.1210/endo.142.12.8556. [DOI] [PubMed] [Google Scholar]
- 49.Masuo K, Mikami H, Ogihara T, Tuck ML. Weight reduction and pharmacologic treatment in obese hypertensives. Am J Hypertens. 2001;14:530–538. doi: 10.1016/s0895-7061(00)01279-6. [DOI] [PubMed] [Google Scholar]
- 50.Mathai ML, Chen N, Cornall L, Weisinger RS. The role of Angiotensin in obesity and metabolic disease. Endocr Metab Immune Disord Drug Targets. 2011;11:198–205. doi: 10.2174/187153011796429853. [DOI] [PubMed] [Google Scholar]
- 51.Mathai ML, Naik S, Sinclair AJ, Weisinger HS, Weisinger RS. Selective reduction in body fat mass and plasma leptin induced by angiotensin-converting enzyme inhibition in rats. Int J Obes (Lond) 2008;32:1576–1584. doi: 10.1038/ijo.2008.126. [DOI] [PubMed] [Google Scholar]
- 52.Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension. 2006;48:1095–1102. doi: 10.1161/01.HYP.0000248211.82232.a7. [DOI] [PubMed] [Google Scholar]
- 53.McClendon KS, Riche DM, Uwaifo GI. Orlistat: current status in clinical therapeutics. Expert Opin Drug Saf. 2009;8:727–744. doi: 10.1517/14740330903321485. [DOI] [PubMed] [Google Scholar]
- 54.Messerli FH, Nunez BD, Ventura HO, Snyder DW. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch Intern Med. 1987;147:1725–1728. doi: 10.1001/archinte.147.10.1725. [DOI] [PubMed] [Google Scholar]
- 55.Mogi M, Iwai M, Horiuchi M. Emerging concept of adipogenesis regulation by the renin-angiotensin system. Hypertension. 2006;48:1020–1022. doi: 10.1161/01.HYP.0000248196.14826.31. [DOI] [PubMed] [Google Scholar]
- 56.Morimoto S, Cassell MD, Sigmund CD. Glial- and neuronal-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277:33235–33241. doi: 10.1074/jbc.M204309200. [DOI] [PubMed] [Google Scholar]
- 57.Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp Physiol. 2008;93:773–797. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park CG, Leenen FH. Effects of centrally administered losartan on deoxycorticosterone-salt hypertension rats. J Korean Med Sci. 2001;16:553–557. doi: 10.3346/jkms.2001.16.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porter JP, Anderson JM, Robison RJ, Phillips AC. Effect of central angiotensin II on body weight gain in young rats. Brain Res. 2003;959:20–28. doi: 10.1016/s0006-8993(02)03676-4. [DOI] [PubMed] [Google Scholar]
- 60.Porter JP, Potratz KR. Effect of intracerebroventricular angiotensin II on body weight and food intake in adult rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R422–R428. doi: 10.1152/ajpregu.00537.2003. [DOI] [PubMed] [Google Scholar]
- 61.Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL, Sigmund CD. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest. 2007;117:1088–1095. doi: 10.1172/JCI31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santos EL, de Picoli SK, Guimaraes PB, Reis FC, Silva SM, Costa-Neto CM, Luz J, Pesquero JB. Effect of angiotensin converting enzyme inhibitor enalapril on body weight and composition in young rats. Int Immunopharmacol. 2008;8:247–253. doi: 10.1016/j.intimp.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 63.Santos SH, Fernandes LR, Mario EG, Ferreira AV, Porto LC, Alvarez-Leite JI, Botion LM, Bader M, Alenina N, Santos RA. Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes. 2008;57:340–347. doi: 10.2337/db07-0953. [DOI] [PubMed] [Google Scholar]
- 64.Satin M. The salt debate--more salacious than salubrious. Nutrition. 2011;27:759–760. doi: 10.1016/j.nut.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 65.Saye JA, Ragsdale NV, Carey RM, Peach MJ. Localization of angiotensin peptide-forming enzymes of 3T3-F442A adipocytes. Am J Physiol. 1993;264:C1570–C1576. doi: 10.1152/ajpcell.1993.264.6.C1570. [DOI] [PubMed] [Google Scholar]
- 66.Sinn PL, Sigmund CD. Identification of three human renin mRNA isoforms from alternative tissue-specific transcriptional initiation. Physiol Genomics. 2000;3:25–31. doi: 10.1152/physiolgenomics.2000.3.1.25. [DOI] [PubMed] [Google Scholar]
- 67.Stec DE, Davisson RL, Haskell RE, Davidson BL, Sigmund CD. Efficient liver-specific deletion of a floxed human angiotensinogen transgene by adenoviral delivery of cre-recombinase in vivo. J Biol Chem. 1999;274:21285–21290. doi: 10.1074/jbc.274.30.21285. [DOI] [PubMed] [Google Scholar]
- 68.Stec DE, Keen HL, Sigmund CD. Lower blood pressure in floxed angiotensinogen mice after adenoviral delivery of cre-recombinase. Hypertension. 2002;39:629–633. doi: 10.1161/hy0202.103418. [DOI] [PubMed] [Google Scholar]
- 69.Stucchi P, Cano V, Ruiz-Gayo M, Fernandez-Alfonso MS. Aliskiren reduces body-weight gain, adiposity and plasma leptin during diet-induced obesity. Br J Pharmacol. 2009;158:771–778. doi: 10.1111/j.1476-5381.2009.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sui Y, Zhao HL, Fan RR, Guan J, He L, Lee HM, Chan JC, Tong PC. Renin-angiotensin system activation in renal adipogenesis. Am J Physiol Renal Physiol. 2010;298:F391–F400. doi: 10.1152/ajprenal.00445.2009. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi N, Li F, Hua K, Deng J, Wang CH, Bowers RR, Bartness TJ, Kim HS, Harp JB. Increased energy expenditure, dietary fat wasting, and resistance to diet-induced obesity in mice lacking renin. Cell Metab. 2007;6:506–512. doi: 10.1016/j.cmet.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thatcher S, Yiannikouris F, Gupte M, Cassis L. The adipose renin-angiotensin system: role in cardiovascular disease. Mol Cell Endocrinol. 2009;302:111–117. doi: 10.1016/j.mce.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vetter ML, Faulconbridge LF, Webb VL, Wadden TA. Behavioral and pharmacologic therapies for obesity. Nat Rev Endocrinol. 2010;6:578–588. doi: 10.1038/nrendo.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang P, Holst C, Wodzig WK, Andersen MR, Astrup A, van Baak MA, Larsen TM, Jebb SA, Kafatos A, Pfeiffer AF, Martinez JA, Handjieva-Darlenska T, Kunesova M, Viguerie N, Langin D, Saris WH, Mariman EC. Circulating ACE is a predictor of weight loss maintenance not only in overweight and obese women, but also in men. Int J Obes (Lond) 2012 doi: 10.1038/ijo.2011.278. [DOI] [PubMed] [Google Scholar]
- 75.Weiland F, Verspohl EJ. Variety of angiotensin receptors in 3T3-L1 preadipose cells and differentiated adipocytes. Horm Metab Res. 2008;40:760–766. doi: 10.1055/s-0028-1082041. [DOI] [PubMed] [Google Scholar]
- 76.Weisinger RS, Stanley TK, Begg DP, Weisinger HS, Spark KJ, Jois M. Angiotensin converting enzyme inhibition lowers body weight and improves glucose tolerance in C57BL/6J mice maintained on a high fat diet. Physiol Behav. 2009;98:192–197. doi: 10.1016/j.physbeh.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 77.Xu D, Borges GR, Davis DR, Agassandian K, Sequeira Lopez ML, Gomez RA, Cassell MD, Grobe JL, Sigmund CD. Neuron- or Glial-Specific Ablation of Secreted Renin Does Not Affect Renal Renin, Baseline Arterial Pressure or Metabolism. Physiol Genomics. 2011;43:286–294. doi: 10.1152/physiolgenomics.00208.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu D, Borges GR, Grobe JL, Pelham CJ, Yang B, Sigmund CD. Preservation of intracellular renin expression is insufficient to compensate for genetic loss of secreted renin. Hypertension. 2009;54:1240–1247. doi: 10.1161/HYPERTENSIONAHA.109.138677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang G, Merrill DC, Thompson MW, Robillard JE, Sigmund CD. Functional expression of the human angiotensinogen gene in transgenic mice. J Biol Chem. 1994;269:32497–32502. [PubMed] [Google Scholar]
- 80.Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, Cassis LA. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302:R244–R251. doi: 10.1152/ajpregu.00323.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida T, Semprun-Prieto L, Wainford RD, Sukhanov S, Kapusta DR, Delafontaine P. Angiotensin II Reduces Food Intake by Altering Orexigenic Neuropeptide Expression in the Mouse Hypothalamus. Endocrinology. 2012 doi: 10.1210/en.2011–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yvan-Charvet L, Even P, Bloch-Faure M, Guerre-Millo M, Moustaid-Moussa N, Ferre P, Quignard-Boulange A. Deletion of the angiotensin type 2 receptor (AT2R) reduces adipose cell size and protects from diet-induced obesity and insulin resistance. Diabetes. 2005;54:991–999. doi: 10.2337/diabetes.54.4.991. [DOI] [PubMed] [Google Scholar]
- 83.Yvan-Charvet L, Massiera F, Lamande N, Ailhaud G, Teboul M, Moustaid-Moussa N, Gasc JM, Quignard-Boulange A. Deficiency of angiotensin type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinology. 2009;150:1421–1428. doi: 10.1210/en.2008-1120. [DOI] [PubMed] [Google Scholar]
- 84.Yvan-Charvet L, Quignard-Boulange A. Role of adipose tissue renin-angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney Int. 2011;79:162–168. doi: 10.1038/ki.2010.391. [DOI] [PubMed] [Google Scholar]
- 85.Zanchi A, Dulloo AG, Perregaux C, Montani JP, Burnier M. Telmisartan prevents the glitazone-induced weight gain without interfering with its insulin-sensitizing properties. Am J Physiol Endocrinol Metab. 2007;293:E91–E95. doi: 10.1152/ajpendo.00024.2007. [DOI] [PubMed] [Google Scholar]
- 86.Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, Saavedra JM. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol. 2006;552:112–122. doi: 10.1016/j.ejphar.2006.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]